
Lactoferrin Protects against Methamphetamine Toxicity by Modulating Autophagy and Mitochondrial Status

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Design
- (i)
- The occurrence of phenotypic changes and altered cell viability produced by LF alone.
- (ii)
- The potential protective effects of LF on METH-induced toxicity. The METH dose used in the present study was 100 μM, which has been recently demonstrated [48] to produce a moderate toxicity in PC12 cells after 72 h of exposure.
- (iii)
- The effects of LF on METH-induced mitochondrial alterations.
- (iv)
- The involvement of specific autophagy steps in the protective effects of LF against METH-induced toxicity.
2.2. Cell Cultures
2.3. Cell Treatments
2.4. WST-1 Assay
2.5. Trypan Blue Staining
2.6. Fluoro Jade B Staining
2.7. Hematoxylin and Eosin (H&E) Histochemistry
2.8. Autophagy and Mitochondria Study
2.9. Mitochondrial Labelling
2.10. Immunocytochemistry at Light Microscopy
2.11. Western Blotting
2.12. Transmission Electron Microscopy
2.13. Post-Embedding Immunocytochemistry
2.14. Ultrastructural Morphometry
2.15. Statistical Analysis
3. Results
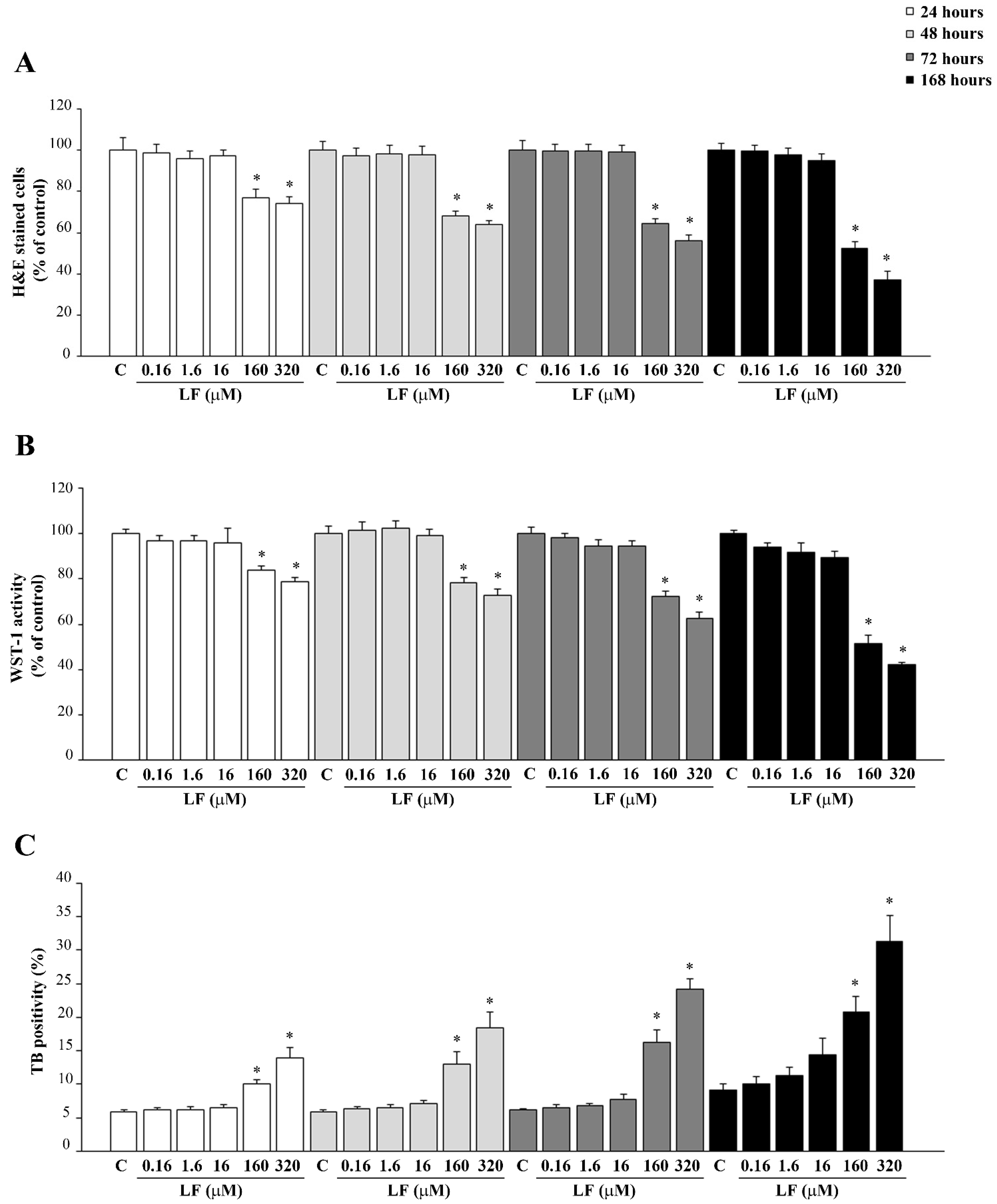
3.1. Dose- and Time-Dependent Effects of LF on PC12 Cell Viability
3.2. LF Induces Elongation of PC12 Cells
3.3. LF Reduces METH-Induced Toxicity in PC12 Cells
3.4. Combined Exposure to LF and METH Enhances Phenotypic Changes in PC12 Cells
3.5. LF Increases Healthy Mitochondria while Counteracting METH-Induced Mithocondrial Alterations
3.6. LF Promotes LC3 Compartmentalization and Reverses METH-Induced LC3 Dissipation
3.7. LF Promotes the Merging of LC3 with Cathepsin D
3.8. LF Promotes the Merging of LC3 with LAMP1 and Promotes the Fusion of Autophagosomes with Lysosomes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lönnerdal, B.; Iyer, S. Lactoferrin: Molecular Structure and Biological Function. Annu. Rev. Nutr. 1995, 15, 93–110. [Google Scholar] [CrossRef]
- Anderson, B.F.; Baker, H.M.; Norris, G.E.; Rice, D.W.; Baker, E.N. Structure of human lactoferrin: Crystallographic structure analysis and refinement at 2.8 A resolution. J. Mol. Biol. 1989, 209, 711–734. [Google Scholar] [CrossRef]
- Moore, S.A.; Anderson, B.F.; Groom, C.R.; Haridas, M.; Baker, E.N. Three-dimensional structure of diferric bovine lactoferrin at 2.8 A resolution. J. Mol. Biol. 1997, 274, 222–236. [Google Scholar] [CrossRef]
- Baker, E.N.; Baker, H.M. Molecular structure, binding properties and dynamics of lactoferrin. Cell. Mol. Life Sci. 2005, 62, 2531–2539. [Google Scholar] [CrossRef]
- Puddu, P.; Valenti, P.; Gessani, S. Immunomodulatory effects of lactoferrin on antigen presenting cells. Biochimie 2009, 91, 11–18. [Google Scholar] [CrossRef]
- Puddu, P.; Latorre, D.; Carollo, M.; Catizone, A.; Ricci, G.; Valenti, P.; Gessani, S. Bovine lactoferrin counteracts Toll-like receptor mediated activation signals in antigen presenting cells. PLoS ONE 2011, 6, e22504. [Google Scholar] [CrossRef]
- Masson, P.; Heremans, J.; Dive, C. An iron-binding protein common to many external secretions. Clin. Chim. Acta 1966, 14, 735–739. [Google Scholar] [CrossRef]
- Vorland, L.H. Lactoferrin: A multifunctional glycoprotein. APMIS 1999, 107, 971–981. [Google Scholar] [CrossRef]
- Masson, P.L.; Heremans, J.F. Lactoferrin in milk from different species. Comp. Biochem. Physiol. 1971, 39, 119–129. [Google Scholar] [CrossRef]
- Masson, P.L.; Heremans, J.F.; Schonne, E. Lactoferrin, an iron-binding protein in neutrophilic leukocytes. J. Exp. Med. 1969, 130, 643–658. [Google Scholar] [CrossRef] [Green Version]
- Huang, R.; Han, L.; Li, J.; Ren, F.; Ke, W.; Jiang, C.; Pei, Y. Neuroprotection in a 6-hydroxydopamine-lesioned Parkinson model using lactoferrin-modified nanoparticles. J. Gene Med. 2009, 11, 754–763. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Ke, W.; Liu, Y.; Wu, D.; Feng, L.; Jiang, C.; Pei, Y. Gene therapy using lactoferrin-modified nanoparticles in a rotenone-induced chronic Parkinson model. J. Neurol. Sci. 2010, 290, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Shi, Y.; Jiang, W.; Han, J.; Huang, S.; Jiang, X. Lactoferrin conjugated PEG-PLGA nanoparticles for brain delivery: Preparation, characterization and efficacy in Parkinson’s disease. Int. J. Pharm. 2011, 415, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Kamalinia, G.; Khodagholi, F.; Atyabi, F.; Amini, M.; Shaerzadeh, F.; Sharifzadeh, M.; Dinarvand, R. Enhanced Brain Delivery of Deferasirox–Lactoferrin Conjugates for Iron Chelation Therapy in Neurodegenerative Disorders: In Vitro and in Vivo Studies. Mol. Pharm. 2013, 10, 4418–4431. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.-C.; Wang, C.-T. Protection of SK-N-MC cells against β-amyloid peptide-induced degeneration using neuron growth factor-loaded liposomes with surface lactoferrin. Biomaterials 2014, 35, 5954–5964. [Google Scholar] [CrossRef]
- Bollimpelli, V.S.; Kumar, P.; Kumari, S.; Kondapi, A.K. Neuroprotective effect of curcumin-loaded lactoferrin nano particles against rotenone induced neurotoxicity. Neurochem. Int. 2016, 95, 37–45. [Google Scholar] [CrossRef]
- Yan, X.; Xu, L.; Bi, C.; Duan, D.; Chu, L.; Yu, X.; Wu, Z.; Wang, A.; Sun, K. Lactoferrin-modified rotigotine nanoparticles for enhanced nose-to-brain delivery: LESA-MS/MS-based drug biodistribution, pharmacodynamics, and neuroprotective effects. Int. J. Nanomed. 2018, 13, 273–281. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.; Wang, A.; Yan, X.; Chu, L.; Yang, X.; Song, Y.; Sun, K.; Yu, X.; Liu, R.; Wu, Z.; et al. Brain-targeted intranasal delivery of dopamine with borneol and lactoferrin co-modified nanoparticles for treating Parkinson’s disease. Drug Deliv. 2019, 26, 700–707. [Google Scholar] [CrossRef] [Green Version]
- Rascón-Cruz, Q.; Espinoza-Sánchez, E.A.; Siqueiros-Cendón, T.S.; Nakamura-Bencomo, S.I.; Arévalo-Gallegos, S.; Iglesias-Figueroa, B.F. Lactoferrin: A Glycoprotein Involved in Immunomodulation, Anticancer, and Antimicrobial Processes. Molecules 2021, 26, 205. [Google Scholar] [CrossRef]
- Kawamata, T.; Tooyama, I.; Yamada, T.; Walker, D.G.; McGeer, P.L. Lactotransferrin immunocytochemistry in Alzheimer and normal human brain. Am. J. Pathol. 1993, 142, 1574–1585. [Google Scholar]
- Shimaoka, S.; Hamaoka, H.; Inoue, J.; Asanuma, M.; Tooyama, I.; Kondo, Y. Lactoferrin-like Immunoreactivity in Distinct Neuronal Populations in the Mouse Central Nervous System. Acta Med. Okayama 2021, 75, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Caccavo, D.; Pellegrino, N.M.; Altamura, M.; Rigon, A.; Amati, L.; Amoroso, A.; Jirillo, E. Antimicrobial and immunoregu-latory functions of lactoferrin and its potential therapeutic application. J. Endotoxin Res. 2002, 8, 403–417. [Google Scholar]
- Legrand, D. Overview of Lactoferrin as a Natural Immune Modulator. J. Pediatr. 2016, 173, S10–S15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osmand, A.P.; Switzer, R.C., III. Differential distribution of lactoferrin and Alz-50 immunoreactivities in neuritic plaques and neurofibrillary tangles in Alzheimer’s disease. In Alzheimer’s Disease: Basic Mechanisms, Diagnosis and Therapeutic Strategies; Iqbal, K., McLachlan, D.R.C., Winblad, B., Wisniewski, H.M., Eds.; Wiley: Hoboken, NJ, USA, 1991; pp. 219–228. [Google Scholar]
- Leveugle, B.; Spik, G.; Perl, D.P.; Bouras, C.; Fillit, H.M.; Hof, P.R. The iron-binding protein lactotransferrin is present in pathologic lesions in a variety of neurodegenerative disorders: A comparative immunohistochemical analysis. Brain Res. 1994, 650, 20–31. [Google Scholar] [CrossRef]
- Leveugle, B.; Faucheux, B.A.; Bouras, C.; Nillesse, N.; Spik, G.; Hirsch, E.; Agid, Y.; Hof, P.R. Cellular distribution of the iron-binding protein lactotransferrin in the mesencephalon of Parkinson’s disease cases. Acta Neuropathol. 1996, 91, 566–572. [Google Scholar] [CrossRef]
- Wang, L.; Sato, H.; Zhao, S.; Tooyama, I. Deposition of lactoferrin in fibrillar-type senile plaques in the brains of transgenic mouse models of Alzheimer’s disease. Neurosci. Lett. 2010, 481, 164–167. [Google Scholar] [CrossRef]
- Fillebeen, C.; Mitchell, V.; Dexter, D.; Benaissa, M.; Beauvillain, J.-C.; Spik, G.; Pierce, A. Lactoferrin is synthesized by mouse brain tissue and its expression is enhanced after MPTP treatment. Mol. Brain Res. 1999, 72, 183–194. [Google Scholar] [CrossRef]
- Fillebeen, C.; Ruchoux, M.-M.; Mitchell, V.; Vincent, S.; Benaïssa, M.; Pierce, A. Lactoferrin is synthesized by activated microglia in the human substantia nigra and its synthesis by the human microglial CHME cell line is upregulated by tumor necrosis factor α or 1-methyl-4-phenylpyridinium treatment. Mol. Brain Res. 2001, 96, 103–113. [Google Scholar] [CrossRef]
- Guo, C.; Yang, Z.-H.; Zhang, S.; Chai, R.; Xue, H.; Zhang, Y.-H.; Li, J.-Y.; Wang, Z.-Y. Intranasal Lactoferrin Enhances α-Secretase-Dependent Amyloid Precursor Protein Processing via the ERK1/2-CREB and HIF-1α Pathways in an Alzheimer’s Disease Mouse Model. Neuropsychopharmacology 2017, 42, 2504–2515. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, W.A.; Salama, R.M.; Schaalan, M.F. A pilot study on the effect of lactoferrin on Alzheimer’s disease pathological sequelae: Impact of the p-Akt/PTEN pathway. Biomed. Pharmacother. 2019, 111, 714–723. [Google Scholar] [CrossRef] [PubMed]
- González-Sánchez, M.; Bartolome, F.; Antequera, D.; Puertas-Martín, V.; González, P.; Gómez-Grande, A.; Llamas-Velasco, S.; Martín, A.H.-S.; Pérez-Martínez, D.; Villarejo-Galende, A.; et al. Decreased salivary lactoferrin levels are specific to Alzheimer’s disease. EBioMedicine 2020, 57, 102834. [Google Scholar] [CrossRef]
- Zakharova, E.T.; Sokolov, A.V.; Pavlichenko, N.N.; Kostevich, V.A.; Abdurasulova, I.N.; Chechushkov, A.V.; Voynova, I.V.; Elizarova, A.Y.; Kolmakov, N.N.; Bass, M.G.; et al. Erythropoietin and Nrf2: Key factors in the neuroprotection provided by apo-lactoferrin. BioMetals 2018, 31, 425–443. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, E.C.; Michel, P.P.; Hirsch, E. The Iron-Binding Protein Lactoferrin Protects Vulnerable Dopamine Neurons from Degeneration by Preserving Mitochondrial Calcium Homeostasis. Mol. Pharmacol. 2013, 84, 888–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Bi, M.; Liu, H.; Song, N.; Xie, J. The protective effect of lactoferrin on ventral mesencephalon neurons against MPP+ is not connected with its iron binding ability. Sci. Rep. 2015, 5, 10729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, S.-F.; Zhang, Y.-H.; Wang, S.; Pang, Z.-Q.; Fan, Y.-G.; Li, J.-Y.; Wang, Z.-Y.; Guo, C. Lactoferrin ameliorates dopaminergic neurodegeneration and motor deficits in MPTP-treated mice. Redox Biol. 2019, 21, 101090. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wu, H.; Zhu, N.; Xu, Z.; Wang, Y.; Qu, Y.; Wang, J. Lactoferrin protects against iron dysregulation, oxidative stress, and apoptosis in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced Parkinson’s disease in mice. J. Neurochem. 2020, 152, 397–415. [Google Scholar] [CrossRef]
- Kopaeva, M.Y.; Cherepov, A.B.; Nesterenko, M.V.; Zarayskaya, I.Y. Pretreatment with Human Lactoferrin Had a Positive Effect on the Dynamics of Mouse Nigrostriatal System Recovery after Acute MPTP Exposure. Biology 2021, 10, 24. [Google Scholar] [CrossRef]
- Burrow, H.; Kanwar, R.K.; Kanwar, J.R. Antioxidant enzyme activities of iron-saturated bovine lactoferrin (Fe-bLf) in human gut epithelial cells under oxidative stress. Med. Chem. 2011, 7, 224–230. [Google Scholar] [CrossRef]
- Aizawa, S.; Hoki, M.; Yamamuro, Y. Lactoferrin promotes autophagy via AMP-activated protein kinase activation through low-density lipoprotein receptor-related protein 1. Biochem. Biophys. Res. Commun. 2017, 493, 509–513. [Google Scholar] [CrossRef]
- Hsu, Y.-H.; Chiu, I.-J.; Lin, Y.-F.; Chen, Y.-J.; Lee, Y.-H.; Chiu, H.-W. Lactoferrin Contributes a Renoprotective Effect in Acute Kidney Injury and Early Renal Fibrosis. Pharmaceutics 2020, 12, 434. [Google Scholar] [CrossRef]
- Huang, L.; Chen, R.; Liu, L.; Zhou, Y.; Chen, Z. Lactoferrin ameliorates pathological cardiac hypertrophy related to mitochondrial quality control in aged mice. Food Funct. 2021, 12, 7514–7526. [Google Scholar] [CrossRef]
- Ravikumar, B.; Rubinsztein, D.C. Can autophagy protect against neurodegeneration caused by aggregate-prone proteins? NeuroReport 2004, 15, 2443–2445. [Google Scholar] [CrossRef]
- Nixon, R.A.; Yang, D.-S.; Lee, J.-H. Neurodegenerative lysosomal disorders: A continuum from development to late age. Autophagy 2008, 4, 590–599. [Google Scholar] [CrossRef] [Green Version]
- Madeo, F.; Eisenberg, T.; Kroemer, G. Autophagy for the avoidance of neurodegeneration. Genes Dev. 2009, 23, 2253–2259. [Google Scholar] [CrossRef] [Green Version]
- Castino, R.; Lazzeri, G.; Lenzi, P.; Bellio, N.; Follo, C.; Ferrucci, M.; Fornai, F.; Isidoro, C. Suppression of autophagy precipitates neuronal cell death following low doses of methamphetamine. J. Neurochem. 2008, 106, 1426–1439. [Google Scholar] [CrossRef]
- Lenzi, P.; Marongiu, R.; Falleni, A.; Gelmetti, V.; Busceti, C.L.; Michiorri, S.; Valente, E.M.; Fornai, F. A subcellular analysis of genetic modulation of PINK1 on mitochondrial alterations, autophagy and cell death. Arch. Ital. Biol. 2012, 150, 194–217. [Google Scholar] [PubMed]
- Ryskalin, L.; Puglisi-Allegra, S.; Lazzeri, G.; Biagioni, F.; Busceti, C.; Balestrini, L.; Fornasiero, A.; Leone, S.; Pompili, E.; Ferrucci, M.; et al. Neuroprotective Effects of Curcumin in Methamphetamine-Induced Toxicity. Molecules 2021, 26, 2493. [Google Scholar] [CrossRef] [PubMed]
- Fleckenstein, A.E.; Metzger, R.R.; Wilkins, D.G.; Gibb, J.W.; Hanson, G.R. Rapid and reversible effects of methamphetamine on dopamine transporters. J. Pharmacol. Exp. Ther. 1997, 282, 834–838. [Google Scholar] [PubMed]
- Kokoshka, J.M.; Vaughan, R.A.; Hanson, G.R.; Fleckenstein, A.E. Nature of methamphetamine-induced rapid and reversible changes in dopamine transporters. Eur. J. Pharmacol. 1998, 361, 269–275. [Google Scholar] [CrossRef]
- Brown, J.M.; Hanson, G.R.; Fleckenstein, A.E. Methamphetamine Rapidly Decreases Vesicular Dopamine Uptake. J. Neurochem. 2000, 74, 2221–2223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandoval, V.; Riddle, E.L.; Hanson, G.R.; Fleckenstein, A.E. Methylphenidate Redistributes Vesicular Monoamine Transporter-2: Role of Dopamine Receptors. J. Neurosci. 2002, 22, 8705–8710. [Google Scholar] [CrossRef] [Green Version]
- Sandoval, V.; Riddle, E.L.; Hanson, G.R.; Fleckenstein, A.E. Methylphenidate Alters Vesicular Monoamine Transport and Prevents Methamphetamine-Induced Dopaminergic Deficits. J. Pharmacol. Exp. Ther. 2003, 304, 1181–1187. [Google Scholar] [CrossRef] [Green Version]
- Cubells, J.F.; Rayport, S.; Rajendran, G.; Sulzer, D. Methamphetamine neurotoxicity involves vacuolation of endocytic organelles and dopamine-dependent intracellular oxidative stress. J. Neurosci. 1994, 14, 2260–2271. [Google Scholar] [CrossRef]
- Sulzer, D.; Rayport, S. Amphetamine and other psychostimulants reduce pH gradients in midbrain dopaminergic neurons and chromaffin granules: A mechanism of action. Neuron 1990, 5, 797–808. [Google Scholar] [CrossRef]
- Riddle, E.L.; Topham, M.K.; Haycock, J.W.; Hanson, G.R.; Fleckenstein, A.E. Differential trafficking of the vesicular monoamine transporter-2 by methamphetamine and cocaine. Eur. J. Pharmacol. 2002, 449, 71–74. [Google Scholar] [CrossRef]
- Kogan, F.J.; Nichols, W.K.; Gibb, J.W. Influence of methamphetamine on nigral and striatal tyrosine hydroxylase activity and on striatal dopamine levels. Eur. J. Pharmacol. 1976, 36, 363–371. [Google Scholar] [CrossRef]
- Wagner, G.C.; Ricaurte, G.A.; Seiden, L.S.; Schuster, C.R.; Miller, R.J.; Westley, J. Long-lasting depletions of striatal dopamine and loss of dopamine uptake sites following repeated administration of methamphetamine. Brain Res. 1980, 181, 151–160. [Google Scholar] [CrossRef]
- O’Dell, S.J.; Weihmuller, F.B.; Marshall, J.F. Multiple methamphetamine injections induce marked increases in extracellular striatal dopamine which correlate with subsequent neurotoxicity. Brain Res. 1991, 564, 256–260. [Google Scholar] [CrossRef]
- Green, A.L.; EL Hait, M.A.S. p-Methoxyamphetamine, a potent reversible inhibitor of type-A monoamine oxidase in vitro and in vivo. J. Pharm. Pharmacol. 1980, 32, 262–266. [Google Scholar] [CrossRef]
- Suzuki, O.; Hattori, H.; Asano, M.; Oya, M.; Katsumata, Y. Inhibition of monoamine oxidase by d-methamphetamine. Biochem. Pharmacol. 1980, 29, 2071–2073. [Google Scholar] [CrossRef]
- Graham, D.G.; Tiffany, S.M.; Vogel, F.S. The Toxicity of Melanin Precursors. J. Investig. Dermatol. 1978, 70, 113–116. [Google Scholar] [CrossRef] [Green Version]
- Cohen, G. Oxy-radical toxicity in catecholamine neurons. Neurotoxicology 1984, 5, 77–82. [Google Scholar]
- Hastings, T.G.; Lewis, D.A.; Zigmond, M.J. Role of oxidation in the neurotoxic effects of intrastriatal dopamine injections. Proc. Natl. Acad. Sci. USA 1996, 93, 1956–1961. [Google Scholar] [CrossRef] [Green Version]
- LaVoie, M.; Hastings, T.G. Dopamine Quinone Formation and Protein Modification Associated with the Striatal Neurotoxicity of Methamphetamine: Evidence against a Role for Extracellular Dopamine. J. Neurosci. 1999, 19, 1484–1491. [Google Scholar] [CrossRef] [PubMed]
- Fornai, F.; Lenzi, P.; Gesi, M.; Soldani, P.; Ferrucci, M.; Lazzeri, G.; Capobianco, L.; Battaglia, G.; De Blasi, A.; Nicoletti, F.; et al. Methamphetamine produces neuronal inclusions in the nigrostriatal system and in PC12 cells. J. Neurochem. 2003, 88, 114–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hastings, T.G.; Zigmond, M.J. Identification of Catechol-Protein Conjugates in Neostriatal Slices Incubated with [3H]Dopamine: Impact of Ascorbic Acid and Glutathione. J. Neurochem. 2002, 63, 1126–1132. [Google Scholar] [CrossRef]
- Lazzeri, G.; Biagioni, F.; Fulceri, F.; Busceti, C.L.; Scavuzzo, M.C.; Ippolito, C.; Salvetti, A.; Lenzi, P.; Fornai, F. mTOR Modulates Methamphetamine-Induced Toxicity through Cell Clearing Systems. Oxidative Med. Cell. Longev. 2018, 2018, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iacovelli, L.; Fulceri, F.; De Blasi, A.; Nicoletti, F.; Ruggieri, S.; Fornai, F. The neurotoxicity of amphetamines: Bridging drugs of abuse and neurodegenerative disorders. Exp. Neurol. 2006, 201, 24–31. [Google Scholar] [CrossRef]
- Wilson, J.M.; Kalasinsky, K.S.; Levey, A.I.; Bergeron, C.; Reiber, G.; Anthony, R.M.; Schmunk, G.A.; Shannak, K.; Haycock, J.W.; Kish, S.J. Striatal dopamine nerve terminal markers in human, chronic methamphetamine users. Nat. Med. 1996, 2, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Chang, L.; Wang, G.-J.; Fowler, J.S.; Leonido-Yee, M.; Franceschi, D.; Sedler, M.J.; Gatley, S.J.; Hitzemann, R.; Ding, Y.-S.; et al. Association of Dopamine Transporter Reduction With Psychomotor Impairment in Methamphetamine Abusers. Am. J. Psychiatry 2001, 158, 377–382. [Google Scholar] [CrossRef]
- Wood, S.; Sage, J.R.; Shuman, T.; Anagnostaras, S.G. Psychostimulants and Cognition: A Continuum of Behavioral and Cognitive Activation. Pharmacol. Rev. 2013, 66, 193–221. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-K.; Lin, S.-K.; Chen, Y.-C.; Huang, M.-C.; Chen, T.-T.; Ree, S.C.; Wang, L.-J. Persistence of psychotic symptoms as an indicator of cognitive impairment in methamphetamine users. Drug Alcohol Depend. 2015, 148, 158–164. [Google Scholar] [CrossRef] [PubMed]
- London, E.D.; Kohno, M.; Morales, A.M.; Ballard, M.E. Chronic methamphetamine abuse and corticostriatal deficits revealed by neuroimaging. Brain Res. 2015, 1628, 174–185. [Google Scholar] [CrossRef] [Green Version]
- Potvin, S.; Pelletier, J.; Grot, S.; Hébert, C.; Barr, A.M.; Lecomte, T. Cognitive deficits in individuals with methamphetamine use disorder: A meta-analysis. Addict. Behav. 2018, 80, 154–160. [Google Scholar] [CrossRef]
- Larsen, K.E.; Fon, E.A.; Hastings, T.G.; Edwards, R.H.; Sulzer, D. Methamphetamine-Induced Degeneration of Dopaminergic Neurons Involves Autophagy and Upregulation of Dopamine Synthesis. J. Neurosci. 2002, 22, 8951–8960. [Google Scholar] [CrossRef]
- Ginet, V.; van de Looij, Y.; Petrenko, V.; Toulotte, A.; Kiss, J.; Hüppi, P.S.; Sizonenko, S.V. Lactoferrin during lactation reduces lipopolysaccharide-induced brain injury. BioFactors 2016, 42, 323–336. [Google Scholar] [PubMed]
- Zhao, X.; Ting, S.-M.; Liu, C.-H.; Sun, G.; Kruzel, M.; Roy-O’Reilly, M.; Aronowski, J. Neutrophil polarization by IL-27 as a therapeutic target for intracerebral hemorrhage. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohrabi, S.M.; Niazi, A.; Chahardoli, M.; Hortamani, A.; Setoodeh, P. In silico investigation of lactoferrin protein character-izations for the prediction of anti-microbial properties. Mol. Biol. Res. Commun. 2014, 3, 85–100. [Google Scholar]
- Taylor, S.; Brock, J.; Kruger, C.; Berner, T.; Murphy, M. Safety determination for the use of bovine milk-derived lactoferrin as a component of an antimicrobial beef carcass spray. Regul. Toxicol. Pharmacol. 2004, 39, 12–24. [Google Scholar] [CrossRef] [PubMed]
- Superti, F. Lactoferrin from Bovine Milk: A Protective Companion for Life. Nutrients 2020, 12, 2562. [Google Scholar] [CrossRef]
- Song, X.; Violin, J.D.; Seidler, F.J.; Slotkin, T.A. Modeling the Developmental Neurotoxicity of Chlorpyrifosin Vitro:Macromolecule Synthesis in PC12 Cells. Toxicol. Appl. Pharmacol. 1998, 151, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Qiao, D.; Seidler, F.J.; Slotkin, T.A. Developmental neurotoxicity of chlorpyrifos modeled in vitro: Comparative effects of metabolites and other cholinesterase inhibitors on DNA synthesis in PC12 and C6 cells. Environ. Health Perspect. 2001, 109, 909–913. [Google Scholar] [CrossRef]
- Ferese, R.; Lenzi, P.; Fulceri, F.; Biagioni, F.; Fabrizi, C.; Gambardella, S.; Familiari, P.; Frati, A.; Limanaqi, F.; Fornai, F. Quantitative Ultrastructural Morphometry and Gene Expression of mTOR-Related Mitochondriogenesis within Glioblastoma Cells. Int. J. Mol. Sci. 2020, 21, 4570. [Google Scholar] [CrossRef] [PubMed]
- Pendergrass, W.; Wolf, N.; Poot, M. Efficacy of MitoTracker Green? and CMXrosamine to measure changes in mitochondrial membrane potentials in living cells and tissues. Cytom. A 2004, 61, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Gautam, N.; Sankaran, S.; Yason, J.A.; Tan, K.S.W.; Gascoigne, N.R.J. A high content imaging flow cytometry approach to study mitochondria in T cells: MitoTracker Green FM dye concentration optimization. Methods 2018, 134–135, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Bendayan, M.; Zollinger, M. Ultrastructural localization of antigenic sites on osmium-fixed tissues applying the protein A-gold technique. J. Histochem. Cytochem. 1983, 31, 101–109. [Google Scholar] [CrossRef] [Green Version]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef]
- Lou, G.; Palikaras, K.; Lautrup, S.; Scheibye-Knudsen, M.; Tavernarakis, N.; Fang, E.F. Mitophagy and Neuroprotection. Trends Mol. Med. 2020, 26, 8–20. [Google Scholar] [CrossRef]
- Lin, T.-Y.; Chiou, S.-H.; Chen, M.; Kuo, C.-D. Human lactoferrin exerts bi-directional actions on PC12 cell survival via ERK1/2 pathway. Biochem. Biophys. Res. Commun. 2005, 337, 330–336. [Google Scholar] [CrossRef]
- Postuma, R.B.; Martins, R.N.; Cappai, R.; Beyreuther, K.; Masters, C.L.; Strickland, D.K.; Mok, S.S.; Small, D.H. Effects of the amyloid protein precursor of Alzheimer’s disease and other ligands of the LDL receptor-related protein on neurite outgrowth from sympathetic neurons in culture. FEBS Lett. 1998, 428, 13–16. [Google Scholar] [CrossRef] [Green Version]
- Sriramoju, B.; Kanwar, R.K.; Kanwar, J.R. Lactoferrin induced neuronal differentiation: A boon for brain tumours. Int. J. Dev. Neurosci. 2015, 41, 28–36. [Google Scholar] [CrossRef]
- Izumo, N.; Yukiko, I.; Kagaya, N.; Furukawa, M.; Iwasaki, R.; Sumino, A.; Hayamizu, K.; Nakano, M.; Hoshino, T.; Kurono, H.; et al. Lactoferrin Suppresses Decreased Locomotor Activities by Improving Dopamine and Serotonin Release in the Amygdala of Ovariectomized Rats. Curr. Mol. Pharmacol. 2020, 14, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, K.F. Structure and Utilization of Tubulin Isotypes. Annu. Rev. Cell Biol. 1988, 4, 687–716. [Google Scholar] [CrossRef] [PubMed]
- Eddé, B.; Jakob, H.; Darmon, M. Two specific markers for neural differentiation of embryonal carcinoma cells. EMBO J. 1983, 2, 1473–1478. [Google Scholar] [CrossRef] [PubMed]
- Denoulet, P.; Eddé, B.; Gros, F. Differential expression of several neurospecific β-tubulin mRNAs in the mouse brain during development. Gene 1986, 50, 289–297. [Google Scholar] [CrossRef]
- Matus, A. Microtubule-associated proteins: Their potential role in determining neuronal morphology. Ann. Rev. Neurosci. 1988, 11, 29–44. [Google Scholar] [CrossRef]
- Katsetos, C.D.; Herman, M.M.; Mork, S.J. Class III beta-tubulin in human development and cancer. Cell Motil. Cytoskelet. 2003, 55, 77–96. [Google Scholar] [CrossRef]
- Tischfield, M.A.; Baris, H.N.; Wu, C.; Rudolph, G.; Van Maldergem, L.; He, W.; Chan, W.-M.; Andrews, C.; Demer, J.L.; Robertson, R.L.; et al. Human TUBB3 Mutations Perturb Microtubule Dynamics, Kinesin Interactions, and Axon Guidance. Cell 2010, 140, 74–87. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.Q.; Oblinger, M.M. Differential regulation of beta III and other tubulin genes during peripheral and central neuron development. J. Cell Sci. 1992, 103, 643–651. [Google Scholar] [CrossRef]
- Arcella, A.; Oliva, M.A.; Staffieri, S.; Aalberti, S.; Grillea, G.; Madonna, M.; Bartolo, M.; Pavone, L.; Giangaspero, F.; Cantore, G.; et al. In vitro and in vivo effect of human lactoferrin on glioblastoma growth. J. Neurosurg. 2015, 123, 1026–1035. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.-G.; Jeong, J.-K.; Lee, J.-H.; Lee, Y.-J.; Seol, J.-W.; Kim, S.-J.; Hur, T.-Y.; Jung, Y.-H.; Kang, S.-J.; Park, S.-Y. Lactoferrin protects against prion protein-induced cell death in neuronal cells by preventing mitochondrial dysfunction. Int. J. Mol. Med. 2013, 31, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Tsubota, A.; Yoshikawa, T.; Nariai, K.; Mitsunaga, M.; Yumoto, Y.; Fukushima, K.; Hoshina, S.; Fujise, K. Bovine lactoferrin potently inhibits liver mitochondrial 8-OHdG levels and retrieves hepatic OGG1 activities in Long-Evans Cinnamon rats. J. Hepatol. 2008, 48, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Meng, X.; Zhang, F.; Xiang, Y.; Wang, J. The in vitro antiviral activity of lactoferrin against common human coronaviruses and SARS-CoV-2 is mediated by targeting the heparan sulfate co-receptor. Emerg. Microbes Infect. 2021, 10, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Coupling mitogenesis and mitophagy for longevity. Autophagy 2015, 11, 1428–1430. [Google Scholar] [CrossRef] [Green Version]
- Fillebeen, C.; Descamps, L.; Dehouck, M.-P.; Fenart, L.; Benaıssa, M.; Spik, G.; Cecchelli, R.; Pierce, A. Receptor-mediated Transcytosis of Lactoferrin through the Blood-Brain Barrier. J. Biol. Chem. 1999, 274, 7011–7017. [Google Scholar] [CrossRef] [Green Version]
- Faucheux, B.A.; Nillesse, N.; Damier, P.; Spik, G.; Mouatt-Prigent, A.; Pierce, A.; Leveugle, B.; Kubis, N.; Hauw, J.J.; Agid, Y. Expression of lactoferrin receptors is increased in the mesencephalon of patients with Parkinson disease. Proc. Natl. Acad. Sci. USA 1995, 92, 9603–9607. [Google Scholar] [CrossRef] [Green Version]
- Ji, B.; Maeda, J.; Higuchi, M.; Inoue, K.; Akita, H.; Harashima, H.; Suhara, T. Pharmacokinetics and brain uptake of lactoferrin in rats. Life Sci. 2006, 78, 851–855. [Google Scholar] [CrossRef]
- Jiang, R.; Lopez, V.; Kelleher, S.L.; Lönnerdal, B. Apo- and holo-lactoferrin are both internalized by lactoferrin receptor via clathrin-mediated endocytosis but differentially affect ERK-signaling and cell proliferation in caco-2 cells. J. Cell. Physiol. 2011, 226, 3022–3031. [Google Scholar] [CrossRef] [Green Version]
- Bu, G.; Sun, Y.; Schwartz, A.L.; Holtzman, D.M. Nerve Growth Factor Induces Rapid Increases in Functional Cell Surface Low Density Lipoprotein Receptor-related Protein. J. Biol. Chem. 1998, 273, 13359–13365. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, E. Iron transport in Parkinson’s disease. Parkinsonism Relat. Disord. 2009, 15, S209–S211. [Google Scholar] [CrossRef]
- Weinreb, O.; Mandel, S.; Youdim, M.B.H.; Amit, T. Targeting dysregulation of brain iron homeostasis in Parkinson’s disease by iron chelators. Free Radic. Biol. Med. 2013, 62, 52–64. [Google Scholar] [CrossRef]
- Yan, N.; Zhang, J. Iron Metabolism, Ferroptosis, and the Links With Alzheimer’s Disease. Front. Neurosci. 2020, 13, 1443. [Google Scholar] [CrossRef] [PubMed]
- Jouini, N.; Saied, Z.; Ben Sassi, S.; Nebli, F.; Messaoud, T.; Hentati, F.; Belal, S. Impacts of Iron Metabolism Dysregulation on Alzheimer’s Disease. J. Alzheimer’s Dis. 2021, 80, 1439–1450. [Google Scholar] [CrossRef] [PubMed]

















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ryskalin, L.; Biagioni, F.; Busceti, C.L.; Polzella, M.; Lenzi, P.; Frati, A.; Ferrucci, M.; Fornai, F. Lactoferrin Protects against Methamphetamine Toxicity by Modulating Autophagy and Mitochondrial Status. Nutrients 2021, 13, 3356. https://doi.org/10.3390/nu13103356
Ryskalin L, Biagioni F, Busceti CL, Polzella M, Lenzi P, Frati A, Ferrucci M, Fornai F. Lactoferrin Protects against Methamphetamine Toxicity by Modulating Autophagy and Mitochondrial Status. Nutrients. 2021; 13(10):3356. https://doi.org/10.3390/nu13103356
Chicago/Turabian StyleRyskalin, Larisa, Francesca Biagioni, Carla L. Busceti, Maico Polzella, Paola Lenzi, Alessandro Frati, Michela Ferrucci, and Francesco Fornai. 2021. "Lactoferrin Protects against Methamphetamine Toxicity by Modulating Autophagy and Mitochondrial Status" Nutrients 13, no. 10: 3356. https://doi.org/10.3390/nu13103356