
Characterization of Gut Microbiome in Korean Patients with Metabolic Associated Fatty Liver Disease


 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design, Setting, and Participants
2.2. Definitions of Variables
2.3. Genomic DNA Extraction and Illumina Sequencing
2.4. Statistical Analysis
3. Results
3.1. Baseline Characteristics
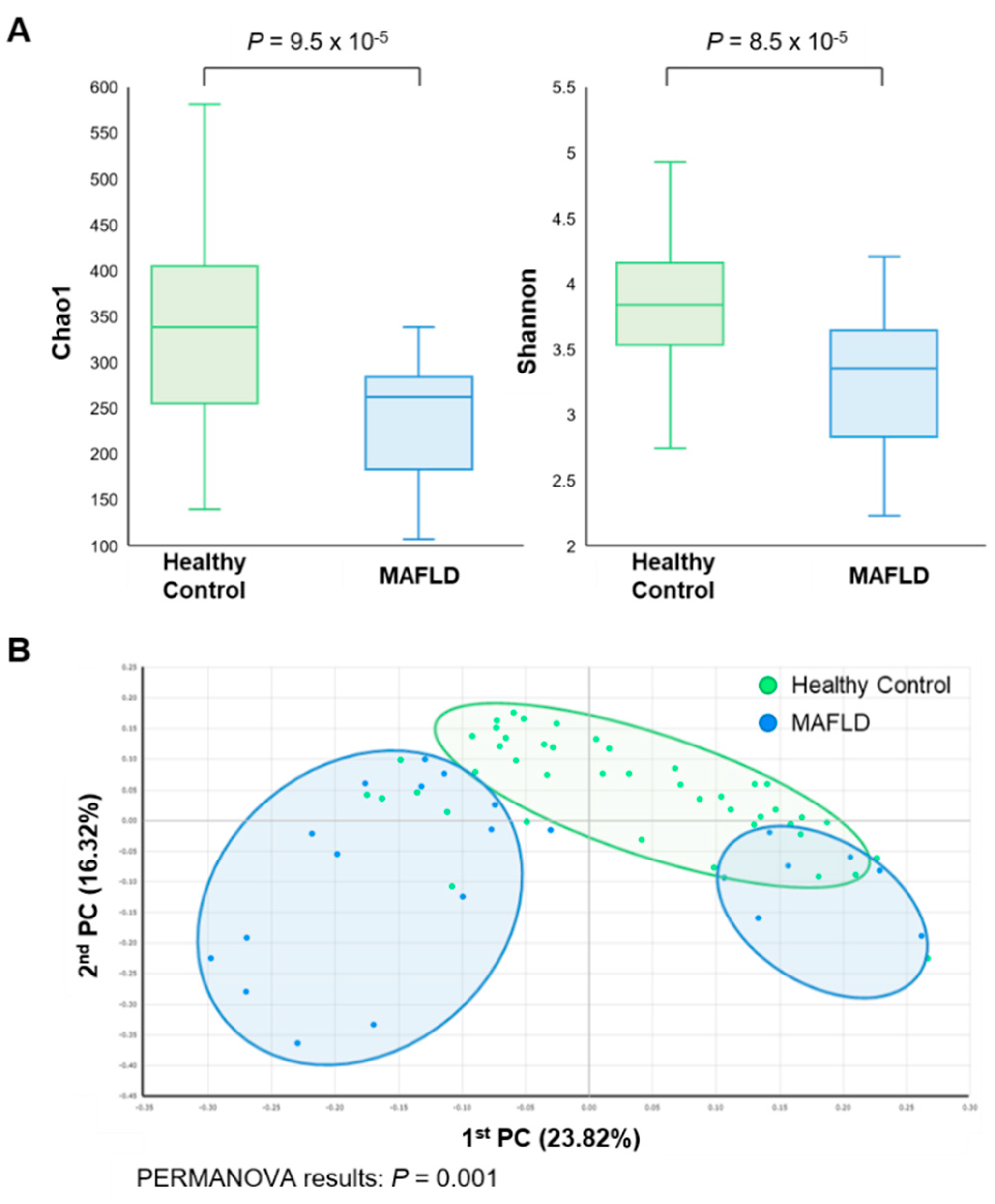
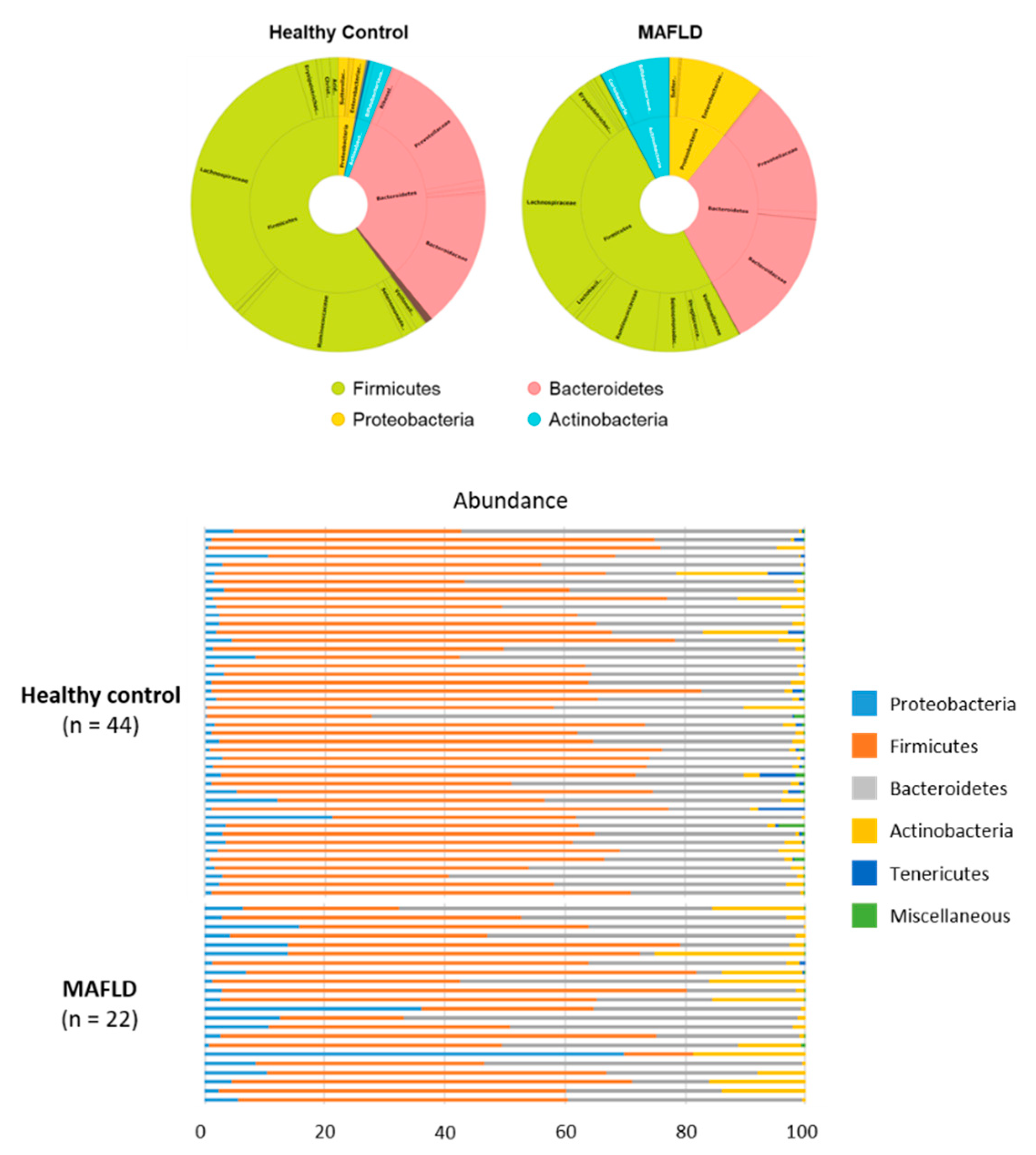
3.2. Comparison of Gut Microbiota
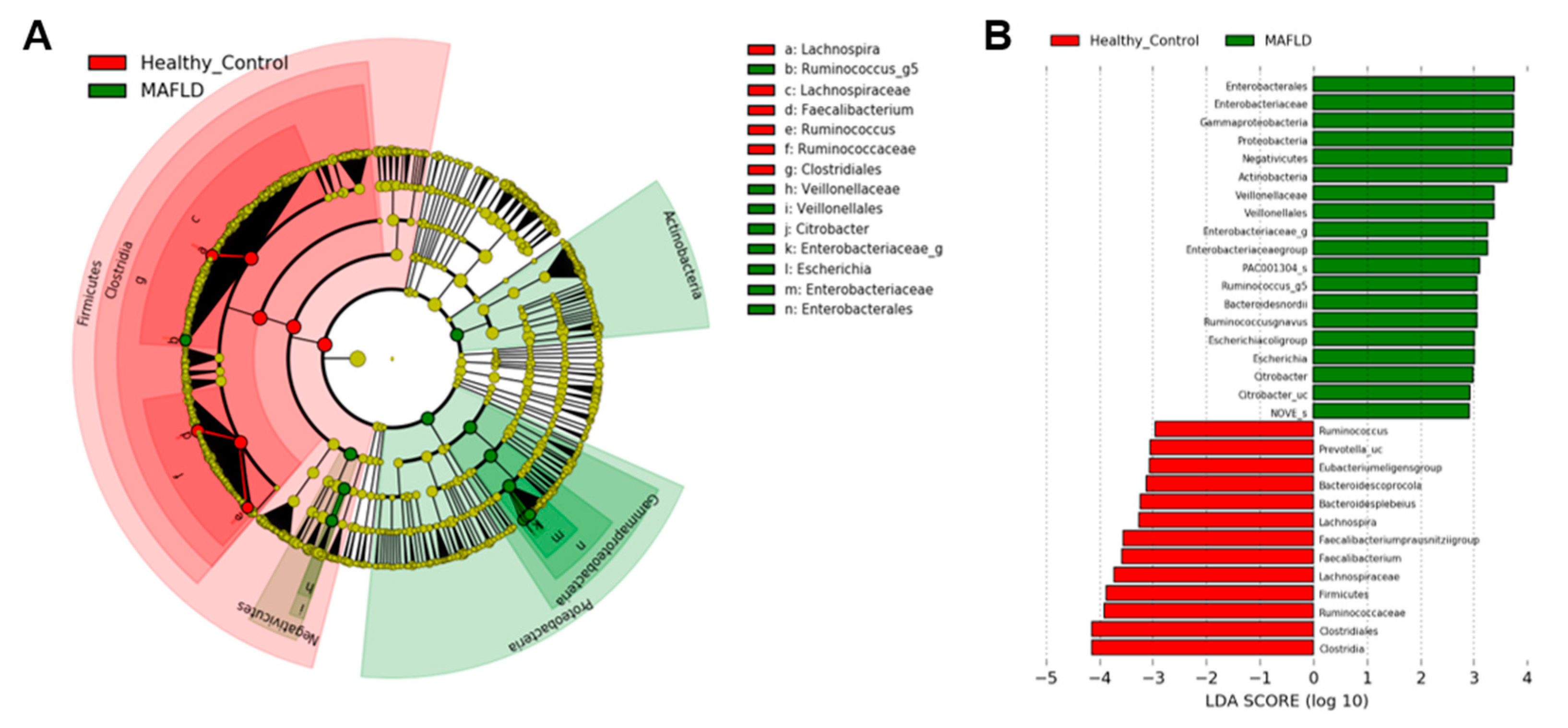
3.3. Butyrate- and Alcohol-Producing Bacteria
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rinella, M.E. Nonalcoholic fatty liver disease: A systematic review. JAMA 2015, 313, 2263–2273. [Google Scholar] [CrossRef]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Sanyal, A.J.; George, J. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1991. [Google Scholar] [CrossRef]
- Tarantino, G.; Citro, V.; Capone, D. Nonalcoholic Fatty Liver Disease: A Challenge from Mechanisms to Therapy. J. Clin. Med. 2019, 9, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trépo, E.; Romeo, S.; Zucman-Rossi, J.; Nahon, P. PNPLA3 gene in liver diseases. J. Hepatol. 2016, 65, 399–412. [Google Scholar] [CrossRef] [Green Version]
- Leung, C.; Rivera, L.; Furness, J.B.; Angus, P.W. The role of the gut microbiota in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Aron-Wisnewsky, J.; Gaborit, B.; Dutour, A.; Clement, K. Gut microbiota and non-alcoholic fatty liver disease: New insights. Clin. Microbiol. Infect. 2013, 19, 338–348. [Google Scholar] [CrossRef] [Green Version]
- Aron-Wisnewsky, J.; Vigliotti, C.; Witjes, J.; Le, P.; Holleboom, A.G.; Verheij, J.; Nieuwdorp, M.; Clément, K. Gut microbiota and human NAFLD: Disentangling microbial signatures from metabolic disorders. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 279–297. [Google Scholar] [CrossRef] [PubMed]
- Vrieze, A.; Holleman, F.; Zoetendal, E.G.; De Vos, W.M.; Hoekstra, J.B.; Nieuwdorp, M. The environment within: How gut microbiota may influence metabolism and body composition. Diabetologia 2010, 53, 606–613. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef]
- Kim, H.N.; Joo, E.J.; Cheong, H.S.; Kim, Y.; Kim, H.L.; Shin, H.; Chang, Y.; Ryu, S. Gut Microbiota and Risk of Persistent Nonalcoholic Fatty Liver Diseases. J. Clin. Med. 2019, 8, 1089. [Google Scholar] [CrossRef] [Green Version]
- Boursier, J.; Mueller, O.; Barret, M.; Machado, M.; Fizanne, L.; Araujo-Perez, F.; Guy, C.D.; Seed, P.C.; Rawls, J.F.; David, L.A.; et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 2016, 63, 764–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mouzaki, M.; Comelli, E.M.; Arendt, B.M.; Bonengel, J.; Fung, S.K.; Fischer, S.E.; McGilvray, I.D.; Allard, J.P. Intestinal microbiota in patients with nonalcoholic fatty liver disease. Hepatology 2013, 58, 120–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aronow, W.S.; Fleg, J.L.; Pepine, C.J.; Artinian, N.T.; Bakris, G.; Brown, A.S.; Ferdinand, K.C.; Ann Forciea, M.; Frishman, W.H.; Jaigobin, C.; et al. ACCF/AHA 2011 expert consensus document on hypertension in the elderly: A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus documents developed in collaboration with the American Academy of Neurology, American Geriatrics Society, American Society for Preventive Cardiology, American Society of Hypertension, American Society of Nephrology, Association of Black Cardiologists, and European Society of Hypertension. J. Am. Coll. Cardiol. 2011, 57, 2037–2114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- American Diabetes Association. (2) Classification and diagnosis of diabetes. Diabetes Care 2015, 38 (Suppl. S1), S8–S16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, N.J.; Robinson, J.G.; Lichtenstein, A.H.; Bairey Merz, C.N.; Blum, C.B.; Eckel, R.H.; Goldberg, A.C.; Gordon, D.; Levy, D.; Lloyd-Jones, D.M.; et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. J. Am. Coll. Cardiol. 2014, 63, 2889–2934. [Google Scholar] [CrossRef] [Green Version]
- Seong, G.; Kim, N.; Joung, J.G.; Kim, E.R.; Chang, D.K.; Chun, J.; Hong, S.N.; Kim, Y.H. Changes in the Intestinal Microbiota of Patients with Inflammatory Bowel Disease with Clinical Remission during an 8-Week Infliximab Infusion Cycle. Microorganisms 2020, 8, 874. [Google Scholar] [CrossRef]
- Yoon, S.H.; Ha, S.M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Vital, M.; Karch, A.; Pieper, D.H. Colonic Butyrate-Producing Communities in Humans: An Overview Using Omics Data. mSystems 2017, 2, e00130-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jennison, E.; Byrne, C.D. The role of the gut microbiome and diet in the pathogenesis of non-alcoholic fatty liver disease. Clin. Mol. Hepatol. 2021, 27, 22–43. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willing, B.P.; Dicksved, J.; Halfvarson, J.; Andersson, A.F.; Lucio, M.; Zheng, Z.; Järnerot, G.; Tysk, C.; Jansson, J.K.; Engstrand, L. A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology 2010, 139, 1844–1854.e1841. [Google Scholar] [CrossRef]
- Chang, J.Y.; Antonopoulos, D.A.; Kalra, A.; Tonelli, A.; Khalife, W.T.; Schmidt, T.M.; Young, V.B. Decreased diversity of the fecal Microbiome in recurrent Clostridium difficile-associated diarrhea. J. Infect. Dis. 2008, 197, 435–438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Canfora, E.E.; Meex, R.C.R.; Venema, K.; Blaak, E.E. Gut microbial metabolites in obesity, NAFLD and T2DM. Nat. Rev. Endocrinol. 2019, 15, 261–273. [Google Scholar] [CrossRef]
- Xie, C.; Halegoua-DeMarzio, D. Role of Probiotics in Non-alcoholic Fatty Liver Disease: Does Gut Microbiota Matter? Nutrients 2019, 11, 2837. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.; Fan, J.G. Microbial metabolites in non-alcoholic fatty liver disease. World J. Gastroenterol. 2019, 25, 2019–2028. [Google Scholar] [CrossRef]
- Chen, J.; Vitetta, L. Gut Microbiota Metabolites in NAFLD Pathogenesis and Therapeutic Implications. Int. J. Mol. Sci. 2020, 21, 5214. [Google Scholar] [CrossRef]
- Ma, J.; Zhou, Q.; Li, H. Gut Microbiota and Nonalcoholic Fatty Liver Disease: Insights on Mechanisms and Therapy. Nutrients 2017, 9, 1124. [Google Scholar] [CrossRef] [PubMed]
- Dao, M.C.; Everard, A.; Aron-Wisnewsky, J.; Sokolovska, N.; Prifti, E.; Verger, E.O.; Kayser, B.D.; Levenez, F.; Chilloux, J.; Hoyles, L.; et al. Akkermansia muciniphila and improved metabolic health during a dietary intervention in obesity: Relationship with gut microbiome richness and ecology. Gut 2016, 65, 426–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zhao, F.; Wang, Y.; Chen, J.; Tao, J.; Tian, G.; Wu, S.; Liu, W.; Cui, Q.; Geng, B.; et al. Gut microbiota dysbiosis contributes to the development of hypertension. Microbiome 2017, 5, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Yassour, M.; Lim, M.Y.; Yun, H.S.; Tickle, T.L.; Sung, J.; Song, Y.M.; Lee, K.; Franzosa, E.A.; Morgan, X.C.; Gevers, D.; et al. Sub-clinical detection of gut microbial biomarkers of obesity and type 2 diabetes. Genome Med. 2016, 8, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.T.; Nieuwdorp, M.; Bäckhed, F. Microbial modulation of insulin sensitivity. Cell Metab. 2014, 20, 753–760. [Google Scholar] [CrossRef] [Green Version]
- Lukovac, S.; Belzer, C.; Pellis, L.; Keijser, B.J.; De Vos, W.M.; Montijn, R.C.; Roeselers, G. Differential modulation by Akkermansia muciniphila and Faecalibacterium prausnitzii of host peripheral lipid metabolism and histone acetylation in mouse gut organoids. mBio 2014, 5, e01438-14. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.; You, H.J.; Bajaj, J.S.; Joo, S.K.; Yu, J.; Park, S.; Kang, H.; Park, J.H.; Kim, J.H.; Lee, D.H.; et al. Distinct signatures of gut microbiome and metabolites associated with significant fibrosis in non-obese NAFLD. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Kara, D.; Luppens, S.B.; Cate, J.M. Differences between single- and dual-species biofilms of Streptococcus mutans and Veillonella parvula in growth, acidogenicity and susceptibility to chlorhexidine. Eur. J. Oral Sci. 2006, 114, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N. Metabolic disease as a risk of hepatocellular carcinoma. Clin. Mol. Hepatol. 2021, 27, 87–90. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Cope, K.; Risby, T.H.; Diehl, A.M. Obesity and female gender increase breath ethanol concentration: Potential implications for the pathogenesis of nonalcoholic steatohepatitis. Am. J. Gastroenterol. 2001, 96, 1200–1204. [Google Scholar] [CrossRef]
- Baker, S.S.; Baker, R.D.; Liu, W.; Nowak, N.J.; Zhu, L. Role of alcohol metabolism in non-alcoholic steatohepatitis. PLoS ONE 2010, 5, e9570. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Healthy Control (n = 44) | MAFLD (n = 22) | p Value | |
|---|---|---|---|
| Age (year) | 51.0 (47.0–55.0) | 46.0 (33.8–58.7) | 0.19 |
| Sex, male | 12 (27.3) | 6 (27.3) | 1.00 |
| BMI (kg/m2) | 20.8 (20.2–22.5) | 28.7 (26.6–30.8) | <0.001 |
| T2DM | 0 (0.0) | 4 (18.2) | 0.01 |
| Hypertension | 0 (0.0) | 3 (13.6) | 0.034 |
| Dyslipidemia | 0 (0.0) | 6 (27.3) | 0.001 |
| AST (IU/L) | 20.0 (17.0–23.3) | 63.5 (42.2–105.7) | <0.001 |
| ALT (IU/L) | 15.5 (13.0–20.0) | 106.0 (53.5–126.5) | <0.001 |
| ALP (IU/L) | 56.0 (45.7–69.2) | 72.5 (58.7–85.5) | <0.001 |
| Total cholesterol (mg/dL) | 179.5 (167.7–192.0) | 169.0 (135.0–198.0) | 0.48 |
| FIB-4 | 1.02 (0.92–1.51) | 1.83 (0.92–2.41) | 0.001 |
| Healthy Control (n = 44) | MAFLD (n = 22) | p Value | |
|---|---|---|---|
| Firmicutes | 60.15 | 50.08 | 0.045 |
| Clostridiales | 53.46 | 35.84 | <0.001 |
| Lachnospiraceae | 32.59 | 25.70 | 0.01 |
| Coprococcus | 0.90 | 0.32 | 0.014 |
| Eubacterium eligens | 1.91 | 0.43 | <0.001 |
| Ruminococcaceae | 18.56 | 8.49 | <0.001 |
| Faecalibacterium | 8.45 | 3.75 | <0.001 |
| Ruminococcus | 1.38 | 0.31 | <0.001 |
| Oscillibacter | 1.31 | 0.82 | 0.015 |
| Agathobaculum | 0.68 | 0.30 | <0.001 |
| Lactobacillales | 1.239 | 2.961 | n.s. |
| Lactobacillaceae | 0.568 | 1.067 | n.s. |
| Lactobacillus | 0.56 | 1.06 | n.s. |
| Enterococcaceae | 0.031 | 0.290 | 0.033 |
| Enterococcus | 0.03 | 0.28 | 0.016 |
| Streptococcaceae | 0.570 | 1.164 | n.s. |
| Streptococcus | 0.55 | 1.10 | n.s. |
| Selenomonadales | 1.242 | 4.463 | n.s. |
| Veillonellaceae | 1.043 | 3.832 | 0.015 |
| Megamonas | 1.16 | 3.53 | n.s. |
| Veillonella | <1 | 2.40 | n.s. |
| Bacteroidetes | 33.07 | 31.36 | n.s. |
| Bacteroidales | 33.064 | 31.362 | n.s. |
| Bacteroidaceae | 15.245 | 15.367 | n.s. |
| Bacteroides | 15.24 | 15.36 | n.s. |
| Prevotellaceae | 15.145 | 14.893 | n.s. |
| Prevotella | 13.88 | 14.14 | n.s. |
| Proteobacteria | 3.09 | 10.69 | 0.001 |
| Enterobacterales | 1.28 | 9.34 | <0.001 |
| Enterobacteriaceae | 1.27 | 9.18 | <0.001 |
| Enterobacter | 0.016 | 1.01 | 0.04 |
| Escherichia | 0.81 | 2.09 | 0.004 |
| Citrobacter | 0.005 | 1.13 | <0.001 |
| Acinectobacteria | 2.54 | 7.68 | 0.021 |
| Bifidobacteriales | 2.069 | 6.437 | n.s. |
| Bifidobacteriaceae | 2.069 | 6.437 | n.s. |
| Bifidobacterium | 2.07 | 6.38 | n.s. |
| Verrucomicrobia | 0.14 | 0.004 | 0.024 |
| Verrucomicrobiales | 0.138 | 0.004 | n.s. |
| Akkermansiaceae | 0.138 | 0.004 | n.s. |
| Akkermansia | 0.13 | 0.004 | n.s. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, J.H.; Lee, J.H.; Cho, M.S.; Kim, H.; Chun, J.; Lee, J.H.; Yoon, Y.; Kang, W. Characterization of Gut Microbiome in Korean Patients with Metabolic Associated Fatty Liver Disease. Nutrients 2021, 13, 1013. https://doi.org/10.3390/nu13031013
Oh JH, Lee JH, Cho MS, Kim H, Chun J, Lee JH, Yoon Y, Kang W. Characterization of Gut Microbiome in Korean Patients with Metabolic Associated Fatty Liver Disease. Nutrients. 2021; 13(3):1013. https://doi.org/10.3390/nu13031013
Chicago/Turabian StyleOh, Joo Hyun, Je Hee Lee, Min Seok Cho, Hyeree Kim, Jongsik Chun, Joon Hyeok Lee, Yeup Yoon, and Wonseok Kang. 2021. "Characterization of Gut Microbiome in Korean Patients with Metabolic Associated Fatty Liver Disease" Nutrients 13, no. 3: 1013. https://doi.org/10.3390/nu13031013