Congenital Lactase Deficiency: Mutations, Functional and Biochemical Implications, and Future Perspectives


Abstract
:1. Introduction
2. Carbohydrate Malabsorption
3. Congenital Lactose Intolerance
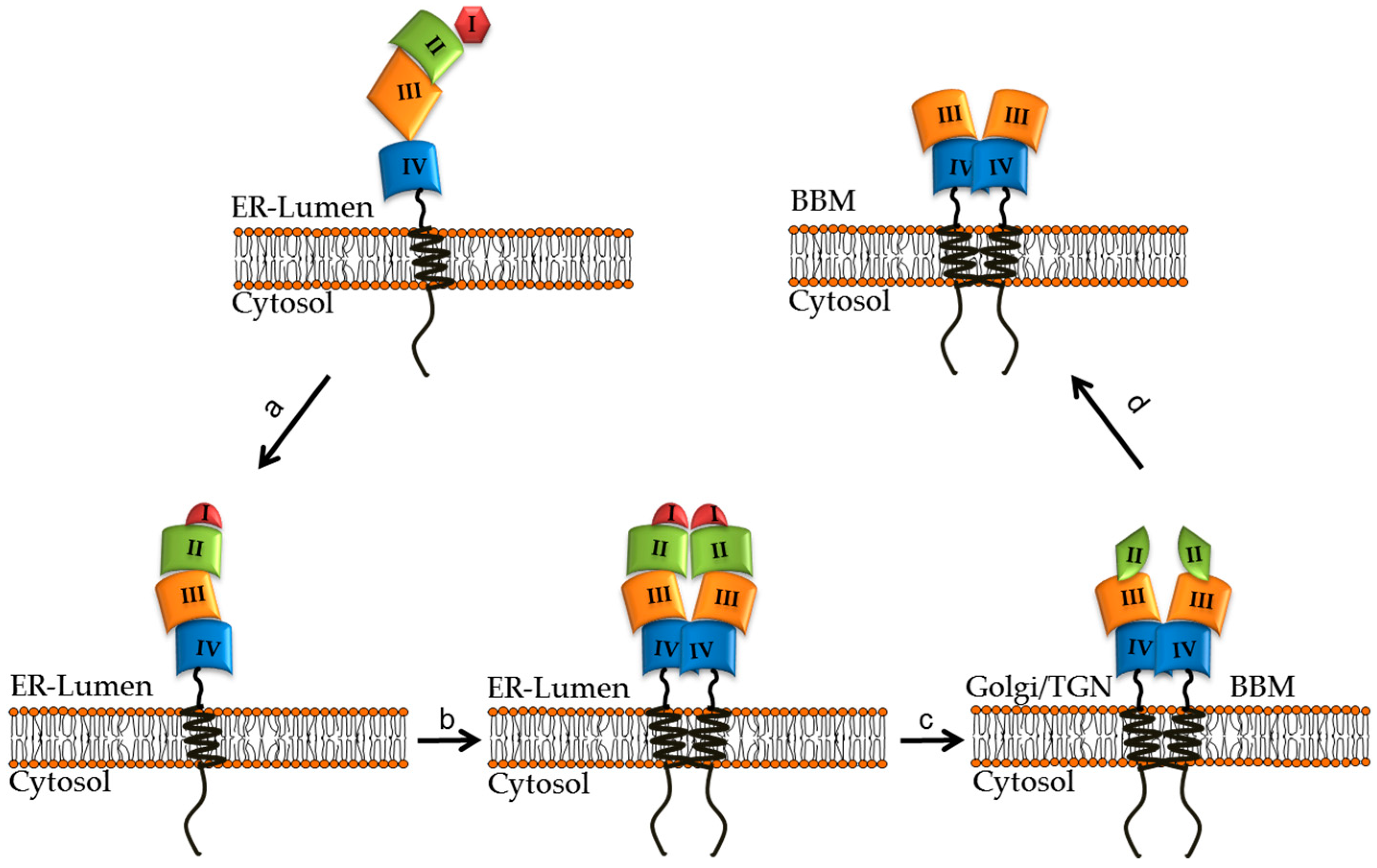
4. Structural and Biosynthetic Features of LPH
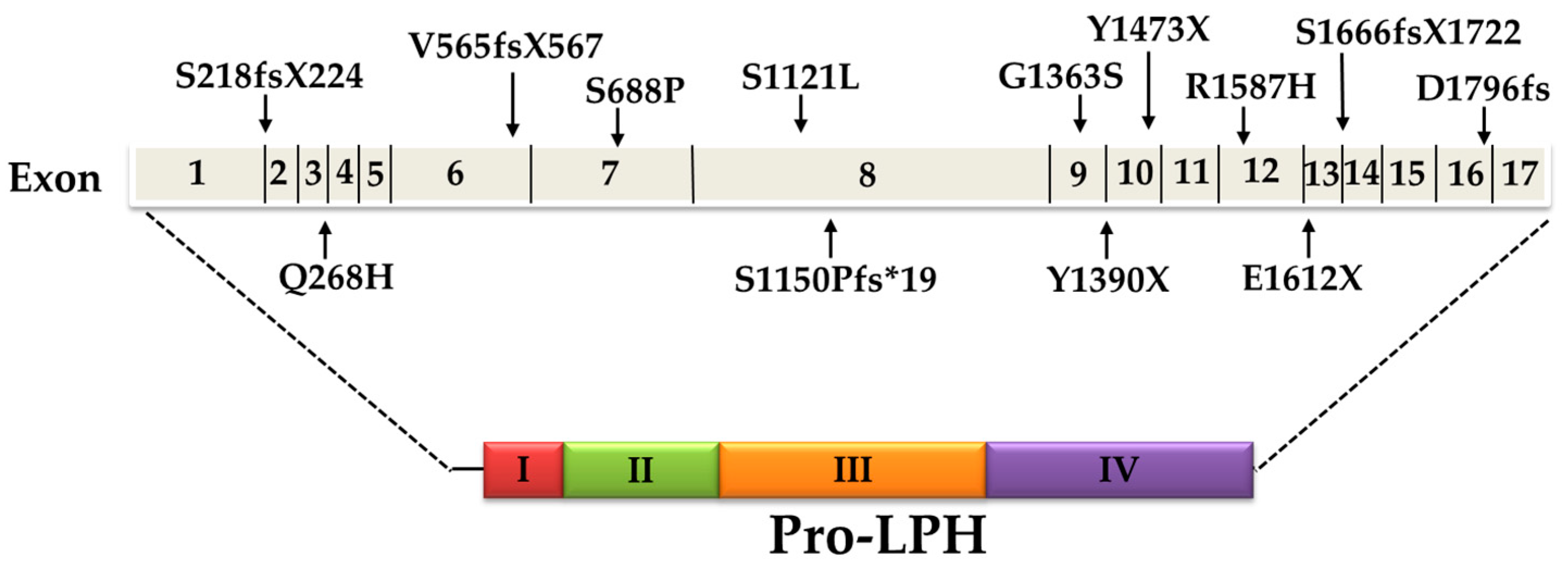
5. Current Knowledge of CLD Mutations
6. Impact of CLD Mutations on the Structural and Biosynthetic Features of LPH
7. Are CLD Mutations Symptomatic in Heterozygote Carriers?
8. Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Nishida, C.; Nocito, F.M. FAO/WHO Scientific Update on carbohydrates in human nutrition: Introduction. Eur. J. Clin. Nutr. 2007, 61, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Vining, E.M.; Hadley, E.C.; Farnham, S.A.; Schneider, E.L. Recommended Dietary Allowances and the Health of the Elderly. N. Engl. J. Med. 1986, 314, 157–160. [Google Scholar]
- Wright, E.M.; Loo, D.D.; Hirayama, B.A. Biology of human sodium glucose transporters. Physiol. Rev. 2011, 91, 733–794. [Google Scholar] [CrossRef] [PubMed]
- Amiri, M.; Naim, H.Y. Characterization of Mucosal Disaccharidases from Human Intestine. Nutrients 2017, 9, 1106. [Google Scholar] [CrossRef] [PubMed]
- Van Beers, E.H.; Büller, H.A.; Grand, R.J.; Einerhand, A.W.C.; Dekker, J. Intestinal Brush Border Glycohydrolases: Structure, Function, and Development. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 197–262. [Google Scholar] [CrossRef] [PubMed]
- Naim, H.Y.; Zimmer, K.-P. Genetically determined disaccharidase deficiency. In Walker’s Pediatric Gastrointestinal Disease, 5th ed.; People Medical Publishing House-USA: Shelton, CT, USA, 2008; pp. 275–287. [Google Scholar]
- Nichols, B.L.; Avery, S.; Sen, P.; Swallow, D.M.; Hahn, D.; Sterchi, E. The maltase-glucoamylase gene: Common ancestry to sucrase-isomaltase with complementary starch digestion activities. Proc. Natl. Acad. Sci. USA 2003, 100, 1432–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballard, O.; Morrow, A.L. Human milk composition: Nutrients and bioactive factors. Pediatr. Clin. 2013, 60, 49–74. [Google Scholar] [CrossRef] [PubMed]
- Daelmans, B.; Dewey, K.; Arimond, M. New and Updated Indicators for Assessing Infant and Young Child Feeding. Food Nutr. Bull. 2009, 30, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Amiri, M.; Diekmann, L.; Von Köckritz-Blickwede, M.; Naim, H. The Diverse Forms of Lactose Intolerance and the Putative Linkage to Several Cancers. Nutrients 2015, 7, 7209–7230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiagarajah, J.R.; Kamin, D.S.; Acra, S.; Goldsmith, J.D.; Roland, J.T.; Lencer, W.I.; Muise, A.M.; Goldenring, J.R.; Avitzur, Y.A.; Martin, M.G. Advances in Evaluation of Chronic Diarrhea in Infants. Gastroenterology 2018, 154, 2045–2059. [Google Scholar] [CrossRef] [PubMed]
- Sander, P.; Alfalah, M.; Keiser, M.; Korponay-Szabo, I.; Kovács, J.B.; Leeb, T.; Naim, H.Y. Novel mutations in the human sucrase-isomaltase gene (SI) that cause congenital carbohydrate malabsorption. Hum. Mutat. 2005, 27, 119. [Google Scholar] [CrossRef] [PubMed]
- Treem, W.R. Congenital Sucrase-Isomaltase Deficiency. J. Pediatr. Gastroenterol. Nutr. 1995, 21, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Gericke, B.; Amiri, M.; Naim, H.Y. The multiple roles of sucrase-isomaltase in the intestinal physiology. Mol. Cell. Pediatr. 2016, 3, 2. [Google Scholar] [CrossRef] [PubMed]
- Jourdan, N.; Brunet, J.P.; Sapin, C.; Blais, A.; Cotte-Laffitte, J.; Forestier, F.; Quero, A.M.; Trugnan, G.; Servin, A.L. Rotavirus infection reduces sucrase-isomaltase expression in human intestinal epithelial cells by perturbing protein targeting and organization of microvillar cytoskeleton. J. Virol. 1998, 72, 7228–7236. [Google Scholar] [PubMed]
- Kuokkanen, M.; Enattah, N.S.; Oksanen, A.; Savilahti, E.; Orpana, A.; Järvelä, I. Transcriptional regulation of the lactase-phlorizin hydrolase gene by polymorphisms associated with adult-type hypolactasia. Gut 2003, 52, 647–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuokkanen, M.; Kokkonen, J.; Enattah, N.S.; Ylisaukko-Oja, T.; Komu, H.; Varilo, T.; Peltonen, L.; Savilahti, E.; Järvelä, I. Mutations in the Translated Region of the Lactase Gene (LCT) Underlie Congenital Lactase Deficiency. Am. J. Hum. Genet. 2006, 78, 339–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heyman, M.B. Lactose Intolerance in Infants, Children, and Adolescents. Pediatrics 2006, 118, 1279–1286. [Google Scholar] [CrossRef] [Green Version]
- Asp, N.-G.; Dahlqvist, A.; Kuitunen, P.; Launiala, K.; Visakorpi, J. Complete deficiency of brush-border lactase in congenital lactose malabsorption. The Lancet 1973, 302, 329–330. [Google Scholar] [CrossRef]
- Freiburghaus, A.U.; Schmitz, J.; Schindler, M.; Rotthauwe, H.W.; Kuitunen, P.; Launiala, K.; Hadorn, B. Protein Patterns of Brush-Border Fragments in Congenital Lactose Malabsorption and in Specific Hypolactasia of the Adult. N. Engl. J. Med. 1976, 294, 1030–1032. [Google Scholar] [CrossRef]
- Savilahti, E.; Launiala, K.; Kuitunen, P. Congenital lactase deficiency. A clinical study on 16 patients. Arch. Dis. Child. 1983, 58, 246–252. [Google Scholar] [CrossRef]
- Naim, H.Y.; Naim, H. Dimerization of lactase-phlorizin hydrolase occurs in the endoplasmic reticulum, involves the putative membrane spanning domain and is required for an efficient transport of the enzyme to the cell surface. Eur. J. Cell Biol. 1996, 70, 198–208. [Google Scholar] [PubMed]
- Mantei, N.; Villa, M.; Enzler, T.; Wacker, H.; Boll, W.; James, P.; Hunziker, W.; Semenza, G. Complete primary structure of human and rabbit lactase-phlorizin hydrolase: Implications for biosynthesis, membrane anchoring and evolution of the enzyme. EMBO J. 1988, 7, 2705–2713. [Google Scholar] [CrossRef] [PubMed]
- Zecca, L.; Mesonero, J.E.; Stutz, A.; Poirée, J.-C.; Giudicelli, J.; Cursio, R.; Gloor, S.M.; Semenza, G. Intestinal lactase-phlorizin hydrolase (LPH): The two catalytic sites; the role of the pancreas in pro-LPH maturation. FEBS Lett. 1998, 435, 225–228. [Google Scholar] [CrossRef]
- Diekmann, L.; Behrendt, M.; Amiri, M.; Naim, H.Y. Structural determinants for transport of lactase phlorizin-hydrolase in the early secretory pathway as a multi-domain membrane glycoprotein. BBA-Gen. Subj. 2017, 1861, 3119–3128. [Google Scholar] [CrossRef] [PubMed]
- Behrendt, M.; Polaina, J.; Naim, H.Y. Structural Hierarchy of Regulatory Elements in the Folding and Transport of an Intestinal Multidomain Protein. J. Biol. Chem. 2009, 285, 4143–4152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacob, R.; Zimmer, K.P.; Naim, H.Y. The apical sorting of lactase-phlorizin hydrolase implicates sorting sequences found in the mature domain. Eur. J. Cell Biol. 1997, 72, 54–60. [Google Scholar] [PubMed]
- Jacob, R.; Peters, K.; Naim, H.Y. The Prosequence of Human Lactase-Phlorizin Hydrolase Modulates the Folding of the Mature Enzyme. J. Biol. Chem. 2001, 277, 8217–8225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panzer, P.; Preuss, U.; Joberty, G.; Naim, H.Y. Protein Domains Implicated in Intracellular Transport and Sorting of Lactase-Phlorizin Hydrolase. J. Biol. Chem. 1998, 273, 13861–13869. [Google Scholar] [CrossRef] [Green Version]
- Jacob, R.; Radebach, I.; Wüthrich, M.; Grünberg, J.; Sterchi, E.E.; Naim, H.Y. Maturation of Human Intestinal Lactase-Phlorizin Hydrolase. Generation of the Brush Border form of the Enzyme Involves at Least Two Proteolytic Cleavage Steps. Eur. J. Biochem. 1996, 236, 789–795. [Google Scholar] [Green Version]
- Naim, H.Y.; Sterchi, E.E.; Lentze, M.J. Biosynthesis and maturation of lactase-phlorizin hydrolase in the human small intestinal epithelial cells. Biochem. J. 1987, 241, 427–434. [Google Scholar] [CrossRef] [Green Version]
- Danielsen, E.M.; Skovbjerg, H.; Norén, O.; Sjöström, H. Biosynthesis of intestinal microvillar proteins intracellular processing of lactase-phlorizin hydrolase. Biochem. Biophys. Res. Commun. 1984, 122, 82–90. [Google Scholar] [CrossRef]
- Jacob, R.; Weiner, J.R.; Stadge, S.; Naim, H.Y. Additional N-Glycosylation and Its Impact on the Folding of Intestinal Lactase-phlorizin Hydrolase. J. Biol. Chem. 2000, 275, 10630–10637. [Google Scholar] [CrossRef] [PubMed]
- Naim, H.Y.; Lentze, M.J. Impact of O-glycosylation on the function of human intestinal lactase-phlorizin hydrolase. Characterization of glycoforms varying in enzyme activity and localization of O-glycoside addition. J. Biol. Chem. 1992, 267, 25494–25504. [Google Scholar]
- Naim, H.Y.; Jacob, R.; Sambrook, J.F.; Gething, M.J. The pro region of human intestinal lactase-phlorizin hydrolase. J. Biol. Chem. 1994, 269, 26933–26943. [Google Scholar]
- Freddara, R.; Routi, T.; Gijsbers, C.; Catassi, C.; Höglund, P.; Torniainen, S.; Savilahti, E.; Järvelä, I. Four novel mutations in the lactase gene (LCT) underlying congenital lactase deficiency (CLD). BMC Gastroenterol. 2009, 9, 8. [Google Scholar]
- Fazeli, W.; Kaczmarek, S.; Kirschstein, M.; Santer, R. A novel mutation within the lactase gene (LCT): The first report of congenital lactase deficiency diagnosed in Central Europe. BMC Gastroenterol. 2015, 15, 706. [Google Scholar] [CrossRef] [PubMed]
- Diekmann, L.; Pfeiffer, K.; Naim, H.Y. Congenital lactose intolerance is triggered by severe mutations on both alleles of the lactase gene. BMC Gastroenterol. 2015, 15, 229. [Google Scholar] [CrossRef]
- Uchida, N.; Sakamoto, O.; Irie, M.; Abukawa, D.; Takeyama, J.; Kure, S.; Tsuchiya, S. Two Novel Mutations in the Lactase Gene in a Japanese Infant with Congenital Lactase Deficiency. Tohoku J. Exp. Med. 2012, 227, 69–72. [Google Scholar] [CrossRef] [Green Version]
- Behrendt, M.; Keiser, M.; Hoch, M.; Naim, H.Y. Impaired Trafficking and Subcellular Localization of a Mutant Lactase Associated with Congenital Lactase Deficiency. Gastroenterology 2009, 136, 2295–2303. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| LPH-Mutation | Location | Mutation Effect | Inheritance Pattern (Genotype) | Ethnic Origin | Reference | |
|---|---|---|---|---|---|---|
| cDNA | Protein | |||||
| c.4170T > A | p. Y1390X | Exon 9 | Nonsense (truncating) | Homozygote | Finland | [1] |
| c.4998_5001delTGAG | p. S1666fsX1722 | Exon 14 | Frameshift (truncating) | Compound heterozygote | ||
| c.653_654delCT | p. S218fsX224 | Exon 2 | Missense | Compound heterozygote | ||
| c.804G > C | p. Q268H | Exon 3 | Missense | Compound heterozygote | ||
| c.4087G > A | p. G1363S | Exon 9 | Missense | Compound heterozygote | ||
| c.1692-1696delAGTGG | p. V565fsX567 | Exon 6 | Frameshift (truncating) | Compound heterozygote | ||
| c.4760G >A | p. R1587H | Exon 12 | Missense | Compound heterozygote | ||
| c.2062T > C | p. S688P | Exon 7 | Missense | Compound heterozygote | Italy | [2] |
| c.4834G > T | p. E1612X | Exon 12 | Nonsense (truncating) | |||
| c.4419C > G | p. Y1473X | Exon 10 | Nonsense (truncating) | Compound heterozygote | Japan | [3] |
| c.5387delA | p. D1796fs | Exon 16 | Frameshift (truncating) | |||
| c.4087G > A | p. G1363S | Exon 9 | Missense | Homozygote | Turkey | [2] |
| c.3448delT | p.S1150Pfs*19 | Exon 8 | Frameshift (truncating) | Homozygote | [4] | |
| c.4087G > A | p. G1363S | Exon 9 | Missense | Homozygote | Iraq | Not published |
| c.3362C > T | p. S1121L | Exon 8 | - | Homozygote | - | Not published |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wanes, D.; Husein, D.M.; Naim, H.Y. Congenital Lactase Deficiency: Mutations, Functional and Biochemical Implications, and Future Perspectives. Nutrients 2019, 11, 461. https://doi.org/10.3390/nu11020461
Wanes D, Husein DM, Naim HY. Congenital Lactase Deficiency: Mutations, Functional and Biochemical Implications, and Future Perspectives. Nutrients. 2019; 11(2):461. https://doi.org/10.3390/nu11020461
Chicago/Turabian StyleWanes, Dalanda, Diab M. Husein, and Hassan Y. Naim. 2019. "Congenital Lactase Deficiency: Mutations, Functional and Biochemical Implications, and Future Perspectives" Nutrients 11, no. 2: 461. https://doi.org/10.3390/nu11020461