Pediatric Non-Alcoholic Fatty Liver Disease: Nutritional Origins and Potential Molecular Mechanisms

 ,
, 
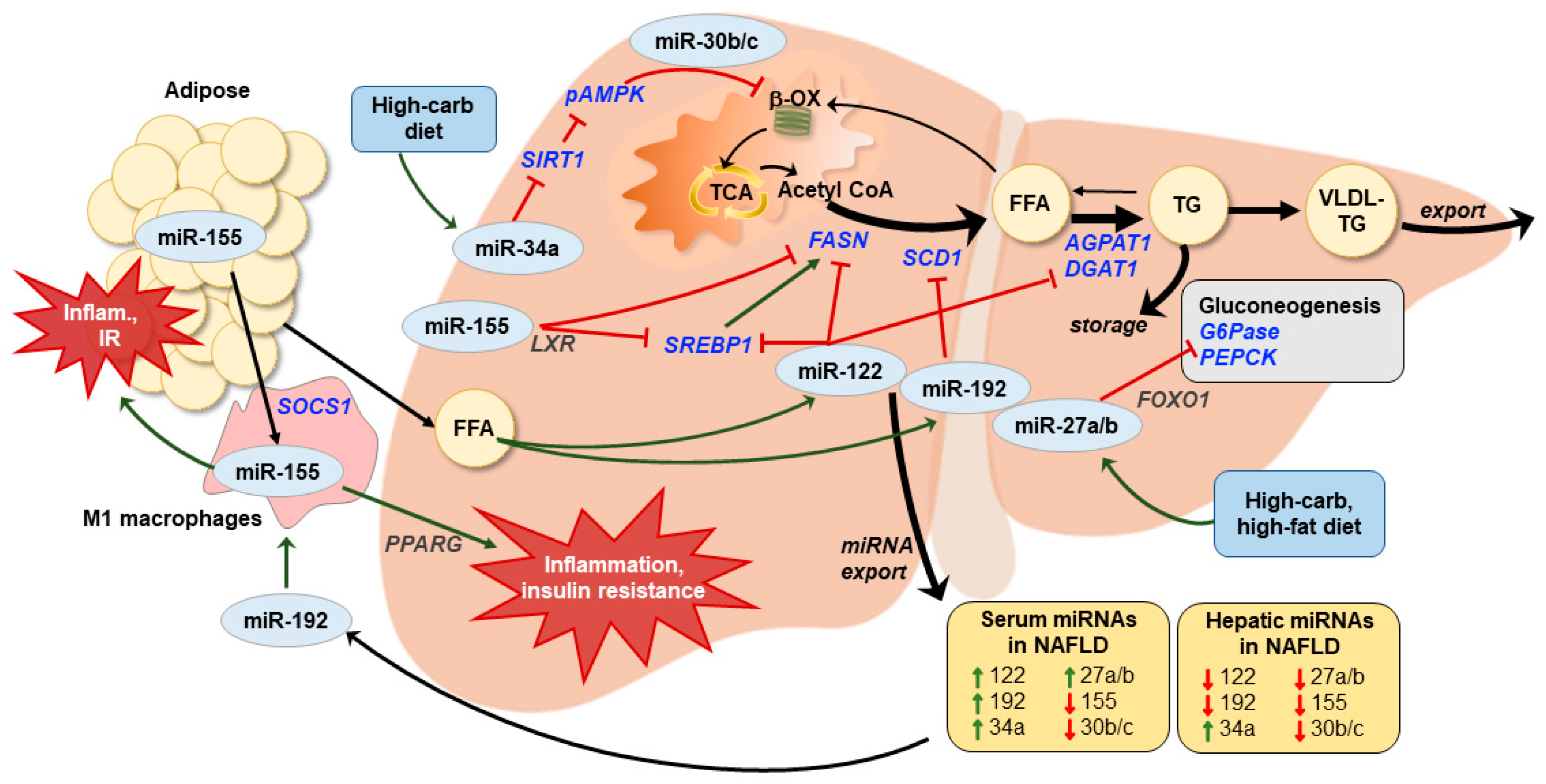{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Clinical Pathophysiology
3. Role of Nutrients in Pediatric NAFLD
4. Mitochondrial Dysfunction and Oxidative Stress in the Progression of Pediatric NAFLD
5. Gut Microbial Dysbiosis in Pediatric NAFLD
6. Micro-RNAs in Fatty Liver Disease: Predictors and Prognosticators
7. Maternal Over-Nutrition and Developmental Programming of NAFLD
8. Programmed/Trained Inflammation in the Pathogenesis of Pediatric NAFLD
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bedossa, P. Pathology of non-alcoholic fatty liver disease. Liver Int. 2017, 37 (Suppl. S1), 85–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younossi, Z.M. Non-alcoholic fatty liver disease—A global public health perspective. J. Hepatol. 2019, 70, 531–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Fazel, Y.; Koenig, A.B.; Sayiner, M.; Goodman, Z.D.; Younossi, Z.M. Epidemiology and natural history of non-alcoholic fatty liver disease. Metabolism 2016, 65, 1017–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, E.L.; Howe, L.D.; Jones, H.E.; Higgins, J.P.; Lawlor, D.A.; Fraser, A. The prevalence of non-alcoholic fatty liver disease in children and adolescents: A systematic review and meta-analysis. PLoS ONE 2015, 10, e0140908. [Google Scholar] [CrossRef] [Green Version]
- Di Sessa, A.; Umano, G.R.; Miraglia Del Giudice, E.; Santoro, N. From the liver to the heart: Cardiac dysfunction in obese children with non-alcoholic fatty liver disease. World J. Hepatol. 2017, 9, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Pacifico, L.; Perla, F.M.; Roggini, M.; Andreoli, G.; D’Avanzo, M.; Chiesa, C. A systematic review of NAFLD-associated extrahepatic disorders in youths. J. Clin. Med. 2019, 8, 868. [Google Scholar] [CrossRef] [Green Version]
- Yodoshi, T.; Arce-Clachar, A.C.; Sun, Q.; Fei, L.; Bramlage, K.; Xanthakos, S.A.; Flores, F.; Mouzaki, M. Glomerular hyperfiltration is associated with liver disease severity in children with nonalcoholic fatty liver disease. J. Pediatr. 2020, 222, 127–133. [Google Scholar] [CrossRef]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Wesolowski, S.R.; El Kasmi, K.C.; Jonscher, K.R.; Friedman, J.E. Developmental origins of NAFLD: A womb with a clue. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 81–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowman, J.K.; Tomlinson, J.W.; Newsome, P.N. Pathogenesis of non-alcoholic fatty liver disease. QJM 2010, 103, 71–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vos, M.B.; Abrams, S.H.; Barlow, S.E.; Caprio, S.; Daniels, S.R.; Kohli, R.; Mouzaki, M.; Sathya, P.; Schwimmer, J.B.; Sundaram, S.S.; et al. NASPGHAN clinical practice guideline for the diagnosis and treatment of nonalcoholic fatty liver disease in children: Recommendations from the Expert Committee on NAFLD (ECON) and the North American Society of Pediatric Gastroenterology, Hepatology and Nutrition (NASPGHAN). J. Pediatr. Gastroenterol. Nutr. 2017, 64, 319–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- AAP Endorses New Guidelines on Non-Alcoholic Fatty Liver Disease. Available online: https://www.aappublications.org/news/2016/12/02/FattyLiver120216 (accessed on 10 September 2020).
- Maximos, M.; Bril, F.; Portillo Sanchez, P.; Lomonaco, R.; Orsak, B.; Biernacki, D.; Suman, A.; Weber, M.; Cusi, K. The role of liver fat and insulin resistance as determinants of plasma aminotransferase elevation in nonalcoholic fatty liver disease. Hepatology 2015, 61, 153–160. [Google Scholar] [CrossRef]
- Alkhouri, N.; Alisi, A.; Okwu, V.; Matloob, A.; Ferrari, F.; Crudele, A.; De Vito, R.; Lopez, R.; Feldstein, A.E.; Nobili, V. Circulating soluble Fas and Fas ligand levels are elevated in children with nonalcoholic steatohepatitis. Dig. Dis. Sci. 2015, 60, 2353–2359. [Google Scholar] [CrossRef]
- Becker, P.P.; Rau, M.; Schmitt, J.; Malsch, C.; Hammer, C.; Bantel, H.; Mullhaupt, B.; Geier, A. Performance of serum microRNAs -122, -192 and -21 as biomarkers in patients with non-alcoholic steatohepatitis. PLoS ONE 2015, 10, e0142661. [Google Scholar] [CrossRef] [PubMed]
- Alkhouri, N.; Feldstein, A.E. Noninvasive diagnosis of nonalcoholic fatty liver disease: Are we there yet? Metabolism 2016, 65, 1087–1095. [Google Scholar] [CrossRef] [Green Version]
- Decaris, M.L.; Li, K.W.; Emson, C.L.; Gatmaitan, M.; Liu, S.; Wang, Y.; Nyangau, E.; Colangelo, M.; Angel, T.E.; Beysen, C.; et al. Identifying nonalcoholic fatty liver disease patients with active fibrosis by measuring extracellular matrix remodeling rates in tissue and blood. Hepatology 2017, 65, 78–88. [Google Scholar] [CrossRef]
- Feng, R.; Luo, C.; Li, C.; Du, S.; Okekunle, A.P.; Li, Y.; Chen, Y.; Zi, T.; Niu, Y. Free fatty acids profile among lean, overweight and obese non-alcoholic fatty liver disease patients: A case—Control study. Lipids Health Dis. 2017, 16, 165. [Google Scholar] [CrossRef] [Green Version]
- He, L.; Deng, L.; Zhang, Q.; Guo, J.; Zhou, J.; Song, W.; Yuan, F. Diagnostic value of CK-18, FGF-21, and related biomarker panel in nonalcoholic fatty liver disease: A systematic review and meta-analysis. BioMed Res. Int. 2017, 2017, 9729107. [Google Scholar] [CrossRef] [PubMed]
- Isokuortti, E.; Zhou, Y.; Peltonen, M.; Bugianesi, E.; Clement, K.; Bonnefont-Rousselot, D.; Lacorte, J.M.; Gastaldelli, A.; Schuppan, D.; Schattenberg, J.M.; et al. Use of HOMA-IR to diagnose non-alcoholic fatty liver disease: A population-based and inter-laboratory study. Diabetologia 2017, 60, 1873–1882. [Google Scholar] [CrossRef] [PubMed]
- Peter, A.; Kovarova, M.; Staiger, H.; Machann, J.; Schick, F.; Königsrainer, A.; Königsrainer, I.; Schleicher, E.; Fritsche, A.; Haring, H.U.; et al. The hepatokines fetuin-A and fetuin-B are upregulated in the state of hepatic steatosis and may differently impact on glucose homeostasis in humans. Am. J. Physiol. Endocrinol. Metab. 2018, 314, E266–E273. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, M.; Imajo, K.; Takahashi, H.; Ogawa, Y.; Eguchi, Y.; Sumida, Y.; Yoneda, M.; Kawanaka, M.; Saito, S.; Tokushige, K.; et al. Clinical strategy of diagnosing and following patients with nonalcoholic fatty liver disease based on invasive and noninvasive methods. J. Gastroenterol. 2018, 53, 181–196. [Google Scholar] [CrossRef] [Green Version]
- Crespo, M.; Lappe, S.; Feldstein, A.E.; Alkhouri, N. Similarities and differences between pediatric and adult nonalcoholic fatty liver disease. Metabolism 2016, 65, 1161–1171. [Google Scholar] [CrossRef]
- Nobili, V.; Alisi, A.; Valenti, L.; Miele, L.; Feldstein, A.E.; Alkhouri, N. NAFLD in children: New genes, new diagnostic modalities and new drugs. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 517–530. [Google Scholar] [CrossRef]
- Mansoor, S.; Collyer, E.; Alkhouri, N. A comprehensive review of noninvasive liver fibrosis tests in pediatric nonalcoholic Fatty liver disease. Curr. Gastroenterol. Rep. 2015, 17, 23. [Google Scholar] [CrossRef]
- Noruegas, M.J.; Matos, H.; Gonçalves, I.; Cipriano, M.A.; Sanches, C. Acoustic radiation force impulse-imaging in the assessment of liver fibrosis in children. Pediatr. Radiol. 2012, 42, 201–204. [Google Scholar] [CrossRef]
- Kamble, R.; Sodhi, K.S.; Thapa, B.R.; Saxena, A.K.; Bhatia, A.; Dayal, D.; Khandelwal, N. Liver acoustic radiation force impulse (ARFI) in childhood obesity: Comparison and correlation with biochemical markers. J. Ultrasound 2017, 20, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Ferraioli, G.; Calcaterra, V.; Lissandrin, R.; Guazzotti, M.; Maiocchi, L.; Tinelli, C.; De Silvestri, A.; Regalbuto, C.; Pelizzo, G.; Larizza, D.; et al. Noninvasive assessment of liver steatosis in children: The clinical value of controlled attenuation parameter. BMC Gastroenterol. 2017, 17, 61. [Google Scholar] [CrossRef]
- Tapper, E.B.; Loomba, R. Noninvasive imaging biomarker assessment of liver fibrosis by elastography in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 274–282. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Jiang, Z.; Zhang, L. Bile acid regulation: A novel therapeutic strategy in non-alcoholic fatty liver disease. Pharmacol. Ther. 2018, 190, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Kalhan, S.C.; Guo, L.; Edmison, J.; Dasarathy, S.; McCullough, A.J.; Hanson, R.W.; Milburn, M. Plasma metabolomic profile in nonalcoholic fatty liver disease. Metabolism 2011, 60, 404–413. [Google Scholar] [CrossRef] [Green Version]
- Burgert, T.S.; Taksali, S.E.; Dziura, J.; Goodman, T.R.; Yeckel, C.W.; Papademetris, X.; Constable, R.T.; Weiss, R.; Tamborlane, W.V.; Savoye, M.; et al. Alanine aminotransferase levels and fatty liver in childhood obesity: Associations with insulin resistance, adiponectin, and visceral fat. J. Clin. Endocrinol. Metab. 2006, 91, 4287–4294. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Wiklund, P.; Autio, R.; Borra, R.; Ojanen, X.; Xu, L.; Törmäkangas, T.; Alen, M. Adipose tissue dysfunction and altered systemic amino acid metabolism are associated with non-alcoholic fatty liver disease. PLoS ONE 2015, 10, e0138889. [Google Scholar] [CrossRef]
- Sabin, M.A.; De Hora, M.; Holly, J.M.; Hunt, L.P.; Ford, A.L.; Williams, S.R.; Baker, J.S.; Retallick, C.J.; Crowne, E.C.; Shield, J.P. Fasting nonesterified fatty acid profiles in childhood and their relationship with adiposity, insulin sensitivity, and lipid levels. Pediatrics 2007, 120, e1426–e1433. [Google Scholar] [CrossRef]
- Fletcher, J.A.; Deja, S.; Satapati, S.; Fu, X.; Burgess, S.C.; Browning, J.D. Impaired ketogenesis and increased acetyl-CoA oxidation promote hyperglycemia in human fatty liver. JCI Insight 2019, 5, e127737. [Google Scholar] [CrossRef]
- Goran, M.I.; Walker, R.; Le, K.A.; Mahurkar, S.; Vikman, S.; Davis, J.N.; Spruijt-Metz, D.; Weigensberg, M.J.; Allayee, H. Effects of PNPLA3 on liver fat and metabolic profile in Hispanic children and adolescents. Diabetes 2010, 59, 3127–3130. [Google Scholar] [CrossRef] [Green Version]
- Santoro, N.; Feldstein, A.E.; Enoksson, E.; Pierpont, B.; Kursawe, R.; Kim, G.; Caprio, S. The association between hepatic fat content and liver injury in obese children and adolescents: Effects of ethnicity, insulin resistance, and common gene variants. Diabetes Care 2013, 36, 1353–1360. [Google Scholar] [CrossRef] [Green Version]
- Santoro, N.; Caprio, S.; Pierpont, B.; Van Name, M.; Savoye, M.; Parks, E.J. Hepatic de novo lipogenesis in obese youth is modulated by a common variant in the GCKR gene. J. Clin. Endocrinol. Metab. 2015, 100, E1125–E1132. [Google Scholar] [CrossRef] [Green Version]
- Goffredo, M.; Caprio, S.; Feldstein, A.E.; D’Adamo, E.; Shaw, M.M.; Pierpont, B.; Savoye, M.; Zhao, H.; Bale, A.E.; Santoro, N. Role of TM6SF2 rs58542926 in the pathogenesis of nonalcoholic pediatric fatty liver disease: A multiethnic study. Hepatology 2016, 63, 117–125. [Google Scholar] [CrossRef] [Green Version]
- Selvakumar, P.K.; Kabbany, M.N.; Lopez, R.; Tozzi, G.; Alisi, A.; Alkhouri, N.; Nobili, V. Reduced lysosomal acid lipase activity-A potential role in the pathogenesis of non alcoholic fatty liver disease in pediatric patients. Dig. Liver Dis. 2016, 48, 909–913. [Google Scholar] [CrossRef]
- Tovoli, F.; Napoli, L.; Negrini, G.; D’Addato, S.; Tozzi, G.; D’Amico, J.; Piscaglia, F.; Bolondi, L. A relative deficiency of lysosomal acid lypase activity characterizes non-alcoholic fatty liver disease. Int. J. Mol. Sci. 2017, 18, 1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baratta, F.; Pastori, D.; Tozzi, G.; D’Erasmo, L.; Di Costanzo, A.; Arca, M.; Ettorre, E.; Ginanni Corradini, S.; Violi, F.; Angelico, F.; et al. Lysosomal acid lipase activity and liver fibrosis in the clinical continuum of non-alcoholic fatty liver disease. Liver Int. 2019, 39, 2301–2308. [Google Scholar] [CrossRef] [PubMed]
- Baratta, F.; Pastori, D.; Ferro, D.; Carluccio, G.; Tozzi, G.; Angelico, F.; Violi, F.; Del Ben, M. Reduced lysosomal acid lipase activity: A new marker of liver disease severity across the clinical continuum of non-alcoholic fatty liver disease? World J. Gastroenterol. 2019, 25, 4172–4180. [Google Scholar] [CrossRef]
- Van Name, M.A.; Savoye, M.; Chick, J.M.; Galuppo, B.T.; Feldstein, A.E.; Pierpont, B.; Johnson, C.; Shabanova, V.; Ekong, U.; Valentino, P.L.; et al. A low ω-6 to ω-3 PUFA ratio (n-6:n-3 PUFA) diet to treat fatty liver disease in obese youth. J. Nutr. 2020, 150, 2314–2321. [Google Scholar] [CrossRef] [PubMed]
- Starling, A.P.; Sauder, K.A.; Kaar, J.L.; Shapiro, A.L.; Siega-Riz, A.M.; Dabelea, D. Maternal dietary patterns during pregnancy are associated with newborn body composition. J. Nutr. 2017, 147, 1334–1339. [Google Scholar] [CrossRef] [Green Version]
- Goran, M.I.; Dumke, K.; Bouret, S.G.; Kayser, B.; Walker, R.W.; Blumberg, B. The obesogenic effect of high fructose exposure during early development. Nat. Rev. Endocrinol. 2013, 9, 494–500. [Google Scholar] [CrossRef] [Green Version]
- Sloboda, D.M.; Li, M.; Patel, R.; Clayton, Z.E.; Yap, C.; Vickers, M.H. Early life exposure to fructose and offspring phenotype: Implications for long term metabolic homeostasis. J. Obes. 2014, 2014, 203474. [Google Scholar] [CrossRef]
- Lee, W.C.; Wu, K.L.H.; Leu, S.; Tain, Y.L. Translational insights on developmental origins of metabolic syndrome: Focus on fructose consumption. Biomed. J. 2018, 41, 96–101. [Google Scholar] [CrossRef]
- Hudgins, L.C.; Parker, T.S.; Levine, D.M.; Hellerstein, M.K. A dual sugar challenge test for lipogenic sensitivity to dietary fructose. J. Clin. Endocrinol. Metab. 2011, 96, 861–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beysen, C.; Ruddy, M.; Stoch, A.; Mixson, L.; Rosko, K.; Riiff, T.; Turner, S.M.; Hellerstein, M.K.; Murphy, E.J. Dose-dependent quantitative effects of acute fructose administration on hepatic de novo lipogenesis in healthy humans. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E126–E132. [Google Scholar] [CrossRef] [PubMed]
- Softic, S.; Gupta, M.K.; Wang, G.X.; Fujisaka, S.; O’Neill, B.T.; Rao, T.N.; Willoughby, J.; Harbison, C.; Fitzgerald, K.; Ilkayeva, O.; et al. Divergent effects of glucose and fructose on hepatic lipogenesis and insulin signaling. J. Clin. Investig. 2017, 127, 4059–4074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Softic, S.; Meyer, J.G.; Wang, G.X.; Gupta, M.K.; Batista, T.M.; Lauritzen, H.P.M.M.; Fujisaka, S.; Serra, D.; Herrero, L.; Willoughby, J.; et al. Dietary sugars alter hepatic fatty acid oxidation via transcriptional and post-translational modifications of mitochondrial proteins. Cell Metab. 2019, 30, 735–753.e734. [Google Scholar] [CrossRef] [PubMed]
- Lustig, R.H.; Mulligan, K.; Noworolski, S.M.; Tai, V.W.; Wen, M.J.; Erkin-Cakmak, A.; Gugliucci, A.; Schwarz, J.M. Isocaloric fructose restriction and metabolic improvement in children with obesity and metabolic syndrome. Obesity 2016, 24, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.M.; Noworolski, S.M.; Erkin-Cakmak, A.; Korn, N.J.; Wen, M.J.; Tai, V.W.; Jones, G.M.; Palii, S.P.; Velasco-Alin, M.; Pan, K.; et al. Effects of dietary fructose restriction on liver fat, de novo lipogenesis, and insulin kinetics in children with obesity. Gastroenterology 2017, 153, 743–752. [Google Scholar] [CrossRef] [Green Version]
- Schwimmer, J.B.; Ugalde-Nicalo, P.; Welsh, J.A.; Angeles, J.E.; Cordero, M.; Harlow, K.E.; Alazraki, A.; Durelle, J.; Knight-Scott, J.; Newton, K.P.; et al. Effect of a low free sugar diet vs usual diet on nonalcoholic fatty liver disease in adolescent boys: A randomized clinical trial. JAMA 2019, 321, 256–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goss, A.M.; Dowla, S.; Pendergrass, M.; Ashraf, A.; Bolding, M.; Morrison, S.; Amerson, A.; Soleymani, T.; Gower, B. Effects of a carbohydrate-restricted diet on hepatic lipid content in adolescents with non-alcoholic fatty liver disease: A pilot, randomized trial. Pediatr. Obes. 2020, 15, e12630. [Google Scholar] [CrossRef]
- Musso, G.; Gambino, R.; De Michieli, F.; Cassader, M.; Rizzetto, M.; Durazzo, M.; Fagà, E.; Silli, B.; Pagano, G. Dietary habits and their relations to insulin resistance and postprandial lipemia in nonalcoholic steatohepatitis. Hepatology 2003, 37, 909–916. [Google Scholar] [CrossRef]
- Araya, J.; Rodrigo, R.; Videla, L.A.; Thielemann, L.; Orellana, M.; Pettinelli, P.; Poniachik, J. Increase in long-chain polyunsaturated fatty acid n − 6/n − 3 ratio in relation to hepatic steatosis in patients with non-alcoholic fatty liver disease. Clin. Sci. 2004, 106, 635–643. [Google Scholar] [CrossRef] [Green Version]
- Jump, D.B.; Lytle, K.A.; Depner, C.M.; Tripathy, S. Omega-3 polyunsaturated fatty acids as a treatment strategy for nonalcoholic fatty liver disease. Pharmacol. Ther. 2018, 181, 108–125. [Google Scholar] [CrossRef]
- Jump, D.B. N-3 polyunsaturated fatty acid regulation of hepatic gene transcription. Curr. Opin. Lipidol. 2008, 19, 242–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nobili, V.; Bedogni, G.; Alisi, A.; Pietrobattista, A.; Risé, P.; Galli, C.; Agostoni, C. Docosahexaenoic acid supplementation decreases liver fat content in children with non-alcoholic fatty liver disease: Double-blind randomised controlled clinical trial. Arch. Dis. Child 2011, 96, 350–353. [Google Scholar] [CrossRef]
- Janczyk, W.; Lebensztejn, D.; Wierzbicka-Rucińska, A.; Mazur, A.; Neuhoff-Murawska, J.; Matusik, P.; Socha, P. Omega-3 fatty acids therapy in children with nonalcoholic fatty liver disease: A randomized controlled trial. J. Pediatr. 2015, 166, 1358–1363.e1351–1358–e1353. [Google Scholar] [CrossRef] [PubMed]
- Boyraz, M.; Pirgon, Ö.; Dündar, B.; Çekmez, F.; Hatipoğlu, N. Long-term treatment with n-3 polyunsaturated fatty acids as a monotherapy in children with nonalcoholic fatty liver disease. J. Clin. Res. Pediatr. Endocrinol. 2015, 7, 121–127. [Google Scholar] [CrossRef]
- Lowell, B.B.; Shulman, G.I. Mitochondrial dysfunction and type 2 diabetes. Science 2005, 307, 384–387. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, T.; Darley-Usmar, V. Metabolic syndrome and mitochondrial dysfunction: Insights from preclinical studies with a mitochondrially targeted antioxidant. Free Radic. Biol. Med. 2012, 52, 838–840. [Google Scholar] [CrossRef] [Green Version]
- Jha, S.K.; Jha, N.K.; Kumar, D.; Ambasta, R.K.; Kumar, P. Linking mitochondrial dysfunction, metabolic syndrome and stress signaling in neurodegeneration. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1132–1146. [Google Scholar] [CrossRef]
- Caldwell, S.H.; Swerdlow, R.H.; Khan, E.M.; Iezzoni, J.C.; Hespenheide, E.E.; Parks, J.K.; Parker, W.D., Jr. Mitochondrial abnormalities in non-alcoholic steatohepatitis. J. Hepatol. 1999, 31, 430–434. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Campbell-Sargent, C.; Mirshahi, F.; Rizzo, W.B.; Contos, M.J.; Sterling, R.K.; Luketic, V.A.; Shiffman, M.L.; Clore, J.N. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001, 120, 1183–1192. [Google Scholar] [CrossRef]
- Lotowska, J.M.; Sobaniec-Lotowska, M.E.; Bockowska, S.B.; Lebensztejn, D.M. Pediatric non-alcoholic steatohepatitis: The first report on the ultrastructure of hepatocyte mitochondria. World J. Gastroenterol. 2014, 20, 4335–4340. [Google Scholar] [CrossRef]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satapati, S.; Kucejova, B.; Duarte, J.A.; Fletcher, J.A.; Reynolds, L.; Sunny, N.E.; He, T.; Nair, L.A.; Livingston, K.A.; Fu, X.; et al. Mitochondrial metabolism mediates oxidative stress and inflammation in fatty liver. J. Clin. Investig. 2015, 125, 4447–4462. [Google Scholar] [CrossRef] [Green Version]
- Sunny, N.E.; Parks, E.J.; Browning, J.D.; Burgess, S.C. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 2011, 14, 804–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, P.; Wei, L.L.; Zhao, S.; Sverdlov, D.Y.; Vaid, K.A.; Miyamoto, M.; Kuramitsu, K.; Lai, M.; Popov, Y.V. Hepatocyte mitochondria-derived danger signals directly activate hepatic stellate cells and drive progression of liver fibrosis. Nat. Commun. 2020, 11, 2362. [Google Scholar] [CrossRef] [PubMed]
- Thyfault, J.P.; Rector, R.S.; Uptergrove, G.M.; Borengasser, S.J.; Morris, E.M.; Wei, Y.; Laye, M.J.; Burant, C.F.; Qi, N.R.; Ridenhour, S.E.; et al. Rats selectively bred for low aerobic capacity have reduced hepatic mitochondrial oxidative capacity and susceptibility to hepatic steatosis and injury. J. Physiol. 2009, 587, 1805–1816. [Google Scholar] [CrossRef] [PubMed]
- Rector, R.S.; Thyfault, J.P.; Uptergrove, G.M.; Morris, E.M.; Naples, S.P.; Borengasser, S.J.; Mikus, C.R.; Laye, M.J.; Laughlin, M.H.; Booth, F.W.; et al. Mitochondrial dysfunction precedes insulin resistance and hepatic steatosis and contributes to the natural history of non-alcoholic fatty liver disease in an obese rodent model. J. Hepatol. 2010, 52, 727–736. [Google Scholar] [CrossRef] [Green Version]
- Ferro, D.; Baratta, F.; Pastori, D.; Cocomello, N.; Colantoni, A.; Angelico, F.; Del Ben, M. New insights into the pathogenesis of non-alcoholic fatty liver disease: Gut-derived lipopolysaccharides and oxidative stress. Nutrients 2020, 12, 2762. [Google Scholar] [CrossRef]
- Nobili, V.; Parola, M.; Alisi, A.; Marra, F.; Piemonte, F.; Mombello, C.; Sutti, S.; Povero, D.; Maina, V.; Novo, E.; et al. Oxidative stress parameters in paediatric non-alcoholic fatty liver disease. Int. J. Mol. Med. 2010, 26, 471–476. [Google Scholar] [CrossRef] [Green Version]
- Nobili, V.; Alisi, A.; Mosca, A.; Crudele, A.; Zaffina, S.; Denaro, M.; Smeriglio, A.; Trombetta, D. The antioxidant effects of hydroxytyrosol and vitamin E on pediatric nonalcoholic fatty liver disease, in a clinical trial: A new treatment? Antioxid. Redox Signal. 2019, 31, 127–133. [Google Scholar] [CrossRef]
- Negri, R.; Trinchese, G.; Carbone, F.; Caprio, M.G.; Stanzione, G.; di Scala, C.; Micillo, T.; Perna, F.; Tarotto, L.; Gelzo, M.; et al. Randomised clinical trial: Calorie restriction regimen with tomato juice supplementation ameliorates oxidative stress and preserves a proper immune surveillance modulating mitochondrial bioenergetics of T-lymphocytes in obese children affected by non-alcoholic fatty liver disease (NAFLD). J. Clin. Med. 2020, 9, 141. [Google Scholar] [CrossRef] [Green Version]
- Spahis, S.; Alvarez, F.; Ahmed, N.; Dubois, J.; Jalbout, R.; Paganelli, M.; Grzywacz, K.; Delvin, E.; Peretti, N.; Levy, E. Non-alcoholic fatty liver disease severity and metabolic complications in obese children: Impact of omega-3 fatty acids. J. Nutr. Biochem. 2018, 58, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Linhart, K.; Bartsch, H.; Seitz, H.K. The role of reactive oxygen species (ROS) and cytochrome P-450 2E1 in the generation of carcinogenic etheno-DNA adducts. Redox Biol. 2014, 3, 56–62. [Google Scholar] [CrossRef] [Green Version]
- Linhart, K.B.; Glassen, K.; Peccerella, T.; Waldherr, R.; Linhart, H.; Bartsch, H.; Seitz, H.K. The generation of carcinogenic etheno-DNA adducts in the liver of patients with nonalcoholic fatty liver disease. Hepatobiliary Surg. Nutr. 2015, 4, 117–123. [Google Scholar] [CrossRef]
- Teufel, U.; Peccerella, T.; Engelmann, G.; Bruckner, T.; Flechtenmacher, C.; Millonig, G.; Stickel, F.; Hoffmann, G.F.; Schirmacher, P.; Mueller, S.; et al. Detection of carcinogenic etheno-DNA adducts in children and adolescents with non-alcoholic steatohepatitis (NASH). Hepatobiliary Surg. Nutr. 2015, 4, 426–435. [Google Scholar] [CrossRef]
- Grabherr, F.; Grander, C.; Effenberger, M.; Adolph, T.E.; Tilg, H. Gut dysfunction and non-alcoholic fatty liver disease. Front. Endocrinol. 2019, 10, 611. [Google Scholar] [CrossRef] [Green Version]
- Boursier, J.; Diehl, A.M. Nonalcoholic fatty liver disease and the gut microbiome. Clin. Liver Dis. 2016, 20, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Schwimmer, J.B.; Johnson, J.S.; Angeles, J.E.; Behling, C.; Belt, P.H.; Borecki, I.; Bross, C.; Durelle, J.; Goyal, N.P.; Hamilton, G.; et al. Microbiome signatures associated with steatohepatitis and moderate to severe fibrosis in children with nonalcoholic fatty liver disease. Gastroenterology 2019, 157, 1109–1122. [Google Scholar] [CrossRef] [Green Version]
- Fukunishi, S.; Nishio, H.; Fukuda, A.; Takeshita, A.; Hanafusa, T.; Higuchi, K.; Suzuki, K. Development of fibrosis in nonalcoholic steatosis through combination of a synthetic diet rich in disaccharide and low-dose lipopolysaccharides in the livers of Zucker (fa/fa) rats. J. Clin. Biochem. Nutr. 2009, 45, 322–328. [Google Scholar] [CrossRef]
- Imajo, K.; Fujita, K.; Yoneda, M.; Nozaki, Y.; Ogawa, Y.; Shinohara, Y.; Kato, S.; Mawatari, H.; Shibata, W.; Kitani, H.; et al. Hyperresponsivity to low-dose endotoxin during progression to nonalcoholic steatohepatitis is regulated by leptin-mediated signaling. Cell Metab. 2012, 16, 44–54. [Google Scholar] [CrossRef] [Green Version]
- Ilan, Y. Leaky gut and the liver: A role for bacterial translocation in nonalcoholic steatohepatitis. World J. Gastroenterol. 2012, 18, 2609–2618. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, K.; Wylie, A.T.; Tucker, K.L.; Hamp, T.J.; Gharaibeh, R.Z.; Fodor, A.A.; Cullen, J.M. Dietary fructose induces endotoxemia and hepatic injury in calorically controlled primates. Am. J. Clin. Nutr. 2013, 98, 349–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahola, A.J.; Lassenius, M.I.; Forsblom, C.; Harjutsalo, V.; Lehto, M.; Groop, P.H. Dietary patterns reflecting healthy food choices are associated with lower serum LPS activity. Sci. Rep. 2017, 7, 6511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastori, D.; Carnevale, R.; Nocella, C.; Novo, M.; Santulli, M.; Cammisotto, V.; Menichelli, D.; Pignatelli, P.; Violi, F. Gut-derived serum lipopolysaccharide is associated with enhanced risk of major adverse cardiovascular events in atrial fibrillation: Effect of adherence to mediterranean diet. J. Am. Heart Assoc. 2017, 6. [Google Scholar] [CrossRef]
- Spruss, A.; Bergheim, I. Dietary fructose and intestinal barrier: Potential risk factor in the pathogenesis of nonalcoholic fatty liver disease. J. Nutr. Biochem. 2009, 20, 657–662. [Google Scholar] [CrossRef]
- Vos, M.B.; Lavine, J.E. Dietary fructose in nonalcoholic fatty liver disease. Hepatology 2013, 57, 2525–2531. [Google Scholar] [CrossRef]
- Erridge, C.; Attina, T.; Spickett, C.M.; Webb, D.J. A high-fat meal induces low-grade endotoxemia: Evidence of a novel mechanism of postprandial inflammation. Am. J. Clin. Nutr. 2007, 86, 1286–1292. [Google Scholar] [CrossRef]
- Deopurkar, R.; Ghanim, H.; Friedman, J.; Abuaysheh, S.; Sia, C.L.; Mohanty, P.; Viswanathan, P.; Chaudhuri, A.; Dandona, P. Differential effects of cream, glucose, and orange juice on inflammation, endotoxin, and the expression of Toll-like receptor-4 and suppressor of cytokine signaling-3. Diabetes Care 2010, 33, 991–997. [Google Scholar] [CrossRef] [Green Version]
- Amar, J.; Burcelin, R.; Ruidavets, J.B.; Cani, P.D.; Fauvel, J.; Alessi, M.C.; Chamontin, B.; Ferriéres, J. Energy intake is associated with endotoxemia in apparently healthy men. Am. J. Clin. Nutr. 2008, 87, 1219–1223. [Google Scholar] [CrossRef] [Green Version]
- Giorgio, V.; Miele, L.; Principessa, L.; Ferretti, F.; Villa, M.P.; Negro, V.; Grieco, A.; Alisi, A.; Nobili, V. Intestinal permeability is increased in children with non-alcoholic fatty liver disease, and correlates with liver disease severity. Dig. Liver Dis. 2014, 46, 556–560. [Google Scholar] [CrossRef]
- Nier, A.; Engstler, A.J.; Maier, I.B.; Bergheim, I. Markers of intestinal permeability are already altered in early stages of non-alcoholic fatty liver disease: Studies in children. PLoS ONE 2017, 12, e0183282. [Google Scholar] [CrossRef] [Green Version]
- Nier, A.; Brandt, A.; Conzelmann, I.B.; Özel, Y.; Bergheim, I. Non-alcoholic fatty liver disease in overweight children: Role of fructose intake and dietary pattern. Nutrients 2018, 10, 1329. [Google Scholar] [CrossRef] [Green Version]
- Bauer, T.M.; Schwacha, H.; Steinbrückner, B.; Brinkmann, F.E.; Ditzen, A.K.; Aponte, J.J.; Pelz, K.; Berger, D.; Kist, M.; Blum, H.E. Small intestinal bacterial overgrowth in human cirrhosis is associated with systemic endotoxemia. Am. J. Gastroenterol. 2002, 97, 2364–2370. [Google Scholar] [CrossRef]
- Bauer, T.M.; Steinbrückner, B.; Brinkmann, F.E.; Ditzen, A.K.; Schwacha, H.; Aponte, J.J.; Pelz, K.; Kist, M.; Blum, H.E. Small intestinal bacterial overgrowth in patients with cirrhosis: Prevalence and relation with spontaneous bacterial peritonitis. Am. J. Gastroenterol. 2001, 96, 2962–2967. [Google Scholar] [CrossRef] [PubMed]
- Harte, A.L.; da Silva, N.F.; Creely, S.J.; McGee, K.C.; Billyard, T.; Youssef-Elabd, E.M.; Tripathi, G.; Ashour, E.; Abdalla, M.S.; Sharada, H.M.; et al. Elevated endotoxin levels in non-alcoholic fatty liver disease. J. Inflamm. 2010, 7, 15. [Google Scholar] [CrossRef] [Green Version]
- Wong, V.W.; Wong, G.L.; Chan, H.Y.; Yeung, D.K.; Chan, R.S.; Chim, A.M.; Chan, C.K.; Tse, Y.K.; Woo, J.; Chu, W.C.; et al. Bacterial endotoxin and non-alcoholic fatty liver disease in the general population: A prospective cohort study. Aliment. Pharmacol. Ther. 2015, 42, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [Green Version]
- den Besten, G.; Bleeker, A.; Gerding, A.; van Eunen, K.; Havinga, R.; van Dijk, T.H.; Oosterveer, M.H.; Jonker, J.W.; Groen, A.K.; Reijngoud, D.J.; et al. Short-chain fatty acids protect against high-fat diet-induced obesity via a PPARγ-dependent switch from lipogenesis to fat oxidation. Diabetes 2015, 64, 2398–2408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef]
- Albillos, A.; de Gottardi, A.; Rescigno, M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J. Hepatol. 2020, 72, 558–577. [Google Scholar] [CrossRef] [Green Version]
- Koch, M.A.; Reiner, G.L.; Lugo, K.A.; Kreuk, L.S.; Stanbery, A.G.; Ansaldo, E.; Seher, T.D.; Ludington, W.B.; Barton, G.M. Maternal IgG and IgA antibodies dampen mucosal T helper cell responses in early life. Cell 2016, 165, 827–841. [Google Scholar] [CrossRef] [Green Version]
- Zeng, M.Y.; Cisalpino, D.; Varadarajan, S.; Hellman, J.; Warren, H.S.; Cascalho, M.; Inohara, N.; Núñez, G. Gut microbiota-induced immunoglobulin G controls systemic infection by symbiotic bacteria and pathogens. Immunity 2016, 44, 647–658. [Google Scholar] [CrossRef] [Green Version]
- Arrieta, M.C.; Stiemsma, L.T.; Dimitriu, P.A.; Thorson, L.; Russell, S.; Yurist-Doutsch, S.; Kuzeljevic, B.; Gold, M.J.; Britton, H.M.; Lefebvre, D.L.; et al. Early infancy microbial and metabolic alterations affect risk of childhood asthma. Sci. Transl. Med. 2015, 7, 307ra152. [Google Scholar] [CrossRef]
- Nogacka, A.M.; Salazar, N.; Arboleya, S.; Suárez, M.; Fernández, N.; Solís, G.; de Los Reyes-Gavilán, C.G.; Gueimonde, M. Early microbiota, antibiotics and health. Cell. Mol. Life Sci. 2018, 75, 83–91. [Google Scholar] [CrossRef]
- Mirpuri, J.; Raetz, M.; Sturge, C.R.; Wilhelm, C.L.; Benson, A.; Savani, R.C.; Hooper, L.V.; Yarovinsky, F. Proteobacteria-specific IgA regulates maturation of the intestinal microbiota. Gut Microbes 2014, 5, 28–39. [Google Scholar] [CrossRef] [Green Version]
- Al Nabhani, Z.; Eberl, G. Imprinting of the immune system by the microbiota early in life. Mucosal Immunol. 2020, 13, 183–189. [Google Scholar] [CrossRef] [Green Version]
- Vatanen, T.; Kostic, A.D.; d’Hennezel, E.; Siljander, H.; Franzosa, E.A.; Yassour, M.; Kolde, R.; Vlamakis, H.; Arthur, T.D.; Hämäläinen, A.M.; et al. Variation in microbiome LPS immunogenicity contributes to autoimmunity in humans. Cell 2016, 165, 842–853. [Google Scholar] [CrossRef] [Green Version]
- Houghteling, P.D.; Walker, W.A. Why is initial bacterial colonization of the intestine important to infants’ and children’s health? J. Pediatr. Gastroenterol. Nutr. 2015, 60, 294–307. [Google Scholar] [CrossRef] [Green Version]
- Lemas, D.J.; Young, B.E.; Baker, P.R., 2nd; Tomczik, A.C.; Soderborg, T.K.; Hernandez, T.L.; de la Houssaye, B.A.; Robertson, C.E.; Rudolph, M.C.; Ir, D.; et al. Alterations in human milk leptin and insulin are associated with early changes in the infant intestinal microbiome. Am. J. Clin. Nutr. 2016, 103, 1291–1300. [Google Scholar] [CrossRef]
- Soderborg, T.K.; Clark, S.E.; Mulligan, C.E.; Janssen, R.C.; Babcock, L.; Ir, D.; Young, B.E.; Krebs, N.F.; Lemas, D.J.; Johnson, L.K.; et al. The gut microbiota in infants of obese mothers increases inflammation and susceptibility to NAFLD. Nat. Commun. 2018, 9, 4462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soderborg, T.K.; Friedman, J.E. Imbalance in gut microbes from babies born to obese mothers increases gut permeability and myeloid cell adaptations that provoke obesity and NAFLD. Microb. Cell 2018, 6, 102–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayakumar, S.; Loomba, R. Review article: Emerging role of the gut microbiome in the progression of nonalcoholic fatty liver disease and potential therapeutic implications. Aliment. Pharmacol. Ther. 2019, 50, 144–158. [Google Scholar] [CrossRef] [PubMed]
- Willeit, P.; Skroblin, P.; Kiechl, S.; Fernández-Hernando, C.; Mayr, M. Liver microRNAs: Potential mediators and biomarkers for metabolic and cardiovascular disease? Eur. Heart J. 2016, 37, 3260–3266. [Google Scholar] [CrossRef] [Green Version]
- Weiland, M.; Gao, X.H.; Zhou, L.; Mi, Q.S. Small RNAs have a large impact: Circulating microRNAs as biomarkers for human diseases. RNA Biol. 2012, 9, 850–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pirola, C.J.; Fernández Gianotti, T.; Castaño, G.O.; Mallardi, P.; San Martino, J.; Mora Gonzalez Lopez Ledesma, M.; Flichman, D.; Mirshahi, F.; Sanyal, A.J.; Sookoian, S. Circulating microRNA signature in non-alcoholic fatty liver disease: From serum non-coding RNAs to liver histology and disease pathogenesis. Gut 2015, 64, 800–812. [Google Scholar] [CrossRef] [Green Version]
- Chai, C.; Rivkin, M.; Berkovits, L.; Simerzin, A.; Zorde-Khvalevsky, E.; Rosenberg, N.; Klein, S.; Yaish, D.; Durst, R.; Shpitzen, S.; et al. Metabolic circuit involving free fatty acids, microRNA 122, and triglyceride synthesis in liver and muscle tissues. Gastroenterology 2017, 153, 1404–1415. [Google Scholar] [CrossRef]
- Yu, J.; Peng, J.; Luan, Z.; Zheng, F.; Su, W. MicroRNAs as a novel tool in the diagnosis of liver lipid dysregulation and fatty liver disease. Molecules 2019, 24, 230. [Google Scholar] [CrossRef] [Green Version]
- Mori, M.A.; Ludwig, R.G.; Garcia-Martin, R.; Brandão, B.B.; Kahn, C.R. Extracellular miRNAs: From biomarkers to mediators of physiology and disease. Cell Metab. 2019, 30, 656–673. [Google Scholar] [CrossRef]
- Lin, H.Y.; Yang, Y.L.; Wang, P.W.; Wang, F.S.; Huang, Y.H. The emerging role of microRNAs in NAFLD: Highlight of microRNA-29a in modulating oxidative stress, inflammation, and beyond. Cells 2020, 9, 1041. [Google Scholar] [CrossRef]
- Chang, J.; Nicolas, E.; Marks, D.; Sander, C.; Lerro, A.; Buendia, M.A.; Xu, C.; Mason, W.S.; Moloshok, T.; Bort, R.; et al. miR-122, a mammalian liver-specific microRNA, is processed from hcr mRNA and may downregulate the high affinity cationic amino acid transporter CAT-1. RNA Biol. 2004, 1, 106–113. [Google Scholar] [CrossRef] [Green Version]
- Iguchi, T.; Niino, N.; Tamai, S.; Sakurai, K.; Mori, K. Comprehensive analysis of circulating microRNA specific to the liver, heart, and skeletal muscle of cynomolgus monkeys. Int. J. Toxicol. 2017, 36, 220–228. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Ge, G.; Pan, T.; Wen, D.; Gan, J. A pilot study of serum microRNAs panel as potential biomarkers for diagnosis of nonalcoholic fatty liver disease. PLoS ONE 2014, 9, e105192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, J.K.; Dai, W.; Zheng, Y.W.; Zhao, S.P. miR-122 promotes hepatic lipogenesis via inhibiting the LKB1/AMPK pathway by targeting Sirt1 in non-alcoholic fatty liver disease. Mol. Med. 2019, 25, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Povero, D.; Eguchi, A.; Li, H.; Johnson, C.D.; Papouchado, B.G.; Wree, A.; Messer, K.; Feldstein, A.E. Circulating extracellular vesicles with specific proteome and liver microRNAs are potential biomarkers for liver injury in experimental fatty liver disease. PLoS ONE 2014, 9, e113651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandt, S.; Roos, J.; Inzaghi, E.; Kotnik, P.; Kovac, J.; Battelino, T.; Cianfarani, S.; Nobili, V.; Colajacomo, M.; Kratzer, W.; et al. Circulating levels of miR-122 and nonalcoholic fatty liver disease in pre-pubertal obese children. Pediatr. Obes. 2018, 13, 175–182. [Google Scholar] [CrossRef]
- Cheung, O.; Puri, P.; Eicken, C.; Contos, M.J.; Mirshahi, F.; Maher, J.W.; Kellum, J.M.; Min, H.; Luketic, V.A.; Sanyal, A.J. Nonalcoholic steatohepatitis is associated with altered hepatic microRNA expression. Hepatology 2008, 48, 1810–1820. [Google Scholar] [CrossRef] [Green Version]
- Braza-Boïls, A.; Marí-Alexandre, J.; Molina, P.; Arnau, M.A.; Barceló-Molina, M.; Domingo, D.; Girbes, J.; Giner, J.; Martínez-Dolz, L.; Zorio, E. Deregulated hepatic microRNAs underlie the association between non-alcoholic fatty liver disease and coronary artery disease. Liver Int. 2016, 36, 1221–1229. [Google Scholar] [CrossRef]
- Miyaaki, H.; Ichikawa, T.; Kamo, Y.; Taura, N.; Honda, T.; Shibata, H.; Milazzo, M.; Fornari, F.; Gramantieri, L.; Bolondi, L.; et al. Significance of serum and hepatic microRNA-122 levels in patients with non-alcoholic fatty liver disease. Liver Int. 2014, 34, e302–e307. [Google Scholar] [CrossRef]
- Mukherjee, K.; Ghoshal, B.; Ghosh, S.; Chakrabarty, Y.; Shwetha, S.; Das, S.; Bhattacharyya, S.N. Reversible HuR-microRNA binding controls extracellular export of miR-122 and augments stress response. EMBO Rep. 2016, 17, 1184–1203. [Google Scholar] [CrossRef]
- Guo, H.; Xu, M.; Cao, Z.; Li, W.; Chen, L.; Xie, X.; Wang, W.; Liu, J. Ultrasound-assisted miR-122-loaded polymeric nanodroplets for hepatocellular carcinoma gene therapy. Mol. Pharm. 2020, 17, 541–553. [Google Scholar] [CrossRef]
- Ning, Q.; Liu, Y.F.; Ye, P.J.; Gao, P.; Li, Z.P.; Tang, S.Y.; He, D.X.; Tang, S.S.; Wei, H.; Yu, C.Y. Delivery of liver-specific miRNA-122 using a targeted macromolecular prodrug toward synergistic therapy for hepatocellular carcinoma. ACS Appl. Mater. Interfaces 2019, 11, 10578–10588. [Google Scholar] [CrossRef]
- Liu, X.L.; Pan, Q.; Cao, H.X.; Xin, F.Z.; Zhao, Z.H.; Yang, R.X.; Zeng, J.; Zhou, H.; Fan, J.G. Lipotoxic hepatocyte-derived exosomal microRNA 192-5p activates macrophages through rictor/Akt/forkhead box transcription factor O1 signaling in nonalcoholic fatty liver disease. Hepatology 2020, 72, 454–469. [Google Scholar] [CrossRef]
- Párrizas, M.; Brugnara, L.; Esteban, Y.; González-Franquesa, A.; Canivell, S.; Murillo, S.; Gordillo-Bastidas, E.; Cussó, R.; Cadefau, J.A.; García-Roves, P.M.; et al. Circulating miR-192 and miR-193b are markers of prediabetes and are modulated by an exercise intervention. J. Clin. Endocrinol. Metab. 2015, 100, E407–E415. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.L.; Cao, H.X.; Wang, B.C.; Xin, F.Z.; Zhang, R.N.; Zhou, D.; Yang, R.X.; Zhao, Z.H.; Pan, Q.; Fan, J.G. miR-192-5p regulates lipid synthesis in non-alcoholic fatty liver disease through SCD-1. World J. Gastroenterol. 2017, 23, 8140–8151. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, N.; Wang, Z.; Ai, D.M.; Cao, Z.Y.; Pan, H.P. Decreased miR-155 level in the peripheral blood of non-alcoholic fatty liver disease patients may serve as a biomarker and may influence LXR activity. Cell. Physiol. Biochem. 2016, 39, 2239–2248. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Lian, F.; Liu, C.; Hu, K.Q.; Wang, X.D. Isocaloric pair-fed high-carbohydrate diet induced more hepatic steatosis and inflammation than high-fat diet mediated by miR-34a/SIRT1 axis in mice. Sci. Rep. 2015, 5, 16774. [Google Scholar] [CrossRef] [PubMed]
- Latorre, J.; Ortega, F.J.; Liñares-Pose, L.; Moreno-Navarrete, J.M.; Lluch, A.; Comas, F.; Oliveras-Cañellas, N.; Ricart, W.; Höring, M.; Zhou, Y.; et al. Compounds that modulate AMPK activity and hepatic steatosis impact the biosynthesis of microRNAs required to maintain lipid homeostasis in hepatocytes. EBioMedicine 2020, 53, 102697. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Xiong, Y.; Sheng, Q.; Zhao, S.; Wattacheril, J.; Flynn, C.R. A micro-RNA expression signature for human NAFLD progression. J. Gastroenterol. 2016, 51, 1022–1030. [Google Scholar] [CrossRef] [Green Version]
- Cermelli, S.; Ruggieri, A.; Marrero, J.A.; Ioannou, G.N.; Beretta, L. Circulating microRNAs in patients with chronic hepatitis C and non-alcoholic fatty liver disease. PLoS ONE 2011, 6, e23937. [Google Scholar] [CrossRef] [Green Version]
- Zarrinpar, A.; Gupta, S.; Maurya, M.R.; Subramaniam, S.; Loomba, R. Serum microRNAs explain discordance of non-alcoholic fatty liver disease in monozygotic and dizygotic twins: A prospective study. Gut 2016, 65, 1546–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Ai, H.; Liu, L.; Zhang, X.; Gao, F.; Zheng, L.; Yi, J.; Sun, L.; Yu, C.; Zhao, H.; et al. Micro-RNA-27a/b negatively regulates hepatic gluconeogenesis by targeting FOXO1. Am. J. Physiol. Endocrinol. Metab. 2019, 317, E911–E924. [Google Scholar] [CrossRef]
- Alisi, A.; Da Sacco, L.; Bruscalupi, G.; Piemonte, F.; Panera, N.; De Vito, R.; Leoni, S.; Bottazzo, G.F.; Masotti, A.; Nobili, V. Mirnome analysis reveals novel molecular determinants in the pathogenesis of diet-induced nonalcoholic fatty liver disease. Lab. Investig. 2011, 91, 283–293. [Google Scholar] [CrossRef]
- Can, U.; Buyukinan, M.; Yerlikaya, F.H. The investigation of circulating microRNAs associated with lipid metabolism in childhood obesity. Pediatr. Obes. 2016, 11, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Dong, B.; Tian, Y.; Lefebvre, P.; Meng, Z.; Wang, X.; Pattou, F.; Han, W.; Wang, X.; Lou, F.; et al. MicroRNA-26a regulates insulin sensitivity and metabolism of glucose and lipids. J. Clin. Investig. 2015, 125, 2497–2509. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Tian, Y.; Tang, D.; Zou, S.; Liu, G.; Song, J.; Zhang, G.; Du, X.; Huang, W.; He, B.; et al. An endoplasmic reticulum stress-microRNA-26a feedback circuit in nonalcoholic fatty liver disease. Hepatology 2020. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.B.; Chernausek, S.D.; Teague, A.M.; Bard, D.E.; Tryggestad, J.B. Maternal diabetes alters microRNA expression in fetal exosomes, human umbilical vein endothelial cells and placenta. Pediatr. Res. 2020. [Google Scholar] [CrossRef]
- Tryggestad, J.B.; Vishwanath, A.; Jiang, S.; Mallappa, A.; Teague, A.M.; Takahashi, Y.; Thompson, D.M.; Chernausek, S.D. Influence of gestational diabetes mellitus on human umbilical vein endothelial cell miRNA. Clin. Sci. 2016, 130, 1955–1967. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Teague, A.M.; Tryggestad, J.B.; Chernausek, S.D. Role of microRNA-130b in placental PGC-1alpha/TFAM mitochondrial biogenesis pathway. Biochem. Biophys. Res. Commun. 2017, 487, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Du, X.; Li, J.; Lonnerdal, B. Human milk exosomes and their microRNAs survive digestion in vitro and are taken up by human intestinal cells. Mol. Nutr. Food Res. 2017, 61, 1700082. [Google Scholar] [CrossRef]
- Benmoussa, A.; Provost, P. Milk microRNAs in health and disease. Compr. Rev. Food Sci. F 2019, 18, 703–722. [Google Scholar] [CrossRef] [Green Version]
- Shankar, K.; Harrell, A.; Liu, X.; Gilchrist, J.M.; Ronis, M.J.; Badger, T.M. Maternal obesity at conception programs obesity in the offspring. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, R528–R538. [Google Scholar] [CrossRef]
- Modi, N.; Murgasova, D.; Ruager-Martin, R.; Thomas, E.L.; Hyde, M.J.; Gale, C.; Santhakumaran, S.; Dore, C.J.; Alavi, A.; Bell, J.D. The influence of maternal body mass index on infant adiposity and hepatic lipid content. Pediatr. Res. 2011, 70, 287–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brumbaugh, D.E.; Tearse, P.; Cree-Green, M.; Fenton, L.Z.; Brown, M.; Scherzinger, A.; Reynolds, R.; Alston, M.; Hoffman, C.; Pan, Z.; et al. Intrahepatic fat is increased in the neonatal offspring of obese women with gestational diabetes. J. Pediatr. 2013, 162, 930–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newton, K.P.; Feldman, H.S.; Chambers, C.D.; Wilson, L.; Behling, C.; Clark, J.M.; Molleston, J.P.; Chalasani, N.; Sanyal, A.J.; Fishbein, M.H.; et al. Low and high birth weights are risk factors for nonalcoholic fatty liver disease in children. J. Pediatr. 2017, 187, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Gregorio, B.M.; Souza-Mello, V.; Carvalho, J.J.; Mandarim-de-Lacerda, C.A.; Aguila, M.B. Maternal high-fat intake predisposes nonalcoholic fatty liver disease in C57BL/6 offspring. Am. J. Obstet. Gynecol. 2010, 203, 495.e491–e498. [Google Scholar] [CrossRef] [PubMed]
- Oben, J.A.; Mouralidarane, A.; Samuelsson, A.M.; Matthews, P.J.; Morgan, M.L.; McKee, C.; Soeda, J.; Fernandez-Twinn, D.S.; Martin-Gronert, M.S.; Ozanne, S.E.; et al. Maternal obesity during pregnancy and lactation programs the development of offspring non-alcoholic fatty liver disease in mice. J. Hepatol. 2010, 52, 913–920. [Google Scholar] [CrossRef]
- Bruce, K.D.; Cagampang, F.R.; Argenton, M.; Zhang, J.; Ethirajan, P.L.; Burdge, G.C.; Bateman, A.C.; Clough, G.F.; Poston, L.; Hanson, M.A.; et al. Maternal high-fat feeding primes steatohepatitis in adult mice offspring, involving mitochondrial dysfunction and altered lipogenesis gene expression. Hepatology 2009, 50, 1796–1808. [Google Scholar] [CrossRef]
- Mouralidarane, A.; Soeda, J.; Visconti-Pugmire, C.; Samuelsson, A.M.; Pombo, J.; Maragkoudaki, X.; Butt, A.; Saraswati, R.; Novelli, M.; Fusai, G.; et al. Maternal obesity programs offspring nonalcoholic fatty liver disease by innate immune dysfunction in mice. Hepatology 2013, 58, 128–138. [Google Scholar] [CrossRef]
- McCurdy, C.E.; Bishop, J.M.; Williams, S.M.; Grayson, B.E.; Smith, M.S.; Friedman, J.E.; Grove, K.L. Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. J. Clin. Investig. 2009, 119, 323–335. [Google Scholar] [CrossRef] [Green Version]
- Igosheva, N.; Abramov, A.Y.; Poston, L.; Eckert, J.J.; Fleming, T.P.; Duchen, M.R.; McConnell, J. Maternal diet-induced obesity alters mitochondrial activity and redox status in mouse oocytes and zygotes. PLoS ONE 2010, 5, e10074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borengasser, S.J.; Lau, F.; Kang, P.; Blackburn, M.L.; Ronis, M.J.; Badger, T.M.; Shankar, K. Maternal obesity during gestation impairs fatty acid oxidation and mitochondrial SIRT3 expression in rat offspring at weaning. PLoS ONE 2011, 6, e24068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgueño, A.L.; Cabrerizo, R.; Gonzales Mansilla, N.; Sookoian, S.; Pirola, C.J. Maternal high-fat intake during pregnancy programs metabolic-syndrome-related phenotypes through liver mitochondrial DNA copy number and transcriptional activity of liver PPARGC1A. J. Nutr. Biochem. 2013, 24, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.L.; Russell, D.L.; Wong, S.L.; Chen, M.; Tsai, T.S.; St John, J.C.; Norman, R.J.; Febbraio, M.A.; Carroll, J.; Robker, R.L. Mitochondrial dysfunction in oocytes of obese mothers: Transmission to offspring and reversal by pharmacological endoplasmic reticulum stress inhibitors. Development 2015, 142, 681–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorn, S.R.; Baquero, K.C.; Newsom, S.A.; El Kasmi, K.C.; Bergman, B.C.; Shulman, G.I.; Grove, K.L.; Friedman, J.E. Early life exposure to maternal insulin resistance has persistent effects on hepatic NAFLD in juvenile nonhuman primates. Diabetes 2014, 63, 2702–2713. [Google Scholar] [CrossRef] [Green Version]
- Alfaradhi, M.Z.; Fernandez-Twinn, D.S.; Martin-Gronert, M.S.; Musial, B.; Fowden, A.; Ozanne, S.E. Oxidative stress and altered lipid homeostasis in the programming of offspring fatty liver by maternal obesity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R26–R34. [Google Scholar] [CrossRef]
- Ashino, N.G.; Saito, K.N.; Souza, F.D.; Nakutz, F.S.; Roman, E.A.; Velloso, L.A.; Torsoni, A.S.; Torsoni, M.A. Maternal high-fat feeding through pregnancy and lactation predisposes mouse offspring to molecular insulin resistance and fatty liver. J. Nutr. Biochem. 2012, 23, 341–348. [Google Scholar] [CrossRef]
- Sureshchandra, S.; Wilson, R.M.; Rais, M.; Marshall, N.E.; Purnell, J.Q.; Thornburg, K.L.; Messaoudi, I. Maternal pregravid obesity remodels the DNA methylation landscape of cord blood monocytes disrupting their inflammatory program. J. Immunol. 2017, 199, 2729–2744. [Google Scholar] [CrossRef] [Green Version]
- Wankhade, U.D.; Zhong, Y.; Kang, P.; Alfaro, M.; Chintapalli, S.V.; Thakali, K.M.; Shankar, K. Enhanced offspring predisposition to steatohepatitis with maternal high-fat diet is associated with epigenetic and microbiome alterations. PLoS ONE 2017, 12, e0175675. [Google Scholar] [CrossRef] [Green Version]
- De Jesus, D.F.; Orime, K.; Kaminska, D.; Kimura, T.; Basile, G.; Wang, C.H.; Haertle, L.; Riemens, R.; Brown, N.K.; Hu, J.; et al. Parental metabolic syndrome epigenetically reprograms offspring hepatic lipid metabolism in mice. J. Clin. Investig. 2020, 130, 2391–2407. [Google Scholar] [CrossRef] [Green Version]
- Suter, M.A.; Chen, A.; Burdine, M.S.; Choudhury, M.; Harris, R.A.; Lane, R.H.; Friedman, J.E.; Grove, K.L.; Tackett, A.J.; Aagaard, K.M. A maternal high-fat diet modulates fetal SIRT1 histone and protein deacetylase activity in nonhuman primates. FASEB J. 2012, 26, 5106–5114. [Google Scholar] [CrossRef] [Green Version]
- de Oliveira, F.L.; Panera, N.; De Stefanis, C.; Mosca, A.; D’Oria, V.; Crudele, A.; De Vito, R.; Nobili, V.; Alisi, A. The number of liver galectin-3 positive cells is dually correlated with NAFLD severity in children. Int. J. Mol. Sci. 2019, 20, 3460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpino, G.; Nobili, V.; Renzi, A.; De Stefanis, C.; Stronati, L.; Franchitto, A.; Alisi, A.; Onori, P.; De Vito, R.; Alpini, G.; et al. Macrophage activation in pediatric nonalcoholic fatty liver disease (NAFLD) correlates with hepatic progenitor cell response via Wnt3a pathway. PLoS ONE 2016, 11, e0157246. [Google Scholar] [CrossRef]
- Lotowska, J.M.; Sobaniec-Lotowska, M.E.; Lebensztejn, D.M. The role of Kupffer cells in the morphogenesis of nonalcoholic steatohepatitis - ultrastructural findings. The first report in pediatric patients. Scand. J. Gastroenterol. 2013, 48, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F.; Zimmermann, H.W. Macrophage heterogeneity in liver injury and fibrosis. J. Hepatol. 2014, 60, 1090–1096. [Google Scholar] [CrossRef] [Green Version]
- Pradere, J.P.; Kluwe, J.; De Minicis, S.; Jiao, J.J.; Gwak, G.Y.; Dapito, D.H.; Jang, M.K.; Guenther, N.D.; Mederacke, I.; Friedman, R.; et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology 2013, 58, 1461–1473. [Google Scholar] [CrossRef] [Green Version]
- Stienstra, R.; Saudale, F.; Duval, C.; Keshtkar, S.; Groener, J.E.; van Rooijen, N.; Staels, B.; Kersten, S.; Müller, M. Kupffer cells promote hepatic steatosis via interleukin-1beta-dependent suppression of peroxisome proliferator-activated receptor alpha activity. Hepatology 2010, 51, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Asanuma, T.; Ono, M.; Kubota, K.; Hirose, A.; Hayashi, Y.; Saibara, T.; Inanami, O.; Ogawa, Y.; Enzan, H.; Onishi, S.; et al. Super paramagnetic iron oxide MRI shows defective Kupffer cell uptake function in non-alcoholic fatty liver disease. Gut 2010, 59, 258–266. [Google Scholar] [CrossRef] [Green Version]
- Gadd, V.L.; Skoien, R.; Powell, E.E.; Fagan, K.J.; Winterford, C.; Horsfall, L.; Irvine, K.; Clouston, A.D. The portal inflammatory infiltrate and ductular reaction in human nonalcoholic fatty liver disease. Hepatology 2014, 59, 1393–1405. [Google Scholar] [CrossRef]
- Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef]
- Kazankov, K.; Jørgensen, S.M.D.; Thomsen, K.L.; Møller, H.J.; Vilstrup, H.; George, J.; Schuppan, D.; Grønbaek, H. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 145–159. [Google Scholar] [CrossRef]
- Deng, Z.B.; Liu, Y.; Liu, C.; Xiang, X.; Wang, J.; Cheng, Z.; Shah, S.V.; Zhang, S.; Zhang, L.; Zhuang, X.; et al. Immature myeloid cells induced by a high-fat diet contribute to liver inflammation. Hepatology 2009, 50, 1412–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obstfeld, A.E.; Sugaru, E.; Thearle, M.; Francisco, A.M.; Gayet, C.; Ginsberg, H.N.; Ables, E.V.; Ferrante, A.W., Jr. C-C chemokine receptor 2 (CCR2) regulates the hepatic recruitment of myeloid cells that promote obesity-induced hepatic steatosis. Diabetes 2010, 59, 916–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, R.; Weston, C.J.; Miao, Z.; Corbett, C.; Armstrong, M.J.; Ertl, L.; Ebsworth, K.; Walters, M.J.; Baumart, T.; Newland, D.; et al. CC chemokine receptor 2 promotes recruitment of myeloid cells associated with insulin resistance in nonalcoholic fatty liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 314, G483–G493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, S.K.; Mantovani, A. Macrophage plasticity and interaction with lymphocyte subsets: Cancer as a paradigm. Nat. Immunol. 2010, 11, 889–896. [Google Scholar] [CrossRef]
- Friedman, J.E.; Dobrinskikh, E.; Alfonso-Garcia, A.; Fast, A.; Janssen, R.C.; Soderborg, T.K.; Anderson, A.L.; Reisz, J.A.; D’Alessandro, A.; Frank, D.N.; et al. Pyrroloquinoline quinone prevents developmental programming of microbial dysbiosis and macrophage polarization to attenuate liver fibrosis in offspring of obese mice. Hepatol. Commun. 2018, 2, 313–328. [Google Scholar] [CrossRef] [Green Version]
- Kamimae-Lanning, A.N.; Krasnow, S.M.; Goloviznina, N.A.; Zhu, X.; Roth-Carter, Q.R.; Levasseur, P.R.; Jeng, S.; McWeeney, S.K.; Kurre, P.; Marks, D.L. Maternal high-fat diet and obesity compromise fetal hematopoiesis. Mol. Metab. 2015, 4, 25–38. [Google Scholar] [CrossRef]
- Carter-Kent, C.; Brunt, E.M.; Yerian, L.M.; Alkhouri, N.; Angulo, P.; Kohli, R.; Ling, S.C.; Xanthakos, S.A.; Whitington, P.F.; Charatcharoenwitthaya, P.; et al. Relations of steatosis type, grade, and zonality to histological features in pediatric nonalcoholic fatty liver disease. J. Pediatr. Gastroenterol. Nutr. 2011, 52, 190–197. [Google Scholar] [CrossRef]
- Alkhouri, N.; Sedki, E.; Alisi, A.; Lopez, R.; Pinzani, M.; Feldstein, A.E.; Nobili, V. Combined paediatric NAFLD fibrosis index and transient elastography to predict clinically significant fibrosis in children with fatty liver disease. Liver Int. 2013, 33, 79–85. [Google Scholar] [CrossRef]
- Nash, M.J.; Frank, D.N.; Friedman, J.E. Early microbes modify immune system development and metabolic homeostasis- the “Restaurant” hypothesis revisited. Front. Endocrinol. 2017, 8, 349. [Google Scholar] [CrossRef]
- Negi, S.; Das, D.K.; Pahari, S.; Nadeem, S.; Agrewala, J.N. Potential role of gut microbiota in induction and regulation of innate immune memory. Front. Immunol. 2019, 10, 2441. [Google Scholar] [CrossRef] [Green Version]
- Campisano, S.; La Colla, A.; Echarte, S.M.; Chisari, A.N. Interplay between early-life malnutrition, epigenetic modulation of the immune function and liver diseases. Nutr. Res. Rev. 2019, 32, 128–145. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mandala, A.; Janssen, R.C.; Palle, S.; Short, K.R.; Friedman, J.E. Pediatric Non-Alcoholic Fatty Liver Disease: Nutritional Origins and Potential Molecular Mechanisms. Nutrients 2020, 12, 3166. https://doi.org/10.3390/nu12103166
Mandala A, Janssen RC, Palle S, Short KR, Friedman JE. Pediatric Non-Alcoholic Fatty Liver Disease: Nutritional Origins and Potential Molecular Mechanisms. Nutrients. 2020; 12(10):3166. https://doi.org/10.3390/nu12103166
Chicago/Turabian StyleMandala, Ashok, Rachel C. Janssen, Sirish Palle, Kevin R. Short, and Jacob E. Friedman. 2020. "Pediatric Non-Alcoholic Fatty Liver Disease: Nutritional Origins and Potential Molecular Mechanisms" Nutrients 12, no. 10: 3166. https://doi.org/10.3390/nu12103166