
Per- and Polyfluoroalkyl Substance Exposure Combined with High-Fat Diet Supports Prostate Cancer Progression

 , , ,
, , ,
Abstract
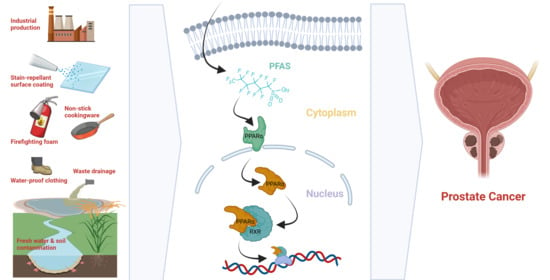
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture and Viability Assays
2.2. In Vivo Prostate Cancer Xenograft Model
2.3. OMICS-Based Metabolic Profiling
2.4. Plate-Based Pyruvate and Acetyl Coa Assays
2.5. Western Blotting for Epigenetic Marker Assessment
3. Results
3.1. PFAS Exposure Increases Cell Proliferation in Malignant Prostate Cancer Cell Lines
3.2. Exposure to PFAS Increases RWPE-kRAS Xenograft Tumor Growth In Vivo
3.3. PFAS Treatment Change Metabolic Phenotype of Prostate Cancer Cells
3.4. PFAS Treatment Increases PPAR Signaling and Histone Acetylation in Prostate Cancer Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Domingo, J.L.; Nadal, M. Human exposure to per- and polyfluoroalkyl substances (PFAS) through drinking water: A review of the recent scientific literature. Environ. Res. 2019, 177, 108648. [Google Scholar] [CrossRef]
- Sunderland, E.M.; Hu, X.C.; Dassuncao, C.; Tokranov, A.K.; Wagner, C.C.; Allen, J.G. A review of the pathways of human exposure to poly- and perfluoroalkyl substances (PFASs) and present understanding of health effects. J. Expo. Sci. Environ. Epidemiol. 2019, 29, 131–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behr, A.C.; Plinsch, C.; Braeuning, A.; Buhrke, T. Activation of human nuclear receptors by perfluoroalkylated substances (PFAS). Toxicol. Vitr. 2020, 62, 104700. [Google Scholar] [CrossRef]
- Ojo, A.F.; Xia, Q.; Peng, C.; Ng, J.C. Evaluation of the individual and combined toxicity of perfluoroalkyl substances to human liver cells using biomarkers of oxidative stress. Chemosphere 2021, 281, 130808. [Google Scholar] [CrossRef]
- Mahinroosta, R.; Senevirathna, L. A review of the emerging treatment technologies for PFAS contaminated soils. J. Environ. Manag. 2020, 255, 109896. [Google Scholar] [CrossRef]
- Temkin, A.M.; Hocevar, B.A.; Andrews, D.Q.; Naidenko, O.V.; Kamendulis, L.M. Application of the Key Characteristics of Carcinogens to Per and Polyfluoroalkyl Substances. Int. J. Environ. Res. Public Health 2020, 17, 1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legoff, L.; D’Cruz, S.C.; Lebosq, M.; Gely-Pernot, A.; Bouchekhchoukha, K.; Monfort, C.; Kernanec, P.Y.; Tevosian, S.; Multigner, L.; Smagulova, F. Developmental exposure to chlordecone induces transgenerational effects in somatic prostate tissue which are associated with epigenetic histone trimethylation changes. Environ. Int. 2021, 152, 106472. [Google Scholar] [CrossRef] [PubMed]
- EPA. Basic Information on PFAS. Available online: https://www.epa.gov/pfas/basic-information-pfas (accessed on 1 July 2021).
- National Institute of Environmental Health Sciences. Perfluoroalkyl and Polyfluoroalkyl Substances (PFAS). Available online: https://www.niehs.nih.gov/health/topics/agents/pfc/index.cfm (accessed on 1 July 2021).
- Costello, L.C.; Franklin, R.B. The clinical relevance of the metabolism of prostate cancer; zinc and tumor suppression: Connecting the dots. Mol. Cancer 2006, 5, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, N.; Wei, S.; Li, M.; Yang, J.; Li, K.; Jin, L.; Xie, Y.; Giesy, J.P.; Zhang, X.; Yu, H. Effects of Perfluorooctanoic Acid on Metabolic Profiles in Brain and Liver of Mouse Revealed by a High-throughput Targeted Metabolomics Approach. Sci. Rep. 2016, 6, 23963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, F.; Jin, Y.; Liu, W.; Quan, X.; Chen, J.; Liang, Z. Global liver proteome analysis using iTRAQ labeling quantitative proteomic technology to reveal biomarkers in mice exposed to perfluorooctane sulfonate (PFOS). Environ. Sci. Technol. 2012, 46, 12170–12177. [Google Scholar] [CrossRef] [PubMed]
- Domazet, S.L.; Grøntved, A.; Timmermann, A.G.; Nielsen, F.; Jensen, T.K. Longitudinal Associations of Exposure to Perfluoroalkylated Substances in Childhood and Adolescence and Indicators of Adiposity and Glucose Metabolism 6 and 12 Years Later: The European Youth Heart Study. Diabetes Care 2016, 39, 1745–1751. [Google Scholar] [CrossRef] [Green Version]
- Alderete, T.L.; Jin, R.; Walker, D.I.; Valvi, D.; Chen, Z.; Jones, D.P.; Peng, C.; Gilliland, F.D.; Berhane, K.; Conti, D.V.; et al. Perfluoroalkyl substances, metabolomic profiling, and alterations in glucose homeostasis among overweight and obese Hispanic children: A proof-of-concept analysis. Environ. Int. 2019, 126, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Qu, Y.Y.; Zhao, R.; Zhang, H.L.; Zhou, Q.; Xu, F.J.; Zhang, X.; Xu, W.H.; Shao, N.; Zhou, S.X.; Dai, B.; et al. Inactivation of the AMPK-GATA3-ECHS1 Pathway Induces Fatty Acid Synthesis That Promotes Clear Cell Renal Cell Carcinoma Growth. Cancer Res. 2020, 80, 319–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, G.; Jacobs, K.B.; Yeager, M.; Kraft, P.; Wacholder, S.; Orr, N.; Yu, K.; Chatterjee, N.; Welch, R.; Hutchinson, A.; et al. Multiple loci identified in a genome-wide association study of prostate cancer. Nat. Genet. 2008, 40, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Kingsley, S.L.; Walker, D.I.; Calafat, A.M.; Chen, A.; Papandonatos, G.D.; Xu, Y.; Jones, D.P.; Lanphear, B.P.; Pennell, K.D.; Braun, J.M. Metabolomics of childhood exposure to perfluoroalkyl substances: A cross-sectional study. Metabolomics 2019, 15, 95. [Google Scholar] [CrossRef]
- The American Cancer Society Medical and Editorial Content Team. Key Statistics for Prostate Cancer. Available online: https://www.cancer.org/cancer/prostate-cancer/about/key-statistics.html (accessed on 1 July 2021).
- Chang, E.T.; Adami, H.O.; Boffetta, P.; Cole, P.; Starr, T.B.; Mandel, J.S. A critical review of perfluorooctanoate and perfluorooctanesulfonate exposure and cancer risk in humans. Crit. Rev. Toxicol. 2014, 44 (Suppl. 1), 1–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vieira, V.M.; Hoffman, K.; Shin, H.M.; Weinberg, J.M.; Webster, T.F.; Fletcher, T. Perfluorooctanoic acid exposure and cancer outcomes in a contaminated community: A geographic analysis. Environ. Health Perspect. 2013, 121, 318–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundin, J.I.; Alexander, B.H.; Olsen, G.W.; Church, T.R. Ammonium perfluorooctanoate production and occupational mortality. Epidemiology 2009, 20, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Eriksen, K.T.; Sørensen, M.; McLaughlin, J.K.; Lipworth, L.; Tjønneland, A.; Overvad, K.; Raaschou-Nielsen, O. Perfluorooctanoate and perfluorooctanesulfonate plasma levels and risk of cancer in the general Danish population. J. Natl. Cancer Inst. 2009, 101, 605–609. [Google Scholar] [CrossRef]
- Gilliland, F.D.; Mandel, J.S. Mortality among employees of a perfluorooctanoic acid production plant. J. Occup. Med. Off. Publ. Ind. Med Assoc. 1993, 35, 950–954. [Google Scholar] [CrossRef]
- Barry, V.; Winquist, A.; Steenland, K. Perfluorooctanoic acid (PFOA) exposures and incident cancers among adults living near a chemical plant. Environ. Health Perspect. 2013, 121, 1313–1318. [Google Scholar] [CrossRef] [Green Version]
- Hardell, E.; Kärrman, A.; van Bavel, B.; Bao, J.; Carlberg, M.; Hardell, L. Case-control study on perfluorinated alkyl acids (PFAAs) and the risk of prostate cancer. Environ. Int. 2014, 63, 35–39. [Google Scholar] [CrossRef]
- Labbé, D.P.; Zadra, G.; Yang, M.; Reyes, J.M.; Lin, C.Y.; Cacciatore, S.; Ebot, E.M.; Creech, A.L.; Giunchi, F.; Fiorentino, M.; et al. High-fat diet fuels prostate cancer progression by rewiring the metabolome and amplifying the MYC program. Nat. Commun. 2019, 10, 4358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Priolo, C.; Pyne, S.; Rose, J.; Regan, E.R.; Zadra, G.; Photopoulos, C.; Cacciatore, S.; Schultz, D.; Scaglia, N.; McDunn, J.; et al. AKT1 and MYC induce distinctive metabolic fingerprints in human prostate cancer. Cancer Res. 2014, 74, 7198–7204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, Q.; Mogol, A.N.; Liu, Y.-J.; Casiano, A.S.; Chien, C.; Drnevich, J.; Imir, O.B.; Kulkoyluoglu-Cotul, E.; Park, N.H.; Shapiro, D.J.; et al. Targeting metabolic adaptations in the breast cancer–liver metastatic niche using dietary approaches to improve endocrine therapy efficacy. bioRxiv 2021. [Google Scholar] [CrossRef]
- Cotul, E.K.; Zuo, Q.; Santaliz-Casiano, A.; Imir, O.B.; Mogol, A.N.; Tunc, E.; Duong, K.; Lee, J.K.; Ramesh, R.; Odukoya, E.; et al. Combined Targeting of Estrogen Receptor Alpha and Exportin 1 in Metastatic Breast Cancers. Cancers 2020, 12, 2397. [Google Scholar] [CrossRef]
- Kulkoyluoglu-Cotul, E.; Arca, A.; Madak-Erdogan, Z. Crosstalk between Estrogen Signaling and Breast Cancer Metabolism. Trends Endocrinol. Metab. 2019, 30, 25–38. [Google Scholar] [CrossRef]
- Kulkoyluoglu-Cotul, E.; Smith, B.P.; Wrobel, K.; Zhao, Y.C.; Chen, K.L.A.; Hieronymi, K.; Imir, O.B.; Duong, K.; O’Callaghan, C.; Mehta, A.; et al. Combined Targeting of Estrogen Receptor Alpha and XPO1 Prevent Akt Activation, Remodel Metabolic Pathways and Induce Autophagy to Overcome Tamoxifen Resistance. Cancers 2019, 11, 479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuo, Q.; Chen, K.L.; Arredondo Eve, A.; Liu, Y.-J.; Kim, S.H.; Katzenellenbogen, B.S.; Katzenellenbogen, J.A.; Madak-Erdogan, Z. Pathway Preferential Estrogens Prevent Hepatosteatosis Due to Ovariectomy and High−Fat Diets. Nutrients 2021, 13, 3334. [Google Scholar] [CrossRef] [PubMed]
- Madak-Erdogan, Z.; Gong, P.; Zhao, Y.C.; Xu, L.; Wrobel, K.U.; Hartman, J.A.; Wang, M.; Cam, A.; Iwaniec, U.T.; Turner, R.T.; et al. Dietary licorice root supplementation reduces diet-induced weight gain, lipid deposition, and hepatic steatosis in ovariectomized mice without stimulating reproductive tissues and mammary gland. Mol. Nutr. Food Res. 2016, 60, 369–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomayko, M.M.; Reynolds, C.P. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother. Pharmacol. 1989, 24, 148–154. [Google Scholar] [CrossRef]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.-É.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef]
- Madak-Erdogan, Z.; Band, S.; Zhao, Y.C.; Smith, B.P.; Kulkoyluoglu-Cotul, E.; Zuo, Q.; Santaliz Casiano, A.; Wrobel, K.; Rossi, G.; Smith, R.L.; et al. Free fatty acids rewire cancer metabolism in obesity-associated breast cancer via estrogen receptor and mTOR signaling. Cancer Res. 2019, 79, 2494–2510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madak-Erdogan, Z.; Charn, T.H.; Jiang, Y.; Liu, E.T.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Integrative genomics of gene and metabolic regulation by estrogen receptors alpha and beta, and their coregulators. Mol. Syst. Biol. 2013, 9, 676. [Google Scholar] [CrossRef] [PubMed]
- Madak-Erdogan, Z.; Kim, S.H.; Gong, P.; Zhao, Y.C.; Zhang, H.; Chambliss, K.L.; Carlson, K.E.; Mayne, C.G.; Shaul, P.W.; Korach, K.S.; et al. Design of pathway preferential estrogens that provide beneficial metabolic and vascular effects without stimulating reproductive tissues. Sci. Signal. 2016, 9, ra53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madak-Erdogan, Z.; Lupien, M.; Stossi, F.; Brown, M.; Katzenellenbogen, B.S. Genomic collaboration of estrogen receptor alpha and extracellular signal-regulated kinase 2 in regulating gene and proliferation programs. Mol. Cell. Biol. 2011, 31, 226–236. [Google Scholar] [CrossRef] [Green Version]
- Madak-Erdogan, Z.; Ventrella, R.; Petry, L.; Katzenellenbogen, B.S. Novel roles for ERK5 and cofilin as critical mediators linking ERalpha-driven transcription, actin reorganization, and invasiveness in breast cancer. Mol. Cancer Res. 2014, 12, 714–727. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.C.; Madak Erdogan, Z. Systems Biology of Metabolic Regulation by Estrogen Receptor Signaling in Breast Cancer. J. Vis. Exp. 2016, 109, e53832. [Google Scholar] [CrossRef] [Green Version]
- Leav, I.; Plescia, J.; Goel, H.L.; Li, J.; Jiang, Z.; Cohen, R.J.; Languino, L.R.; Altieri, D.C. Cytoprotective mitochondrial chaperone TRAP-1 as a novel molecular target in localized and metastatic prostate cancer. Am. J. Pathol. 2010, 176, 393–401. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.L.; Hsu, S.C.; Chung, T.Y.; Chu, C.Y.; Wang, H.J.; Hsiao, P.W.; Yeh, S.D.; Ann, D.K.; Yen, Y.; Kung, H.J. Arginine is an epigenetic regulator targeting TEAD4 to modulate OXPHOS in prostate cancer cells. Nat. Commun. 2021, 12, 2398. [Google Scholar] [CrossRef] [PubMed]
- Vidal, A.C.; Oyekunle, T.; Howard, L.E.; De Hoedt, A.M.; Kane, C.J.; Terris, M.K.; Cooperberg, M.R.; Amling, C.L.; Klaassen, Z.; Freedland, S.J.; et al. Obesity, race, and long-term prostate cancer outcomes. Cancer 2020, 126, 3733–3741. [Google Scholar] [CrossRef] [PubMed]
- Freire, P.F.; Martin, J.P.; Herrero, O.; Peropadre, A.; de la Pena, E.; Hazen, M. In vitro assessment of the cytotoxic and mutagenic potential of perfluorooctanoic acid. Toxicol. Vitr. 2008, 22, 1228–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eriksen, K.T.; Raaschou-Nielsen, O.; Sørensen, M.; Roursgaard, M.; Loft, S.; Møller, P. Genotoxic potential of the perfluorinated chemicals PFOA, PFOS, PFBS, PFNA and PFHxA in human HepG2 cells. Mutat. Res. Genet. Toxicol. Environ. Mutagenes. 2010, 700, 39–43. [Google Scholar] [CrossRef]
- Lindeman, B.; Maass, C.; Duale, N.; Gützkow, K.B.; Brunborg, G.; Andreassen, Å. Effects of per-and polyfluorinated compounds on adult rat testicular cells following in vitro exposure. Reprod. Toxicol. 2012, 33, 531–537. [Google Scholar] [CrossRef]
- Wan, Y.-J.; Li, Y.-Y.; Xia, W.; Chen, J.; Lv, Z.-Q.; Zeng, H.-C.; Zhang, L.; Yang, W.-J.; Chen, T.; Lin, Y. Alterations in tumor biomarker GSTP gene methylation patterns induced by prenatal exposure to PFOS. Toxicology 2010, 274, 57–64. [Google Scholar] [CrossRef]
- Tian, M.; Peng, S.; Martin, F.L.; Zhang, J.; Liu, L.; Wang, Z.; Dong, S.; Shen, H. Perfluorooctanoic acid induces gene promoter hypermethylation of glutathione-S-transferase Pi in human liver L02 cells. Toxicology 2012, 296, 48–55. [Google Scholar] [CrossRef]
- Rashid, F.; Ramakrishnan, A.; Fields, C.; Irudayaraj, J. Acute PFOA exposure promotes epigenomic alterations in mouse kidney tissues. Toxicol. Rep. 2020, 7, 125–132. [Google Scholar] [CrossRef]
- Wen, Y.; Mirji, N.; Irudayaraj, J. Epigenetic toxicity of PFOA and GenX in HepG2 cells and their role in lipid metabolism. Toxicol. Vitr. 2020, 65, 104797. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Preston, R.; Goldman, L.R.; Brebi-Mieville, P.; Ili-Gangas, C.; LeBron, C.; Witter, F.R.; Apelberg, B.J.; Hernández-Roystacher, M.; Jaffe, A.; Halden, R.U. Global DNA hypomethylation is associated with in utero exposure to cotinine and perfluorinated alkyl compounds. Epigenetics 2010, 5, 539–546. [Google Scholar] [CrossRef] [Green Version]
- Watkins, D.J.; Wellenius, G.A.; Butler, R.A.; Bartell, S.M.; Fletcher, T.; Kelsey, K.T. Associations between serum perfluoroalkyl acids and LINE-1 DNA methylation. Environ. Int. 2014, 63, 71–76. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.; Azumi, K.; Goudarzi, H.; Araki, A.; Miyashita, C.; Kobayashi, S.; Itoh, S.; Sasaki, S.; Ishizuka, M.; Nakazawa, H. Effects of prenatal perfluoroalkyl acid exposure on cord blood IGF2/H19 methylation and ponderal index: The Hokkaido Study. J. Expo. Sci. Environ. Epidemiol. 2017, 27, 251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patsouris, D.; Reddy, J.K.; Müller, M.; Kersten, S. Peroxisome proliferator-activated receptor alpha mediates the effects of high-fat diet on hepatic gene expression. Endocrinology 2006, 147, 1508–1516. [Google Scholar] [CrossRef] [Green Version]
- DeWitt, J.C.; Shnyra, A.; Badr, M.Z.; Loveless, S.E.; Hoban, D.; Frame, S.R.; Cunard, R.; Anderson, S.E.; Meade, B.J.; Peden-Adams, M.M.; et al. Immunotoxicity of perfluorooctanoic acid and perfluorooctane sulfonate and the role of peroxisome proliferator-activated receptor alpha. Crit. Rev. Toxicol. 2009, 39, 76–94. [Google Scholar] [CrossRef] [PubMed]
- Evans, N.; Cardon, M.; Hartig, P.; Medlock Kakaley, E.; Wilson, V.; Conley, J.; Gray, L.E. In vitro binding of human and rat PPAR alpha, beta/delta, and gamma receptors to PFAS, fatty acids, and clofibric acid. In Proceedings of the VIRTUAL-Society of Toxicology Annual Meeting, Anaheim, CA, USA, 30 April 2020. [Google Scholar]
- Wolf, D.C.; Moore, T.; Abbott, B.D.; Rosen, M.B.; Das, K.P.; Zehr, R.D.; Lindstrom, A.B.; Strynar, M.J.; Lau, C. Comparative Hepatic Effects of Perfluorooctanoic Acid and WY 14,643 in PPAR-α Knockout and Wild-type Mice. Toxicol. Pathol. 2008, 36, 632–639. [Google Scholar] [CrossRef] [Green Version]
- Stanifer, J.W.; Stapleton, H.M.; Souma, T.; Wittmer, A.; Zhao, X.; Boulware, L.E. Perfluorinated Chemicals as Emerging Environmental Threats to Kidney Health. A Scoping Review. Clin. J. Am. Soc. Nephrol. 2018, 13, 1479–1492. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Imir, O.B.; Kaminsky, A.Z.; Zuo, Q.-Y.; Liu, Y.-J.; Singh, R.; Spinella, M.J.; Irudayaraj, J.; Hu, W.-Y.; Prins, G.S.; Madak Erdogan, Z. Per- and Polyfluoroalkyl Substance Exposure Combined with High-Fat Diet Supports Prostate Cancer Progression. Nutrients 2021, 13, 3902. https://doi.org/10.3390/nu13113902
Imir OB, Kaminsky AZ, Zuo Q-Y, Liu Y-J, Singh R, Spinella MJ, Irudayaraj J, Hu W-Y, Prins GS, Madak Erdogan Z. Per- and Polyfluoroalkyl Substance Exposure Combined with High-Fat Diet Supports Prostate Cancer Progression. Nutrients. 2021; 13(11):3902. https://doi.org/10.3390/nu13113902
Chicago/Turabian StyleImir, Ozan Berk, Alanna Zoe Kaminsky, Qian-Ying Zuo, Yu-Jeh Liu, Ratnakar Singh, Michael J. Spinella, Joseph Irudayaraj, Wen-Yang Hu, Gail S. Prins, and Zeynep Madak Erdogan. 2021. "Per- and Polyfluoroalkyl Substance Exposure Combined with High-Fat Diet Supports Prostate Cancer Progression" Nutrients 13, no. 11: 3902. https://doi.org/10.3390/nu13113902