Asaronic Acid Inhibited Glucose-Triggered M2-Phenotype Shift Through Disrupting the Formation of Coordinated Signaling of IL-4Rα-Tyk2-STAT6 and GLUT1-Akt-mTOR-AMPK

 and
and
Abstract
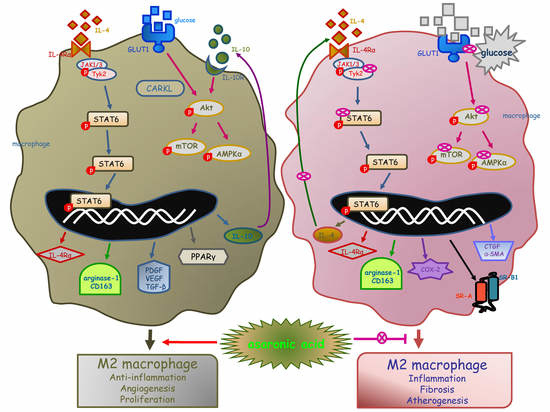
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Western Blot Analysis
2.4. Enzyme-Linked Immunosorbent Assay (ELISA)
2.5. Immunocytochemical Analysis
2.6. Dihydroethidium (DHE) Staining for ROS Production
2.7. Data Analysis
3. Results
3.1. IL-4-Mediated IL-4Rα Induction by Asaronic Acid
3.2. IL-4-Mediated M2 Macrophage Activation by Asaronic Acid
3.3. Angiogenic and Proliferative Capacity of Asaronic Acid in M2-Polarized Macrophages
3.4. Effects of Asaronic Acid on IL-4-Mediated M2 Macrophage Metabolism
3.5. Inhibition of Glucose-Triggered M2 Macrophage Activation by Asaronic Acid
3.6. Glucose-Triggered Metabolic Alterations in M2 Macrophages
3.7. Blockade of Diabetic M2 Macrophage Dysfunction by Asaronic Acid
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Parisi, L.; Gini, E.; Baci, D.; Tremolati, M.; Fanuli, M.; Bassani, B.; Farronato, G.; Bruno, A.; Mortara, L. Macrophage polarization in chronic inflammatory diseases: Killers or builders? J. Immunol. Res. 2018, 2018, 8917804. [Google Scholar] [CrossRef] [PubMed]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 macrophages and the Th1/Th2 Paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.C.; Zou, X.B.; Chai, Y.F.; Yao, Y.M. Macrophage polarization in inflammatory diseases. Int. J. Biol. Sci. 2014, 10, 520–529. [Google Scholar] [CrossRef] [PubMed]
- Rőszer, T. Understanding the mysterious M2 macrophage through activation markers and effector mechanisms. Mediat. Inflamm. 2015, 2015, 816460. [Google Scholar] [CrossRef] [Green Version]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef] [PubMed]
- Lissner, D.; Schumann, M.; Batra, A.; Kredel, L.I.; Kühl, A.A.; Erben, U.; Siegmund, B. Monocyte and M1 macrophage-induced barrier defect contributes to chronic intestinal inflammation in IBD. Inflamm. Bowel Dis. 2015, 21, 1297–1305. [Google Scholar] [CrossRef] [Green Version]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Krzyszczyk, P.; Schloss, R.; Palmer, A.; Berthiaume, F. The role of macrophages in acute and chronic wound healing and interventions to promote pro-wound healing phenotypes. Front. Physiol. 2018, 9, 419. [Google Scholar] [CrossRef]
- Vannella, K.M.; Wynn, T.A. Mechanisms of organ injury and repair by macrophages. Annu. Rev. Physiol. 2017, 79, 593–617. [Google Scholar] [CrossRef]
- Kraakman, M.J.; Murphy, A.J.; Jandeleit-Dahm, K.; Kammoun, H.L. Macrophage polarization in obesity and type 2 diabetes: Weighing down our understanding of macrophage function? Front. Immunol. 2014, 5, 470. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Tu, Y.; Chen, H.; Jackson, A.O.; Patel, V.; Yin, K. Micro-environment and intracellular metabolism modulation of adipose tissue macrophage polarization in relation to chronic inflammatory diseases. Diabetes Metab. Res. Rev. 2018, 34, e2993. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Song, Z.; Zhou, M.; Yang, Y.; Zhao, Y.; Liu, B.; Zhang, X. Infiltrating macrophages in diabetic nephropathy promote podocytes apoptosis via TNF-α-ROS-p38MAPK pathway. Oncotarget 2017, 8, 53276–53287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landis, R.C.; Quimby, K.R.; Greenidge, A.R. M1/M2 macrophages in diabetic nephropathy: Nrf2/HO-1 as therapeutic targets. Curr. Pharmaceut. Design 2018, 24, 2241–2249. [Google Scholar] [CrossRef]
- Kanter, J.E.; Kramer, F.; Barnhart, S.; Averill, M.M.; Vivekanandan-Giri, A.; Vickery, T.; Li, L.O.; Becker, L.; Yuan, W.; Chait, A.; et al. Diabetes promotes an inflammatory macrophage phenotype and atherosclerosis through acyl-CoA synthetase 1. Proc. Natl. Acad. Sci. USA 2012, 109, E715–E724. [Google Scholar] [CrossRef] [Green Version]
- Guiteras, R.; Flaquer, M.; Cruzado, J.M. Macrophage in chronic kidney disease. Clin. Kidney J. 2016, 9, 765–771. [Google Scholar] [CrossRef]
- Kim, M.G.; Kim, S.C.; Ko, Y.S.; Lee, H.Y.; Jo, S.K.; Cho, W. The role of M2 macrophages in the progression of chronic kidney disease following acute kidney injury. PLoS ONE 2015, 10, e0143961. [Google Scholar] [CrossRef] [Green Version]
- Anders, H.J.; Ryu, M. Renal microenvironments and macrophage phenotypes determine progression or resolution of renal inflammation and fibrosis. Kidney Int. 2011, 80, 915–925. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Rong, J. Macrophage polarization as a therapeutic target in myocardial infarction. Curr. Drug Targets 2018, 19, 651–662. [Google Scholar] [CrossRef]
- Peterson, K.R.; Cottam, M.A.; Kennedy, A.J.; Hasty, A.H. Macrophage-targeted therapeutics for metabolic disease. Trends Pharmacol. Sci. 2018, 39, 536–546. [Google Scholar] [CrossRef]
- Wang, Y.; Smith, W.; Hao, D.; He, B.; Kong, L. M1 and M2 macrophage polarization and potentially therapeutic naturally occurring compounds. Int. Immunopharmacol. 2019, 70, 459–466. [Google Scholar] [CrossRef]
- Saqib, U.; Sarkar, S.; Suk, K.; Mohammad, O.; Baig, M.S.; Savai, R. Phytochemicals as modulators of M1-M2 macrophages in inflammation. Oncotarget 2018, 9, 17937–17950. [Google Scholar] [CrossRef] [Green Version]
- Ni, Y.; Zhuge, F.; Nagashimada, M.; Ota, T. Novel action of carotenoids on non-alcoholic fatty liver disease: Macrophage polarization and liver homeostasis. Nutrients 2016, 8, 391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abron, J.D.; Singh, N.P.; Price, R.L.; Nagarkatti, M.; Nagarkatti, P.S.; Singh, U.P. Genistein induces macrophage polarization and systemic cytokine to ameliorate experimental colitis. PLoS ONE 2018, 13, e0199631. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Paek, J.H.; Shin, D.; Lee, J.Y.; Lim, S.S.; Kang, Y.H. Purple perilla extracts with α-asarone enhance cholesterol efflux from oxidized LDL-exposed macrophages. Int. J. Mol. Med. 2015, 35, 957–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, H.; Park, S.H.; Kang, M.K.; Kim, Y.H.; Lee, E.J.; Kim, D.Y.; Kim, S.I.; Oh, S.; Lim, S.S.; Kang, Y.H. Asaronic acid attenuates macrophage activation toward M1 phenotype through inhibition of NF-κB pathway and JAK-STAT signaling in glucose-loaded murine macrophages. J. Agric. Food Chem. 2019, 67, 10069–10078. [Google Scholar] [CrossRef] [PubMed]
- Pavlou, S.; Lindsay, J.; Ingram, R.; Xu, H.; Chen, M. corresponding author Sustained high glucose exposure sensitizes macrophage responses to cytokine stimuli but reduces their phagocytic activity. BMC Immunol. 2018, 19, 24. [Google Scholar] [CrossRef] [Green Version]
- Ayala, T.S.; Tessaro, F.H.G.; Jannuzzi, G.P.; Bella, L.M.; Ferreira, K.S.; Martins, J.O. High glucose environments interfere with bone marrow-derived macrophage inflammatory mediator release, the tlr4 pathway and glucose metabolism. Sci. Rep. 2019, 9, 11447. [Google Scholar] [CrossRef]
- Bouhlel, M.A.; Derudas, B.; Rigamonti, E.; Dièvart, R.; Brozek, J.; Haulon, S.; Zawadzki, C.; Jude, B.; Torpier, G.; Marx, N.; et al. PPARγ activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell. Metab. 2007, 6, 137–143. [Google Scholar] [CrossRef] [Green Version]
- Szanto, A.; Balint, B.L.; Nagy, Z.S.; Barta, E.; Dezso, B.; Pap, A.; Szeles, L.; Poliska, S.; Oros, M.; Evans, R.M.; et al. STAT6 transcription factor is a facilitator of the nuclear receptor PPARγ-regulated gene expression in macrophages and dendritic cells. Immunity 2010, 33, 699–712. [Google Scholar] [CrossRef] [Green Version]
- Freemerman, A.J.; Johnson, A.R.; Sacks, G.N.; Milner, J.J.; Kirk, E.L.; Troester, M.A.; Macintyre, A.N.; Goraksha-Hicks, P.; Rathmell, J.C.; Makowski, L. Metabolic reprogramming of macrophages: Glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J. Biol. Chem. 2014, 289, 7884–7896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langston, P.K.; Shibata, M.; Horng, T. Metabolism supports macrophage activation. Front. Immunol. 2017, 8, 61. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Zhao, L.; Liu, J.; Liu, A.; Jia, C.; Ma, D.; Jiang, Y.; Bai, X. Multi-mechanisms are involved in reactive oxygen species regulation of mTORC1 signaling. Cell. Signal. 2010, 22, 1469–1476. [Google Scholar] [CrossRef]
- Hinchy, E.C.; Gruszczyk, A.V.; Willows, R.; Navaratnam, N.; Hall, A.R.; Bates, G.; Bright, T.P.; Krieg, T.; Carling, D.; Murphy, M.P. Mitochondria-derived ROS activate AMP-activated protein kinase (AMPK) indirectly. J. Biol. Chem. 2018, 293, 17208–17217. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Yang, B.; Huang, J.; Lin, Y.; Xiang, T.; Wan, J.; Li, H.; Chouaib, S.; Ren, G. Cyclooxygenase-2 in tumor-associated macrophages promotes breast cancer cell survival by triggering a positive-feedback loop between macrophages and cancer cells. Oncotarget 2015, 6, 29637–29650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, J.; Shi, J.; Chen, L.; Lv, Z.; Chen, X.; Cao, H.; Xiang, Z.; Han, X. M2 macrophages promote myofibroblast differentiation of LR-MSCs and are associated with pulmonary fibrogenesis. Cell. Commun. Signal. 2018, 16, 89. [Google Scholar] [CrossRef] [Green Version]
- Braga, T.T.; Agudelo, J.S.H.; Camara, N.O.S. Macrophages during the fibrotic process: M2 as friend and foe. Front. Immunol. 2015, 6, 602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuharatakaki, K.; Sakai, M.; Sakamoto, Y.; Takeya, M.; Horiuchi, S. Expression of class A scavenger receptor is enhanced by high glucose in vitro and under diabetic conditions in vivo: One mechanism for an increased rate of atherosclerosis in diabetes. J. Biol. Chem. 2005, 280, 3355–3364. [Google Scholar] [CrossRef] [Green Version]
- Michael, D.R.; Ashlin, T.G.; Buckley, M.L.; Ramji, D.P. Macrophages, lipid metabolism and geneexpression in atherogenesis: A therapeutic target of the future? Clin. Lipidol. 2012, 7, 37–48. [Google Scholar] [CrossRef]
- Mounayar, M.; Kefaloyianni, E.; Smith, B.; Solhjou, Z.; Maarouf, O.H.; Azzi, J.; Chabtini, L.; Fiorina, P.; Kraus, M.; Briddell, R.; et al. PI3kα and STAT1 interplay regulates human mesenchymal stem cell immune polarization. Stem Cells 2015, 33, 1892–1901. [Google Scholar] [CrossRef] [Green Version]
- Kreider, T.; Anthony, R.M.; Urban, J.F.; Gause, W.C. Alternatively activated macrophages in helminth infections. Curr. Opin. Immunol. 2007, 19, 448–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ban, C.R.; Twigg, S.M. Fibrosis in diabetes complications: Pathogenic mechanisms and circulating and urinary markers. Vasc. Health Risk Manag. 2008, 4, 575–596. [Google Scholar] [PubMed] [Green Version]
- Fujiwara, Y.; Komohara, Y.; Ikeda, T.; Takeya, M. Corosolic acid inhibits glioblastoma cell proliferation by suppressing the activation of signal transducer and activator of transcription-3 and nuclear factor-kappa B in tumor cells and tumor-associated macrophages. Cancer Sci. 2011, 102, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Gui, Z.; Zhou, Y.; Xia, L.; Lin, K.; Xu, Y. Quercetin alleviates rat osteoarthritis by inhibiting inflammation and apoptosis of chondrocytes, modulating synovial macrophages polarization to M2 macrophages. Free Radic. Biol. Med. 2019, 145, 146–160. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Zhang, X.; Chen, X.; Li, Y.; Ke, Z.; Tang, T.; Chai, H.; Guo, A.M.; Chen, H.; Yang, J. Isoliquiritigenin, a flavonoid from licorice, blocks M2 macrophage polarization in colitis-associated tumorigenesis through downregulating PGE2 and IL-6. Toxicol. Appl. Pharmacol. 2014, 279, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Huang, J.; Lin, M.; Xie, T.; You, T. Quercetin promotes diabetic wound healing via switching macrophages from M1 to M2 polarization. J. Surg. Res. 2019, 246, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Carmona, M.D.; Plaza-Seron, M.C.; Vargas-Candela, A.; Tinahones, F.J.; Gomez-Huelgas, R.; Bernal-Lopez, M.R. CD36 overexpression: A possible etiopathogenic mechanism of atherosclerosis in patients with prediabetes and diabetes. Diabetol. Metab. Syndr. 2017, 9, 55. [Google Scholar] [CrossRef] [Green Version]
- Wei, H.; Tarling, E.J.; McMillen, T.S.; Tang, C.; LeBoeuf, R.C. ABCG1 regulates mouse adipose tissue macrophage cholesterol levels and ratio of M1 to M2 cells in obesity and caloric restriction. J. Lipid Res. 2015, 56, 2337–2347. [Google Scholar] [CrossRef] [Green Version]
- Sag, D.; Cekic, C.; Wu, R.; Linden, J.; Hedrick, C.C. The cholesterol transporter ABCG1 links cholesterol homeostasis and tumour immunity. Nat. Commun. 2015, 6, 6354. [Google Scholar] [CrossRef] [Green Version]
- Guo, M.; Xiao, J.; Sheng, X.; Zhang, X.; Tie, Y.; Wang, L.; Zhao, L.; Ji, X. Ginsenoside Rg3 mitigates atherosclerosis progression in diabetic apoE-/- mice by skewing macrophages to the M2 phenotype. Front. Pharmacol. 2018, 9, 464. [Google Scholar] [CrossRef] [Green Version]
- Tian, S.; Chen, S.Y. Macrophage polarization in kidney diseases. Macrophage 2015, 2, e679. [Google Scholar] [PubMed] [Green Version]
- Thapa, B.; Lee, K. Metabolic influence on macrophage polarization and pathogenesis. BMB Rep. 2019, 52, 360–372. [Google Scholar] [CrossRef]
- Nouwen, L.V.; Everts, B. Pathogens menTORing macrophages and dendritic cells: Manipulation of mTOR and cellular Metabolism to promote immune escape. Cells 2020, 9, 161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.; Huang, Y.; Zheng, W.; Yan, J.; Cheng, M.; Zhao, R.; Chen, L.; Hu, C.; Jia, W. Resveratrol reduces intracellular reactive oxygen species levels by inducing autophagy through the AMPK-mTOR pathway. Front. Med. 2018, 12, 697–706. [Google Scholar] [CrossRef] [PubMed]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, H.; Park, S.-H.; Kang, M.-K.; Kim, Y.-H.; Lee, E.-J.; Kim, D.Y.; Kim, S.-I.; Oh, S.Y.; Na, W.; Lim, S.S.; et al. Asaronic Acid Inhibited Glucose-Triggered M2-Phenotype Shift Through Disrupting the Formation of Coordinated Signaling of IL-4Rα-Tyk2-STAT6 and GLUT1-Akt-mTOR-AMPK. Nutrients 2020, 12, 2006. https://doi.org/10.3390/nu12072006
Oh H, Park S-H, Kang M-K, Kim Y-H, Lee E-J, Kim DY, Kim S-I, Oh SY, Na W, Lim SS, et al. Asaronic Acid Inhibited Glucose-Triggered M2-Phenotype Shift Through Disrupting the Formation of Coordinated Signaling of IL-4Rα-Tyk2-STAT6 and GLUT1-Akt-mTOR-AMPK. Nutrients. 2020; 12(7):2006. https://doi.org/10.3390/nu12072006
Chicago/Turabian StyleOh, Hyeongjoo, Sin-Hye Park, Min-Kyung Kang, Yun-Ho Kim, Eun-Jung Lee, Dong Yeon Kim, Soo-Il Kim, Su Yeon Oh, Woojin Na, Soon Sung Lim, and et al. 2020. "Asaronic Acid Inhibited Glucose-Triggered M2-Phenotype Shift Through Disrupting the Formation of Coordinated Signaling of IL-4Rα-Tyk2-STAT6 and GLUT1-Akt-mTOR-AMPK" Nutrients 12, no. 7: 2006. https://doi.org/10.3390/nu12072006