
Bile Acid–Gut Microbiota Axis in Inflammatory Bowel Disease: From Bench to Bedside

 ,
,
Abstract
:1. Introduction
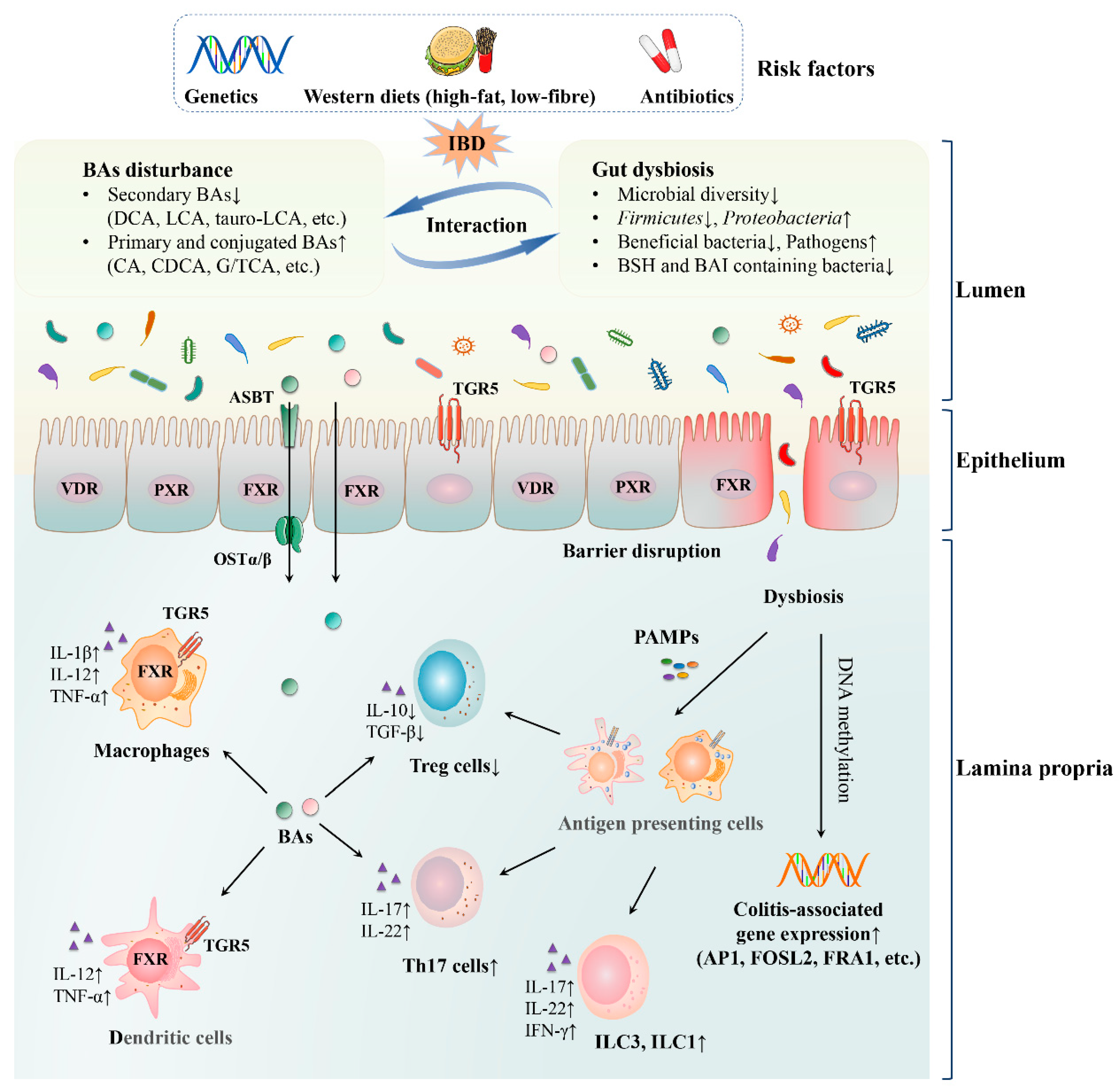
2. Bile Acid–Gut Microbiota Axis in IBD
2.1. Gut Dysbiosis in IBD
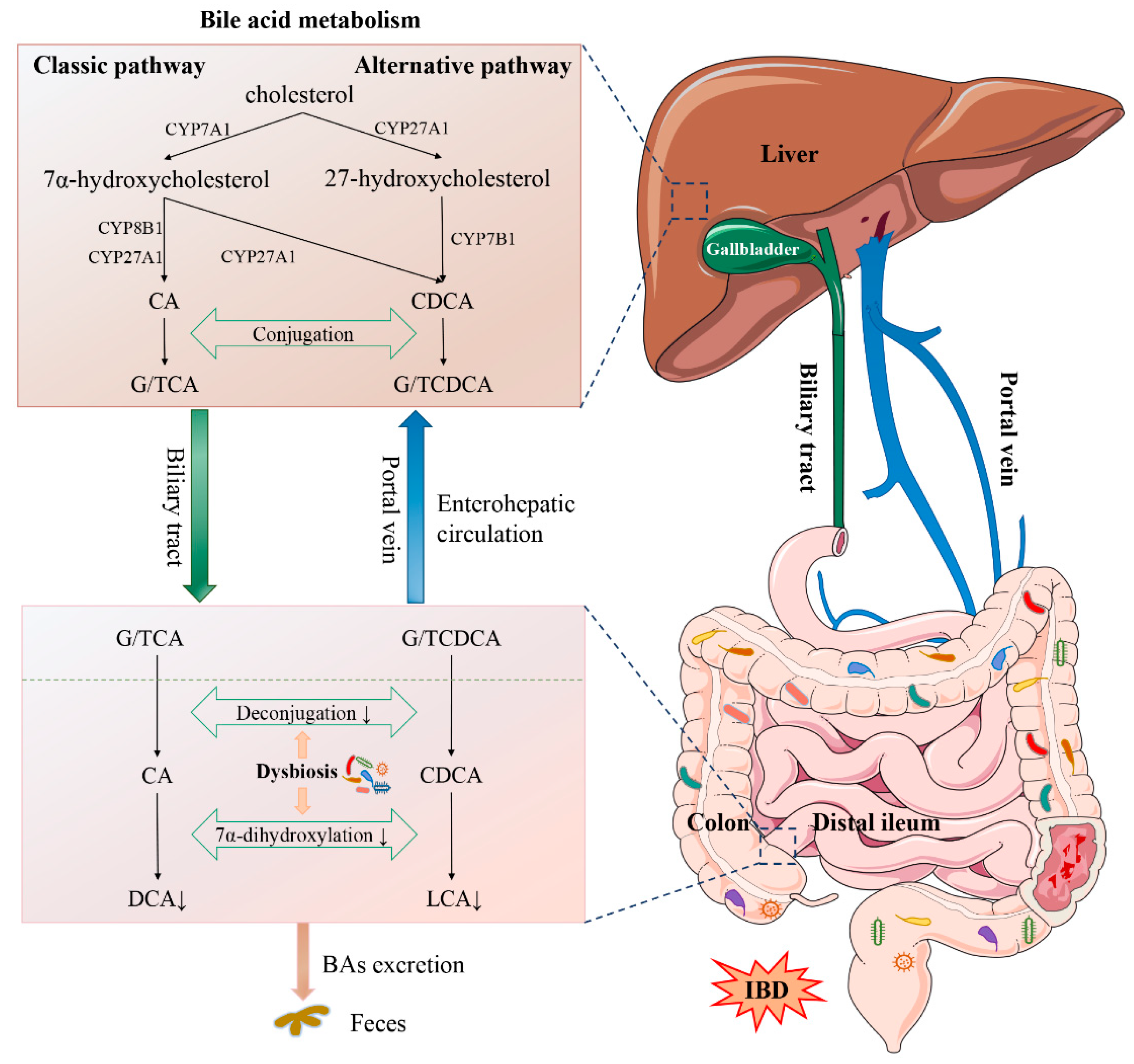
2.2. Bile Acid Synthesis and Metabolism
2.3. Microbial Modulation of Bile Acid Synthesis and Metabolism
2.4. Bile Acids Influence the Composition of Gut Microbiota
2.5. Bile Acid-Activated Receptors
2.5.1. FXR
2.5.2. TGR5
2.5.3. PXR
2.5.4. VDR
3. Therapeutic Target of Bile Acid–Gut Microbiota Axis for IBD
3.1. Dietary Therapy
3.2. Probiotics and Prebiotics
3.3. Engineered Bacteria
3.4. Fecal Microbiota Transplantation (FMT)
3.5. UDCA
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2018, 390, 2769–2778. [Google Scholar] [CrossRef]
- Nadeem, M.S.; Kumar, V.; Al-Abbasi, F.A.; Kamal, M.A.; Anwar, F. Risk of colorectal cancer in inflammatory bowel diseases. Semin. Cancer Biol. 2020, 64, 51–60. [Google Scholar] [CrossRef]
- Shouval, D.S.; Rufo, P.A. The Role of Environmental Factors in the Pathogenesis of Inflammatory Bowel Diseases: A Review. JAMA Pediatr. 2017, 171, 999–1005. [Google Scholar] [CrossRef]
- Kaplan, G.G.; Ng, S.C. Understanding and Preventing the Global Increase of Inflammatory Bowel Disease. Gastroenterology 2017, 152, 313–321. [Google Scholar] [CrossRef] [Green Version]
- Weingarden, A.R.; Vaughn, B.P. Intestinal microbiota, fecal microbiota transplantation, and inflammatory bowel disease. Gut Microbes 2017, 8, 238–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, X.; Sun, X.; Oh, S.F.; Wu, M.; Zhang, Y.; Zheng, W.; Geva-Zatorsky, N.; Jupp, R.; Mathis, D.; Benoist, C.; et al. Microbial bile acid metabolites modulate gut RORγ regulatory T cell homeostasis. Nature 2020, 577, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Xie, G.; Jia, W. Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 111–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gérard, P. Metabolism of cholesterol and bile acids by the gut microbiota. Pathogens 2013, 3, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Duboc, H.; Rajca, S.; Rainteau, D.; Benarous, D.; Maubert, M.A.; Quervain, E.; Thomas, G.; Barbu, V.; Humbert, L.; Despras, G.; et al. Connecting dysbiosis, bile-acid dysmetabolism and gut inflammation in inflammatory bowel diseases. Gut 2013, 62, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.R.; Haileselassie, Y.; Nguyen, L.P.; Tropini, C.; Wang, M.; Becker, L.S.; Sim, D.; Jarr, K.; Spear, E.T.; Singh, G.; et al. Dysbiosis-Induced Secondary Bile Acid Deficiency Promotes Intestinal Inflammation. Cell Host Microbe 2020, 27, 659–670. [Google Scholar] [CrossRef]
- Wohlgemuth, S.; Keller, S.; Kertscher, R.; Stadion, M.; Haller, D.; Kisling, S.; Jahreis, G.; Blaut, M.; Loh, G. Intestinal steroid profiles and microbiota composition in colitic mice. Gut Microbes 2011, 2, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Van den Bossche, L.; Hindryckx, P.; Devisscher, L.; Devriese, S.; Van Welden, S.; Holvoet, T.; Vilchez-Vargas, R.; Vital, M.; Pieper, D.H.; Vanden Bussche, J.; et al. Ursodeoxycholic Acid and Its Taurine- or Glycine-Conjugated Species Reduce Colitogenic Dysbiosis and Equally Suppress Experimental Colitis in Mice. Appl. Environ. Microbiol. 2017, 83, e02766-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glassner, K.L.; Abraham, B.P.; Quigley, E.M.M. The microbiome and inflammatory bowel disease. J. Allergy Clin. Immunol. 2020, 145, 16–27. [Google Scholar] [CrossRef] [Green Version]
- Pascal, V.; Pozuelo, M.; Borruel, N.; Casellas, F.; Campos, D.; Santiago, A.; Martinez, X.; Varela, E.; Sarrabayrouse, G.; Machiels, K.; et al. A microbial signature for Crohn’s disease. Gut 2017, 66, 813–822. [Google Scholar] [CrossRef]
- Franzosa, E.A.; Sirota-Madi, A.; Avila-Pacheco, J.; Fornelos, N.; Haiser, H.J.; Reinker, S.; Vatanen, T.; Hall, A.B.; Mallick, H.; McIver, L.J.; et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 2019, 4, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.P.; Goudarzi, M.; Singh, N.; Tong, M.; McHardy, I.H.; Ruegger, P.; Asadourian, M.; Moon, B.-H.; Ayson, A.; Borneman, J.; et al. A Disease-Associated Microbial and Metabolomics State in Relatives of Pediatric Inflammatory Bowel Disease Patients. Cell. Mol. Gastroenterol. Hepatol. 2016, 2, 750–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gevers, D.; Kugathasan, S.; Denson, L.A.; Vázquez-Baeza, Y.; Van Treuren, W.; Ren, B.; Schwager, E.; Knights, D.; Song, S.J.; Yassour, M.; et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 2014, 15, 382–392. [Google Scholar] [CrossRef] [Green Version]
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E.; Ajami, N.J.; Bonham, K.S.; Brislawn, C.J.; et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, X.; Zhang, X.; Xiao, F.; Hu, H.; Li, X.; Dong, F.; Sun, M.; Xiao, Y.; Ge, T.; et al. Microbial and metabolic features associated with outcome of infliximab therapy in pediatric Crohn’s disease. Gut Microbes 2021, 13. [Google Scholar] [CrossRef]
- Machiels, K.; Joossens, M.; Sabino, J.; De Preter, V.; Arijs, I.; Eeckhaut, V.; Ballet, V.; Claes, K.; Van Immerseel, F.; Verbeke, K.; et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut 2014, 63, 1275–1283. [Google Scholar] [CrossRef]
- Knoll, R.L.; Forslund, K.; Kultima, J.R.; Meyer, C.U.; Kullmer, U.; Sunagawa, S.; Bork, P.; Gehring, S. Gut microbiota differs between children with Inflammatory Bowel Disease and healthy siblings in taxonomic and functional composition: A metagenomic analysis. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G327–G339. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.-H.; Liu, F.; Zhu, X.-R.; Suo, F.-Y.; Jia, Z.-J.; Yao, S.-K. Altered profiles of fecal bile acids correlate with gut microbiota and inflammatory responses in patients with ulcerative colitis. World J. Gastroenterol. 2021, 27, 3609–3629. [Google Scholar] [CrossRef]
- Ansari, I.; Raddatz, G.; Gutekunst, J.; Ridnik, M.; Cohen, D.; Abu-Remaileh, M.; Tuganbaev, T.; Shapiro, H.; Pikarsky, E.; Elinav, E.; et al. The microbiota programs DNA methylation to control intestinal homeostasis and inflammation. Nat. Microbiol. 2020, 5, 610–619. [Google Scholar] [CrossRef]
- Kelly, C.J.; Zheng, L.; Campbell, E.L.; Saeedi, B.; Scholz, C.C.; Bayless, A.J.; Wilson, K.E.; Glover, L.E.; Kominsky, D.J.; Magnuson, A.; et al. Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host Microbe 2015, 17, 662–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Gury-BenAri, M.; Thaiss, C.A.; Serafini, N.; Winter, D.R.; Giladi, A.; Lara-Astiaso, D.; Levy, M.; Salame, T.M.; Weiner, A.; David, E.; et al. The Spectrum and Regulatory Landscape of Intestinal Innate Lymphoid Cells Are Shaped by the Microbiome. Cell 2016, 166, 1231–1246. [Google Scholar] [CrossRef] [PubMed]
- Castellanos, J.G.; Longman, R.S. Innate lymphoid cells link gut microbes with mucosal T cell immunity. Gut Microbes 2020, 11, 231–236. [Google Scholar] [CrossRef]
- Bernink, J.H.; Krabbendam, L.; Germar, K.; de Jong, E.; Gronke, K.; Kofoed-Nielsen, M.; Munneke, J.M.; Hazenberg, M.D.; Villaudy, J.; Buskens, C.J.; et al. Interleukin-12 and -23 Control Plasticity of CD127(+) Group 1 and Group 3 Innate Lymphoid Cells in the Intestinal Lamina Propria. Immunity 2015, 43, 146–160. [Google Scholar] [CrossRef] [Green Version]
- Zeng, B.; Shi, S.; Ashworth, G.; Dong, C.; Liu, J.; Xing, F. ILC3 function as a double-edged sword in inflammatory bowel diseases. Cell Death Dis. 2019, 10, 315. [Google Scholar] [CrossRef] [Green Version]
- Britton, G.J.; Contijoch, E.J.; Mogno, I.; Vennaro, O.H.; Llewellyn, S.R.; Ng, R.; Li, Z.; Mortha, A.; Merad, M.; Das, A.; et al. Microbiotas from Humans with Inflammatory Bowel Disease Alter the Balance of Gut Th17 and RORγt Regulatory T Cells and Exacerbate Colitis in Mice. Immunity 2019, 50, 212–224. [Google Scholar] [CrossRef] [Green Version]
- Neurath, M.F. Targeting immune cell circuits and trafficking in inflammatory bowel disease. Nat. Immunol. 2019, 20, 970–979. [Google Scholar] [CrossRef] [PubMed]
- Devkota, S.; Wang, Y.; Musch, M.W.; Leone, V.; Fehlner-Peach, H.; Nadimpalli, A.; Antonopoulos, D.A.; Jabri, B.; Chang, E.B. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10−/− mice. Nature 2012, 487, 104–108. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.L.; Takeda, K.; Sundrud, M.S. Emerging roles of bile acids in mucosal immunity and inflammation. Mucosal Immunol. 2019, 12, 851–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayin, S.I.; Wahlström, A.; Felin, J.; Jäntti, S.; Marschall, H.-U.; Bamberg, K.; Angelin, B.; Hyötyläinen, T.; Orešič, M.; Bäckhed, F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013, 17, 225–235. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.; Cai, Y.; Lao, X.; Wang, X.; Lin, X.; Cui, Y.; Kalavagunta, P.K.; Liao, J.; Jin, L.; Shang, J.; et al. Taxonomic profiling and populational patterns of bacterial bile salt hydrolase (BSH) genes based on worldwide human gut microbiome. Microbiome 2019, 7, 9. [Google Scholar] [CrossRef] [Green Version]
- Long, S.L.; Gahan, C.G.M.; Joyce, S.A. Interactions between gut bacteria and bile in health and disease. Mol. Asp. Med. 2017, 56, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Weng, Y.J.; Gan, H.Y.; Li, X.; Huang, Y.; Li, Z.C.; Deng, H.M.; Chen, S.Z.; Zhou, Y.; Wang, L.S.; Han, Y.P.; et al. Correlation of diet, microbiota and metabolite networks in inflammatory bowel disease. J. Dig. Dis. 2019, 20, 447–459. [Google Scholar] [CrossRef]
- Murakami, M.; Iwamoto, J.; Honda, A.; Tsuji, T.; Tamamushi, M.; Ueda, H.; Monma, T.; Konishi, N.; Yara, S.; Hirayama, T.; et al. Detection of Gut Dysbiosis due to Reduced Clostridium Subcluster XIVa Using the Fecal or Serum Bile Acid Profile. Inflamm. Bowel Dis. 2018, 24, 1035–1044. [Google Scholar] [CrossRef] [PubMed]
- Diederen, K.; Li, J.V.; Donachie, G.E.; de Meij, T.G.; de Waart, D.R.; Hakvoort, T.B.M.; Kindermann, A.; Wagner, J.; Auyeung, V.; Te Velde, A.A.; et al. Exclusive enteral nutrition mediates gut microbial and metabolic changes that are associated with remission in children with Crohn’s disease. Sci. Rep. 2020, 10, 18879. [Google Scholar] [CrossRef]
- Heinken, A.; Ravcheev, D.A.; Baldini, F.; Heirendt, L.; Fleming, R.M.T.; Thiele, I. Systematic assessment of secondary bile acid metabolism in gut microbes reveals distinct metabolic capabilities in inflammatory bowel disease. Microbiome 2019, 7, 75. [Google Scholar] [CrossRef]
- Hernández-Rocha, C.; Borowski, K.; Turpin, W.; Filice, M.; Nayeri, S.; Raygoza Garay, J.A.; Stempak, J.M.; Silverberg, M.S. Integrative analysis of colonic biopsies from inflammatory bowel disease patients identifies an interaction between microbial bile-acid inducible gene abundance and human Angiopoietin-like 4 gene expression. J. Crohn’s Colitis 2021. [Google Scholar] [CrossRef]
- Hang, S.; Paik, D.; Yao, L.; Kim, E.; Trinath, J.; Lu, J.; Ha, S.; Nelson, B.N.; Kelly, S.P.; Wu, L.; et al. Bile acid metabolites control T17 and T cell differentiation. Nature 2019, 576, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Fiorucci, S.; Biagioli, M.; Zampella, A.; Distrutti, E. Bile Acids Activated Receptors Regulate Innate Immunity. Front. Immunol. 2018, 9, 1853. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Shen, Y.; Cen, M.; Zhu, Y.; Cheng, F.; Tang, L.; Zheng, X.; Kim, J.J.; Dai, N.; Hu, W. Modulation of the Gut Microbiota-Farnesoid X Receptor Axis Improves Deoxycholic Acid-induced Intestinal Inflammation in Mice. J. Crohn’s Colitis 2021. [Google Scholar] [CrossRef]
- Islam, K.B.M.S.; Fukiya, S.; Hagio, M.; Fujii, N.; Ishizuka, S.; Ooka, T.; Ogura, Y.; Hayashi, T.; Yokota, A. Bile acid is a host factor that regulates the composition of the cecal microbiota in rats. Gastroenterology 2011, 141, 1773–1781. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, J.M.; Alves, J.M.; Hylemon, P.B.; Bajaj, J.S. Cirrhosis, bile acids and gut microbiota: Unraveling a complex relationship. Gut Microbes 2013, 4, 382–387. [Google Scholar] [CrossRef] [Green Version]
- Begley, M.; Gahan, C.G.M.; Hill, C. The interaction between bacteria and bile. FEMS Microbiol. Rev. 2005, 29, 625–651. [Google Scholar] [CrossRef] [Green Version]
- De Boever, P.; Wouters, R.; Verschaeve, L.; Berckmans, P.; Schoeters, G.; Verstraete, W. Protective effect of the bile salt hydrolase-active Lactobacillus reuteri against bile salt cytotoxicity. Appl. Microbiol. Biotechnol. 2000, 53, 709–714. [Google Scholar] [CrossRef] [PubMed]
- D’Aldebert, E.; Biyeyeme Bi Mve, M.-J.; Mergey, M.; Wendum, D.; Firrincieli, D.; Coilly, A.; Fouassier, L.; Corpechot, C.; Poupon, R.; Housset, C.; et al. Bile salts control the antimicrobial peptide cathelicidin through nuclear receptors in the human biliary epithelium. Gastroenterology 2009, 136, 1435–1443. [Google Scholar] [CrossRef]
- Inagaki, T.; Moschetta, A.; Lee, Y.-K.; Peng, L.; Zhao, G.; Downes, M.; Yu, R.T.; Shelton, J.M.; Richardson, J.A.; Repa, J.J.; et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 3920–3925. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Chiang, J.Y.L. Bile acid signaling in metabolic disease and drug therapy. Pharm. Rev. 2014, 66, 948–983. [Google Scholar] [CrossRef] [Green Version]
- Postler, T.S.; Ghosh, S. Understanding the Holobiont: How Microbial Metabolites Affect Human Health and Shape the Immune System. Cell Metab. 2017, 26, 110–130. [Google Scholar] [CrossRef] [Green Version]
- Fiorucci, S.; Distrutti, E. Bile Acid-Activated Receptors, Intestinal Microbiota, and the Treatment of Metabolic Disorders. Trends Mol. Med. 2015, 21, 702–714. [Google Scholar] [CrossRef]
- Li, F.; Jiang, C.; Krausz, K.W.; Li, Y.; Albert, I.; Hao, H.; Fabre, K.M.; Mitchell, J.B.; Patterson, A.D.; Gonzalez, F.J. Microbiome remodelling leads to inhibition of intestinal farnesoid X receptor signalling and decreased obesity. Nat. Commun. 2013, 4, 2384. [Google Scholar] [CrossRef]
- Manichanh, C.; Rigottier-Gois, L.; Bonnaud, E.; Gloux, K.; Pelletier, E.; Frangeul, L.; Nalin, R.; Jarrin, C.; Chardon, P.; Marteau, P.; et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut 2006, 55, 205–211. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Xie, C.; Nichols, R.G.; Chan, S.H.J.; Jiang, C.; Hao, R.; Smith, P.B.; Cai, J.; Simons, M.N.; Hatzakis, E.; et al. Farnesoid X Receptor Signaling Shapes the Gut Microbiota and Controls Hepatic Lipid Metabolism. mSystems 2016, 1, e00070-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadaleta, R.M.; van Erpecum, K.J.; Oldenburg, B.; Willemsen, E.C.; Renooij, W.; Murzilli, S.; Klomp, L.W.; Siersema, P.D.; Schipper, M.E.; Danese, S.; et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut 2011, 60, 463–472. [Google Scholar] [CrossRef]
- Vavassori, P.; Mencarelli, A.; Renga, B.; Distrutti, E.; Fiorucci, S. The bile acid receptor FXR is a modulator of intestinal innate immunity. J. Immunol. 2009, 183, 6251–6261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadaleta, R.M.; van Mil, S.W.C.; Oldenburg, B.; Siersema, P.D.; Klomp, L.W.J.; van Erpecum, K.J. Bile acids and their nuclear receptor FXR: Relevance for hepatobiliary and gastrointestinal disease. Biochim. Biophys. Acta 2010, 1801, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Gadaleta, R.M.; Garcia-Irigoyen, O.; Cariello, M.; Scialpi, N.; Peres, C.; Vetrano, S.; Fiorino, G.; Danese, S.; Ko, B.; Luo, J.; et al. Fibroblast Growth Factor 19 modulates intestinal microbiota and inflammation in presence of Farnesoid X Receptor. EBioMedicine 2020, 54, 102719. [Google Scholar] [CrossRef]
- Duboc, H.; Taché, Y.; Hofmann, A.F. The bile acid TGR5 membrane receptor: From basic research to clinical application. Dig. Liver Dis. 2014, 46, 302–312. [Google Scholar] [CrossRef] [Green Version]
- Cipriani, S.; Mencarelli, A.; Chini, M.G.; Distrutti, E.; Renga, B.; Bifulco, G.; Baldelli, F.; Donini, A.; Fiorucci, S. The bile acid receptor GPBAR-1 (TGR5) modulates integrity of intestinal barrier and immune response to experimental colitis. PLoS ONE 2011, 6, e25637. [Google Scholar] [CrossRef]
- Calmus, Y.; Poupon, R. Shaping macrophages function and innate immunity by bile acids: Mechanisms and implication in cholestatic liver diseases. Clin. Res. Hepatol. Gastroenterol. 2014, 38, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; He, C.; Cong, Y.; Liu, Z. Regulatory immune cells in regulation of intestinal inflammatory response to microbiota. Mucosal Immunol. 2015, 8, 969–978. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Haselow, K.; Bode, J.G.; Wammers, M.; Ehlting, C.; Keitel, V.; Kleinebrecht, L.; Schupp, A.-K.; Häussinger, D.; Graf, D. Bile acids PKA-dependently induce a switch of the IL-10/IL-12 ratio and reduce proinflammatory capability of human macrophages. J. Leukoc. Biol. 2013, 94, 1253–1264. [Google Scholar] [CrossRef] [PubMed]
- Yoneno, K.; Hisamatsu, T.; Shimamura, K.; Kamada, N.; Ichikawa, R.; Kitazume, M.T.; Mori, M.; Uo, M.; Namikawa, Y.; Matsuoka, K.; et al. TGR5 signalling inhibits the production of pro-inflammatory cytokines by in vitro differentiated inflammatory and intestinal macrophages in Crohn’s disease. Immunology 2013, 139, 19–29. [Google Scholar] [CrossRef] [Green Version]
- Ichikawa, R.; Takayama, T.; Yoneno, K.; Kamada, N.; Kitazume, M.T.; Higuchi, H.; Matsuoka, K.; Watanabe, M.; Itoh, H.; Kanai, T.; et al. Bile acids induce monocyte differentiation toward interleukin-12 hypo-producing dendritic cells via a TGR5-dependent pathway. Immunology 2012, 136, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Xie, S.; Chi, Z.; Zhang, J.; Liu, Y.; Zhang, L.; Zheng, M.; Zhang, X.; Xia, D.; Ke, Y.; et al. Bile Acids Control Inflammation and Metabolic Disorder through Inhibition of NLRP3 Inflammasome. Immunity 2016, 45, 802–816. [Google Scholar] [CrossRef] [Green Version]
- Oladimeji, P.O.; Chen, T. PXR: More Than Just a Master Xenobiotic Receptor. Mol. Pharm. 2018, 93, 119–127. [Google Scholar] [CrossRef]
- Dempsey, J.L.; Wang, D.; Siginir, G.; Fei, Q.; Raftery, D.; Gu, H.; Yue Cui, J. Pharmacological Activation of PXR and CAR Downregulates Distinct Bile Acid-Metabolizing Intestinal Bacteria and Alters Bile Acid Homeostasis. Toxicol. Sci. 2019, 168, 40–60. [Google Scholar] [CrossRef]
- Caparrós-Martín, J.A.; Lareu, R.R.; Ramsay, J.P.; Peplies, J.; Reen, F.J.; Headlam, H.A.; Ward, N.C.; Croft, K.D.; Newsholme, P.; Hughes, J.D.; et al. Statin therapy causes gut dysbiosis in mice through a PXR-dependent mechanism. Microbiome 2017, 5, 95. [Google Scholar] [CrossRef] [Green Version]
- Venkatesh, M.; Mukherjee, S.; Wang, H.; Li, H.; Sun, K.; Benechet, A.P.; Qiu, Z.; Maher, L.; Redinbo, M.R.; Phillips, R.S.; et al. Symbiotic bacterial metabolites regulate gastrointestinal barrier function via the xenobiotic sensor PXR and Toll-like receptor 4. Immunity 2014, 41, 296–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmann, T.; Moehle, C.; Mauerer, R.; Scharl, M.; Liebisch, G.; Zahn, A.; Stremmel, W.; Schmitz, G. Loss of detoxification in inflammatory bowel disease: Dysregulation of pregnane X receptor target genes. Gastroenterology 2004, 127, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Glas, J.; Seiderer, J.; Fischer, D.; Tengler, B.; Pfennig, S.; Wetzke, M.; Beigel, F.; Olszak, T.; Weidinger, M.; Göke, B.; et al. Pregnane X receptor (PXR/NR1I2) gene haplotypes modulate susceptibility to inflammatory bowel disease. Inflamm. Bowel Dis. 2011, 17, 1917–1924. [Google Scholar] [CrossRef] [PubMed]
- Martínez, A.; Márquez, A.; Mendoza, J.; Taxonera, C.; Fernández-Arquero, M.; Díaz-Rubio, M.; de la Concha, E.G.; Urcelay, E. Role of the PXR gene locus in inflammatory bowel diseases. Inflamm. Bowel Dis. 2007, 13, 1484–1487. [Google Scholar] [CrossRef]
- Shah, Y.M.; Ma, X.; Morimura, K.; Kim, I.; Gonzalez, F.J. Pregnane X receptor activation ameliorates DSS-induced inflammatory bowel disease via inhibition of NF-kappaB target gene expression. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G1114–G1122. [Google Scholar] [CrossRef]
- Zhou, C.; Tabb, M.M.; Nelson, E.L.; Grün, F.; Verma, S.; Sadatrafiei, A.; Lin, M.; Mallick, S.; Forman, B.M.; Thummel, K.E.; et al. Mutual repression between steroid and xenobiotic receptor and NF-kappaB signaling pathways links xenobiotic metabolism and inflammation. J. Clin. Investig. 2006, 116, 2280–2289. [Google Scholar] [CrossRef] [Green Version]
- Makishima, M.; Lu, T.T.; Xie, W.; Whitfield, G.K.; Domoto, H.; Evans, R.M.; Haussler, M.R.; Mangelsdorf, D.J. Vitamin D receptor as an intestinal bile acid sensor. Science 2002, 296, 1313–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Thingholm, L.B.; Skiecevičienė, J.; Rausch, P.; Kummen, M.; Hov, J.R.; Degenhardt, F.; Heinsen, F.-A.; Rühlemann, M.C.; Szymczak, S.; et al. Genome-wide association analysis identifies variation in vitamin D receptor and other host factors influencing the gut microbiota. Nat. Genet. 2016, 48, 1396–1406. [Google Scholar] [CrossRef]
- Simmons, J.D.; Mullighan, C.; Welsh, K.I.; Jewell, D.P. Vitamin D receptor gene polymorphism: Association with Crohn’s disease susceptibility. Gut 2000, 47, 211–214. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Chen, Y.; Golan, M.A.; Annunziata, M.L.; Du, J.; Dougherty, U.; Kong, J.; Musch, M.; Huang, Y.; Pekow, J.; et al. Intestinal epithelial vitamin D receptor signaling inhibits experimental colitis. J. Clin. Investig. 2013, 123, 3983–3996. [Google Scholar] [CrossRef] [Green Version]
- Garg, M.; Royce, S.G.; Tikellis, C.; Shallue, C.; Sluka, P.; Wardan, H.; Hosking, P.; Monagle, S.; Thomas, M.; Lubel, J.S.; et al. The intestinal vitamin D receptor in inflammatory bowel disease: Inverse correlation with inflammation but no relationship with circulating vitamin D status. Ther. Adv. Gastroenterol. 2019, 12, 1756284818822566. [Google Scholar] [CrossRef]
- He, L.; Liu, T.; Shi, Y.; Tian, F.; Hu, H.; Deb, D.K.; Chen, Y.; Bissonnette, M.; Li, Y.C. Gut Epithelial Vitamin D Receptor Regulates Microbiota-Dependent Mucosal Inflammation by Suppressing Intestinal Epithelial Cell Apoptosis. Endocrinology 2018, 159, 967–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaishnava, S.; Behrendt, C.L.; Ismail, A.S.; Eckmann, L.; Hooper, L.V. Paneth cells directly sense gut commensals and maintain homeostasis at the intestinal host-microbial interface. Proc. Natl. Acad. Sci. USA 2008, 105, 20858–20863. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.; Zhang, Y.-G.; Xia, Y.; Sun, J. Imbalance of autophagy and apoptosis in intestinal epithelium lacking the vitamin D receptor. FASEB J. 2019, 33, 11845–11856. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Zhang, Y.-G.; Lu, R.; Xia, Y.; Zhou, D.; Petrof, E.O.; Claud, E.C.; Chen, D.; Chang, E.B.; Carmeliet, G.; et al. Intestinal epithelial vitamin D receptor deletion leads to defective autophagy in colitis. Gut 2015, 64, 1082–1094. [Google Scholar] [CrossRef] [PubMed]
- Paramsothy, S.; Nielsen, S.; Kamm, M.A.; Deshpande, N.P.; Faith, J.J.; Clemente, J.C.; Paramsothy, R.; Walsh, A.J.; van den Bogaerde, J.; Samuel, D.; et al. Specific Bacteria and Metabolites Associated with Response to Fecal Microbiota Transplantation in Patients with Ulcerative Colitis. Gastroenterology 2019, 156, 1440–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Martins, R.; Sullivan, M.C.; Friedman, E.S.; Misic, A.M.; El-Fahmawi, A.; De Martinis, E.C.P.; O’Brien, K.; Chen, Y.; Bradley, C.; et al. Diet-induced remission in chronic enteropathy is associated with altered microbial community structure and synthesis of secondary bile acids. Microbiome 2019, 7, 126. [Google Scholar] [CrossRef] [Green Version]
- Ke, J.; Li, Y.; Han, C.; He, R.; Lin, R.; Qian, W.; Hou, X. Fucose Ameliorate Intestinal Inflammation Through Modulating the Crosstalk Between Bile Acids and Gut Microbiota in a Chronic Colitis Murine Model. Inflamm. Bowel Dis. 2020, 26, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.-Q.; Yuan, Z.-W.; Zhang, X.-S.; Dong, J.-Q.; Liu, X.-N.; Peng, X.-T.; Yao, W.-L.; Ji, P.; Wei, Y.-M.; Hua, Y.-L. Total alkaloids of Sophora alopecuroides L. ameliorated murine colitis by regulating bile acid metabolism and gut microbiota. J. Ethnopharmacol. 2020, 255, 112775. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.K.; Abraham, B.; El-Serag, H. Dietary intake and risk of developing inflammatory bowel disease: A systematic review of the literature. Am. J. Gastroenterol. 2011, 106, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Yuan, J.; Li, J.; Li, H.; Zhang, J.; Tang, J.; Ni, Y.; Huang, T.; Wang, F.; Zhao, F.; et al. Unconjugated and secondary bile acid profiles in response to higher-fat, lower-carbohydrate diet and associated with related gut microbiota: A 6-month randomized controlled-feeding trial. Clin. Nutr. 2020, 39, 395–404. [Google Scholar] [CrossRef] [PubMed]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [Green Version]
- Llewellyn, S.R.; Britton, G.J.; Contijoch, E.J.; Vennaro, O.H.; Mortha, A.; Colombel, J.-F.; Grinspan, A.; Clemente, J.C.; Merad, M.; Faith, J.J. Interactions Between Diet and the Intestinal Microbiota Alter Intestinal Permeability and Colitis Severity in Mice. Gastroenterology 2018, 154. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, S.C.; Escher, J.; Hébuterne, X.; Kłęk, S.; Krznaric, Z.; Schneider, S.; Shamir, R.; Stardelova, K.; Wierdsma, N.; Wiskin, A.E.; et al. ESPEN practical guideline: Clinical Nutrition in inflammatory bowel disease. Clin. Nutr. 2020, 39, 632–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruemmele, F.M.; Veres, G.; Kolho, K.L.; Griffiths, A.; Levine, A.; Escher, J.C.; Amil Dias, J.; Barabino, A.; Braegger, C.P.; Bronsky, J.; et al. Consensus guidelines of ECCO/ESPGHAN on the medical management of pediatric Crohn’s disease. J. Crohn’s Colitis 2014, 8, 1179–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashton, J.J.; Gavin, J.; Beattie, R.M. Exclusive enteral nutrition in Crohn’s disease: Evidence and practicalities. Clin. Nutr. 2019, 38, 80–89. [Google Scholar] [CrossRef]
- Hansen, T.; Duerksen, D.R. Enteral Nutrition in the Management of Pediatric and Adult Crohn’s Disease. Nutrients 2018, 10, 537. [Google Scholar] [CrossRef] [Green Version]
- Leach, S.T.; Mitchell, H.M.; Eng, W.R.; Zhang, L.; Day, A.S. Sustained modulation of intestinal bacteria by exclusive enteral nutrition used to treat children with Crohn’s disease. Aliment. Pharmacol. Ther. 2008, 28, 724–733. [Google Scholar] [CrossRef] [PubMed]
- Quince, C.; Ijaz, U.Z.; Loman, N.; Eren, A.M.; Saulnier, D.; Russell, J.; Haig, S.J.; Calus, S.T.; Quick, J.; Barclay, A.; et al. Extensive Modulation of the Fecal Metagenome in Children With Crohn’s Disease During Exclusive Enteral Nutrition. Am. J. Gastroenterol. 2015, 110, 1718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connors, J.; Dunn, K.A.; Allott, J.; Bandsma, R.; Rashid, M.; Otley, A.R.; Bielawski, J.P.; Van Limbergen, J. The relationship between fecal bile acids and microbiome community structure in pediatric Crohn’s disease. ISME J. 2020, 14, 702–713. [Google Scholar] [CrossRef] [PubMed]
- Tsertsvadze, A.; Gurung, T.; Court, R.; Clarke, A.; Sutcliffe, P. Clinical effectiveness and cost-effectiveness of elemental nutrition for the maintenance of remission in Crohn’s disease: A systematic review and meta-analysis. Health Technol. Assess. 2015, 19, 1–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sartor, R.B.; Wu, G.D. Roles for Intestinal Bacteria, Viruses, and Fungi in Pathogenesis of Inflammatory Bowel Diseases and Therapeutic Approaches. Gastroenterology 2017, 152, 327–339. [Google Scholar] [CrossRef] [Green Version]
- Gareau, M.G.; Sherman, P.M.; Walker, W.A. Probiotics and the gut microbiota in intestinal health and disease. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 503–514. [Google Scholar] [CrossRef] [Green Version]
- Kruis, W.; Fric, P.; Pokrotnieks, J.; Lukás, M.; Fixa, B.; Kascák, M.; Kamm, M.A.; Weismueller, J.; Beglinger, C.; Stolte, M.; et al. Maintaining remission of ulcerative colitis with the probiotic Escherichia coli Nissle 1917 is as effective as with standard mesalazine. Gut 2004, 53, 1617–1623. [Google Scholar] [CrossRef]
- Rembacken, B.J.; Snelling, A.M.; Hawkey, P.M.; Chalmers, D.M.; Axon, A.T. Non-pathogenic Escherichia coli versus mesalazine for the treatment of ulcerative colitis: A randomised trial. Lancet 1999, 354, 635–639. [Google Scholar] [CrossRef]
- Kruis, W.; Schütz, E.; Fric, P.; Fixa, B.; Judmaier, G.; Stolte, M. Double-blind comparison of an oral Escherichia coli preparation and mesalazine in maintaining remission of ulcerative colitis. Aliment. Pharmacol. Ther. 1997, 11, 853–858. [Google Scholar] [CrossRef]
- Kato, K.; Mizuno, S.; Umesaki, Y.; Ishii, Y.; Sugitani, M.; Imaoka, A.; Otsuka, M.; Hasunuma, O.; Kurihara, R.; Iwasaki, A.; et al. Randomized placebo-controlled trial assessing the effect of bifidobacteria-fermented milk on active ulcerative colitis. Aliment. Pharmacol. Ther. 2004, 20, 1133–1141. [Google Scholar] [CrossRef]
- Zocco, M.A.; dal Verme, L.Z.; Cremonini, F.; Piscaglia, A.C.; Nista, E.C.; Candelli, M.; Novi, M.; Rigante, D.; Cazzato, I.A.; Ojetti, V.; et al. Efficacy of Lactobacillus GG in maintaining remission of ulcerative colitis. Aliment. Pharmacol. Ther. 2006, 23, 1567–1574. [Google Scholar] [CrossRef]
- Oliva, S.; Di Nardo, G.; Ferrari, F.; Mallardo, S.; Rossi, P.; Patrizi, G.; Cucchiara, S.; Stronati, L. Randomised clinical trial: The effectiveness of Lactobacillus reuteri ATCC 55730 rectal enema in children with active distal ulcerative colitis. Aliment. Pharmacol. Ther. 2012, 35, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Tursi, A.; Brandimarte, G.; Papa, A.; Giglio, A.; Elisei, W.; Giorgetti, G.M.; Forti, G.; Morini, S.; Hassan, C.; Pistoia, M.A.; et al. Treatment of relapsing mild-to-moderate ulcerative colitis with the probiotic VSL#3 as adjunctive to a standard pharmaceutical treatment: A double-blind, randomized, placebo-controlled study. Am. J. Gastroenterol. 2010, 105, 2218–2227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miele, E.; Pascarella, F.; Giannetti, E.; Quaglietta, L.; Baldassano, R.N.; Staiano, A. Effect of a probiotic preparation (VSL#3) on induction and maintenance of remission in children with ulcerative colitis. Am. J. Gastroenterol. 2009, 104, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Saez-Lara, M.J.; Gomez-Llorente, C.; Plaza-Diaz, J.; Gil, A. The role of probiotic lactic acid bacteria and bifidobacteria in the prevention and treatment of inflammatory bowel disease and other related diseases: A systematic review of randomized human clinical trials. Biomed. Res. Int. 2015, 2015, 505878. [Google Scholar] [CrossRef]
- Madsen, K.; Cornish, A.; Soper, P.; McKaigney, C.; Jijon, H.; Yachimec, C.; Doyle, J.; Jewell, L.; De Simone, C. Probiotic bacteria enhance murine and human intestinal epithelial barrier function. Gastroenterology 2001, 121, 580–591. [Google Scholar] [CrossRef] [Green Version]
- Chapman, T.M.; Plosker, G.L.; Figgitt, D.P. VSL#3 probiotic mixture: A review of its use in chronic inflammatory bowel diseases. Drugs 2006, 66, 1371–1387. [Google Scholar]
- Martoni, C.J.; Labbé, A.; Ganopolsky, J.G.; Prakash, S.; Jones, M.L. Changes in bile acids, FGF-19 and sterol absorption in response to bile salt hydrolase active L. reuteri NCIMB 30242. Gut Microbes 2015, 6, 57–65. [Google Scholar] [CrossRef] [Green Version]
- Zhai, Q.; Liu, Y.; Wang, C.; Qu, D.; Zhao, J.; Zhang, H.; Tian, F.; Chen, W. Lactobacillus plantarum CCFM8661 modulates bile acid enterohepatic circulation and increases lead excretion in mice. Food Funct. 2019, 10, 1455–1464. [Google Scholar] [CrossRef]
- Degirolamo, C.; Rainaldi, S.; Bovenga, F.; Murzilli, S.; Moschetta, A. Microbiota modification with probiotics induces hepatic bile acid synthesis via downregulation of the Fxr-Fgf15 axis in mice. Cell Rep. 2014, 7, 12–18. [Google Scholar] [CrossRef] [Green Version]
- Bourreille, A.; Cadiot, G.; Le Dreau, G.; Laharie, D.; Beaugerie, L.; Dupas, J.-L.; Marteau, P.; Rampal, P.; Moyse, D.; Saleh, A.; et al. Saccharomyces boulardii does not prevent relapse of Crohn’s disease. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2013, 11, 982–987. [Google Scholar] [CrossRef]
- Bjarnason, I.; Sission, G.; Hayee, B.H. A randomised, double-blind, placebo-controlled trial of a multi-strain probiotic in patients with asymptomatic ulcerative colitis and Crohn’s disease. Inflammopharmacology 2019, 27, 465–473. [Google Scholar] [CrossRef] [Green Version]
- Ganji-Arjenaki, M.; Rafieian-Kopaei, M. Probiotics are a good choice in remission of inflammatory bowel diseases: A meta analysis and systematic review. J. Cell. Physiol. 2018, 233, 2091–2103. [Google Scholar] [CrossRef]
- van der Lelie, D.; Oka, A.; Taghavi, S.; Umeno, J.; Fan, T.-J.; Merrell, K.E.; Watson, S.D.; Ouellette, L.; Liu, B.; Awoniyi, M.; et al. Rationally designed bacterial consortia to treat chronic immune-mediated colitis and restore intestinal homeostasis. Nat. Commun. 2021, 12, 3105. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.R.; Hutkins, R.; Sanders, M.E.; Prescott, S.L.; Reimer, R.A.; Salminen, S.J.; Scott, K.; Stanton, C.; Swanson, K.S.; Cani, P.D.; et al. Expert consensus document: The International Scientific Association for Probiotics and Prebiotics (ISAPP) consensus statement on the definition and scope of prebiotics. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 491–502. [Google Scholar] [CrossRef] [Green Version]
- Casellas, F.; Borruel, N.; Torrejón, A.; Varela, E.; Antolin, M.; Guarner, F.; Malagelada, J.R. Oral oligofructose-enriched inulin supplementation in acute ulcerative colitis is well tolerated and associated with lowered faecal calprotectin. Aliment. Pharmacol. Ther. 2007, 25, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Valcheva, R.; Koleva, P.; Martínez, I.; Walter, J.; Gänzle, M.G.; Dieleman, L.A. Inulin-type fructans improve active ulcerative colitis associated with microbiota changes and increased short-chain fatty acids levels. Gut Microbes 2019, 10, 334–357. [Google Scholar] [CrossRef] [PubMed]
- Vandeputte, D.; Falony, G.; Vieira-Silva, S.; Wang, J.; Sailer, M.; Theis, S.; Verbeke, K.; Raes, J. Prebiotic inulin-type fructans induce specific changes in the human gut microbiota. Gut 2017, 66, 1968–1974. [Google Scholar] [CrossRef]
- Scott, K.P.; Martin, J.C.; Duncan, S.H.; Flint, H.J. Prebiotic stimulation of human colonic butyrate-producing bacteria and bifidobacteria, in vitro. FEMS Microbiol. Ecol. 2014, 87, 30–40. [Google Scholar] [CrossRef] [Green Version]
- Alexander, C.; Cross, T.-W.L.; Devendran, S.; Neumer, F.; Theis, S.; Ridlon, J.M.; Suchodolski, J.S.; de Godoy, M.R.C.; Swanson, K.S. Effects of prebiotic inulin-type fructans on blood metabolite and hormone concentrations and faecal microbiota and metabolites in overweight dogs. Br. J. Nutr. 2018, 120, 711–720. [Google Scholar] [CrossRef] [Green Version]
- Steidler, L.; Hans, W.; Schotte, L.; Neirynck, S.; Obermeier, F.; Falk, W.; Fiers, W.; Remaut, E. Treatment of murine colitis by Lactococcus lactis secreting interleukin-10. Science 2000, 289, 1352–1355. [Google Scholar] [CrossRef] [Green Version]
- Hanson, M.L.; Hixon, J.A.; Li, W.; Felber, B.K.; Anver, M.R.; Stewart, C.A.; Janelsins, B.M.; Datta, S.K.; Shen, W.; McLean, M.H.; et al. Oral delivery of IL-27 recombinant bacteria attenuates immune colitis in mice. Gastroenterology 2014, 146, 210–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Praveschotinunt, P.; Duraj-Thatte, A.M.; Gelfat, I.; Bahl, F.; Chou, D.B.; Joshi, N.S. Engineered E. coli Nissle 1917 for the delivery of matrix-tethered therapeutic domains to the gut. Nat. Commun. 2019, 10, 5580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandenbroucke, K.; Hans, W.; Van Huysse, J.; Neirynck, S.; Demetter, P.; Remaut, E.; Rottiers, P.; Steidler, L. Active delivery of trefoil factors by genetically modified Lactococcus lactis prevents and heals acute colitis in mice. Gastroenterology 2004, 127, 502–513. [Google Scholar] [CrossRef]
- Campbell, C.; McKenney, P.T.; Konstantinovsky, D.; Isaeva, O.I.; Schizas, M.; Verter, J.; Mai, C.; Jin, W.-B.; Guo, C.-J.; Violante, S.; et al. Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature 2020, 581, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Allegretti, J.R.; Mullish, B.H.; Kelly, C.; Fischer, M. The evolution of the use of faecal microbiota transplantation and emerging therapeutic indications. Lancet 2019, 394, 420–431. [Google Scholar] [CrossRef]
- Khoruts, A.; Sadowsky, M.J. Understanding the mechanisms of faecal microbiota transplantation. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 508–516. [Google Scholar] [CrossRef] [Green Version]
- Weingarden, A.; González, A.; Vázquez-Baeza, Y.; Weiss, S.; Humphry, G.; Berg-Lyons, D.; Knights, D.; Unno, T.; Bobr, A.; Kang, J.; et al. Dynamic changes in short- and long-term bacterial composition following fecal microbiota transplantation for recurrent Clostridium difficile infection. Microbiome 2015, 3, 10. [Google Scholar] [CrossRef] [Green Version]
- Bojanova, D.P.; Bordenstein, S.R. Fecal Transplants: What Is Being Transferred? PLoS Biol. 2016, 14, e1002503. [Google Scholar] [CrossRef]
- Ng, S.C.; Kamm, M.A.; Yeoh, Y.K.; Chan, P.K.S.; Zuo, T.; Tang, W.; Sood, A.; Andoh, A.; Ohmiya, N.; Zhou, Y.; et al. Scientific frontiers in faecal microbiota transplantation: Joint document of Asia-Pacific Association of Gastroenterology (APAGE) and Asia-Pacific Society for Digestive Endoscopy (APSDE). Gut 2020, 69, 83–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costello, S.; Waters, O.; Bryant, R.; Katsikeros, R.; Makanyanga, J.; Schoeman, M.; Mountifield, R.; Tee, D.; Howell, S.; Hughes, P.; et al. OP036 Short duration, low intensity pooled faecal microbiota transplantation induces remission in patients with mild-moderately active ulcerative colitis: A randomised controlled trial. J. Crohn’s Colitis 2017, 11, S23. [Google Scholar] [CrossRef] [Green Version]
- Paramsothy, S.; Kamm, M.A.; Kaakoush, N.O.; Walsh, A.J.; van den Bogaerde, J.; Samuel, D.; Leong, R.W.L.; Connor, S.; Ng, W.; Paramsothy, R.; et al. Multidonor intensive faecal microbiota transplantation for active ulcerative colitis: A randomised placebo-controlled trial. Lancet 2017, 389, 1218–1228. [Google Scholar] [CrossRef]
- Costello, S.P.; Hughes, P.A.; Waters, O.; Bryant, R.V.; Vincent, A.D.; Blatchford, P.; Katsikeros, R.; Makanyanga, J.; Campaniello, M.A.; Mavrangelos, C.; et al. Effect of Fecal Microbiota Transplantation on 8-Week Remission in Patients With Ulcerative Colitis: A Randomized Clinical Trial. JAMA 2019, 321, 156–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokol, H.; Landman, C.; Seksik, P.; Berard, L.; Montil, M.; Nion-Larmurier, I.; Bourrier, A.; Le Gall, G.; Lalande, V.; De Rougemont, A.; et al. Fecal microbiota transplantation to maintain remission in Crohn’s disease: A pilot randomized controlled study. Microbiome 2020, 8, 12. [Google Scholar] [CrossRef]
- Cui, B.; Feng, Q.; Wang, H.; Wang, M.; Peng, Z.; Li, P.; Huang, G.; Liu, Z.; Wu, P.; Fan, Z.; et al. Fecal microbiota transplantation through mid-gut for refractory Crohn’s disease: Safety, feasibility, and efficacy trial results. J. Gastroenterol. Hepatol. 2015, 30, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Imdad, A.; Nicholson, M.R.; Tanner-Smith, E.E.; Zackular, J.P.; Gomez-Duarte, O.G.; Beaulieu, D.B.; Acra, S. Fecal transplantation for treatment of inflammatory bowel disease. Cochrane Database Syst. Rev. 2018, 11, CD012774. [Google Scholar] [CrossRef] [PubMed]
- DeFilipp, Z.; Bloom, P.P.; Torres Soto, M.; Mansour, M.K.; Sater, M.R.A.; Huntley, M.H.; Turbett, S.; Chung, R.T.; Chen, Y.-B.; Hohmann, E.L. Drug-Resistant Bacteremia Transmitted by Fecal Microbiota Transplant. N. Engl. J. Med. 2019, 381, 2043–2050. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, J.P.; Xie, G.; Raufman, J.-P.; Hogan, S.; Griffin, T.L.; Packard, C.A.; Chatfield, D.A.; Hagey, L.R.; Steinbach, J.H.; Hofmann, A.F. Human cecal bile acids: Concentration and spectrum. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G256–G263. [Google Scholar] [CrossRef] [Green Version]
- Rubin, R.A.; Kowalski, T.E.; Khandelwal, M.; Malet, P.F. Ursodiol for hepatobiliary disorders. Ann. Intern. Med. 1994, 121, 207–218. [Google Scholar] [CrossRef]
- Van den Bossche, L.; Borsboom, D.; Devriese, S.; Van Welden, S.; Holvoet, T.; Devisscher, L.; Hindryckx, P.; De Vos, M.; Laukens, D. Tauroursodeoxycholic acid protects bile acid homeostasis under inflammatory conditions and dampens Crohn’s disease-like ileitis. Lab. Investig. 2017, 97, 519–529. [Google Scholar] [CrossRef]
- Ward, J.B.J.; Lajczak, N.K.; Kelly, O.B.; O’Dwyer, A.M.; Giddam, A.K.; Ní Gabhann, J.; Franco, P.; Tambuwala, M.M.; Jefferies, C.A.; Keely, S.; et al. Ursodeoxycholic acid and lithocholic acid exert anti-inflammatory actions in the colon. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G550–G558. [Google Scholar] [CrossRef] [Green Version]
- Laukens, D.; Devisscher, L.; Van den Bossche, L.; Hindryckx, P.; Vandenbroucke, R.E.; Vandewynckel, Y.-P.; Cuvelier, C.; Brinkman, B.M.; Libert, C.; Vandenabeele, P.; et al. Tauroursodeoxycholic acid inhibits experimental colitis by preventing early intestinal epithelial cell death. Lab. Investig. 2014, 94, 1419–1430. [Google Scholar] [CrossRef] [Green Version]
- Pardi, D.S.; Loftus, E.V.; Kremers, W.K.; Keach, J.; Lindor, K.D. Ursodeoxycholic acid as a chemopreventive agent in patients with ulcerative colitis and primary sclerosing cholangitis. Gastroenterology 2003, 124, 889–893. [Google Scholar] [CrossRef] [Green Version]
- Pearson, T.; Caporaso, J.G.; Yellowhair, M.; Bokulich, N.A.; Padi, M.; Roe, D.J.; Wertheim, B.C.; Linhart, M.; Martinez, J.A.; Bilagody, C.; et al. Effects of ursodeoxycholic acid on the gut microbiome and colorectal adenoma development. Cancer Med. 2019, 8, 617–628. [Google Scholar] [CrossRef] [Green Version]
- Eaton, J.E.; Silveira, M.G.; Pardi, D.S.; Sinakos, E.; Kowdley, K.V.; Luketic, V.A.C.; Harrison, M.E.; McCashland, T.; Befeler, A.S.; Harnois, D.; et al. High-dose ursodeoxycholic acid is associated with the development of colorectal neoplasia in patients with ulcerative colitis and primary sclerosing cholangitis. Am. J. Gastroenterol. 2011, 106, 1638–1645. [Google Scholar] [CrossRef] [Green Version]
- Boonstra, K.; van Erpecum, K.J.; van Nieuwkerk, K.M.J.; Drenth, J.P.H.; Poen, A.C.; Witteman, B.J.M.; Tuynman, H.A.R.E.; Beuers, U.; Ponsioen, C.Y. Primary sclerosing cholangitis is associated with a distinct phenotype of inflammatory bowel disease. Inflamm. Bowel Dis. 2012, 18, 2270–2276. [Google Scholar] [CrossRef]
- Quraishi, M.N.; Acharjee, A.; Beggs, A.D.; Horniblow, R.; Tselepis, C.; Gkoutos, G.; Ghosh, S.; Rossiter, A.E.; Loman, N.; van Schaik, W.; et al. A Pilot Integrative Analysis of Colonic Gene Expression, Gut Microbiota, and Immune Infiltration in Primary Sclerosing Cholangitis-Inflammatory Bowel Disease: Association of Disease With Bile Acid Pathways. J. Crohn’s Colitis 2020, 14, 935–947. [Google Scholar] [CrossRef] [Green Version]
- Dyson, J.K.; Beuers, U.; Jones, D.E.J.; Lohse, A.W.; Hudson, M. Primary sclerosing cholangitis. Lancet 2018, 391, 2547–2559. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Publication | Patients | Samples and Methods | Major Findings |
|---|---|---|---|
| Duboc et al., 2013 [9] | 12 with CD, 30 with UC and 29 HCs | Fecal samples (real-time qPCR and HPLC) Serum samples (HPLC) |
|
| Sinha et al., 2020 [10] | 17 with UC and seven with FAP | Stool samples (metagenomic sequencing and metabolomic analysis) |
|
| Franzosa et al., 2019 [15] | 88 with CD, 76 with UC and 56 non-IBD | Stool samples (metagenomic sequencing and metabolomic analysis) |
|
| Jacobs et al., 2016 [16] | 26 with CD, 10 with UC and 54 healthy first-degree relatives | Stool samples (16S rRNA sequencing and HPLC) |
|
| Lloyd-Price et al., 2019 [18] | 67 with CD, 38 with UC and 27 non-IBDs | Stool samples (metagenomic sequencing and metabolomic analysis) |
|
| Wang et al., 2021 [19] | 29 pediatric patients with CD and 20 HCs | Fecal samples (16S rRNA sequencing and UPLC-MS) |
|
| Weng et al., 2019 [37] | 173 with CD, 107 with UC and 42 HCs | Fecal samples (metagenomic sequencing and metabolomic analysis) Mucosal biopsy samples (16S rRNA sequencing) |
|
| Murakami et al., 2018 [38] | Six with CD, six with UC and 26 HCs | Fecal samples (T-RFLP analysis and HPLC) Serum samples (HPLC) |
|
| Diederen et al., 2020 [39] | 43 pediatric patients with CD and 18 HCs | Fecal samples (16S rRNA sequencing and HPLC) |
|
| Yang et al., 2021 [22] | 32 patients with UC and 23 HCs | Fecal samples (16S rRNA sequencing and UPLC-MS) |
|
| Publication | Subjects | Treatment | Samples and Methods | Major Findings |
|---|---|---|---|---|
| Diederen et al., 2020 [39] | 43 pediatric patients with CD and 18 healthy controls | EEN for 6 weeks followed by 2 weeks of EEN tapering | Fecal samples (16S rRNA sequencing and HPLC) |
|
| Paramsothy et al., 2019 [88] | 81 patients with active UC | FMT or placebo colonoscopic infusion, followed by enemas 5 days per week for 8 weeks | Fecal samples (metagenomic and metabolomic analysis) Colonic biopsy samples (16S rRNA gene and transcript sequencing) |
|
| Wang et al., 2021 [19] | 29 pediatric CDs | Infliximab infusion for 3–6 times | Fecal samples (16S rRNA sequencing and UPLC-MS) |
|
| Wang et al., 2019 [89] | Canine model of chronic inflammatory enteropathy | Hydrolyzed protein diet for 6 weeks | Fecal samples (metagenomic and metabolomic analysis) |
|
| Ke et al., 2020 [90] | Mice with DSS-induced chronic colitis | Fucose gavage for 57 days | Ileal tissue lysates and colonic feces (16S rRNA sequencing and UPLC-MS) |
|
| Jia et al., 2020 [91] | Mice with DSS-induced acute colitis | Oral total alkaloids of Sophora alopecuroides L. for 7 days | Cecum content (16S rDNA gene sequencing) Liver, bile, serum, cecum content and colon samples (UPLC-MS) |
|
| Bossche et al., 2017 [12] | Mice with DSS-induced acute colitis | Daily gavage of UDCA, TUDCA, GUDCA or placebo for 10 days | Fecal samples (16S rRNA sequencing and HPLC) |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, M.; Gu, Y.; Li, L.; Liu, T.; Song, X.; Sun, Y.; Cao, X.; Wang, B.; Jiang, K.; Cao, H. Bile Acid–Gut Microbiota Axis in Inflammatory Bowel Disease: From Bench to Bedside. Nutrients 2021, 13, 3143. https://doi.org/10.3390/nu13093143
Yang M, Gu Y, Li L, Liu T, Song X, Sun Y, Cao X, Wang B, Jiang K, Cao H. Bile Acid–Gut Microbiota Axis in Inflammatory Bowel Disease: From Bench to Bedside. Nutrients. 2021; 13(9):3143. https://doi.org/10.3390/nu13093143
Chicago/Turabian StyleYang, Min, Yu Gu, Lingfeng Li, Tianyu Liu, Xueli Song, Yue Sun, Xiaocang Cao, Bangmao Wang, Kui Jiang, and Hailong Cao. 2021. "Bile Acid–Gut Microbiota Axis in Inflammatory Bowel Disease: From Bench to Bedside" Nutrients 13, no. 9: 3143. https://doi.org/10.3390/nu13093143