
Chlorobenzene Oxidation over Phosphotungstic-Acid-Coated Cerium Oxide: Synergistic Effect of Phosphotungstic and Cerium Oxide and Inhibition Mechanism of Sulfur Dioxide
Abstract
:1. Introduction
2. Experimental Section
2.1. Catalyst Preparation
2.2. Catalytic Performance Evaluation
2.3. Catalyst Characterization
3. Results and Discussion
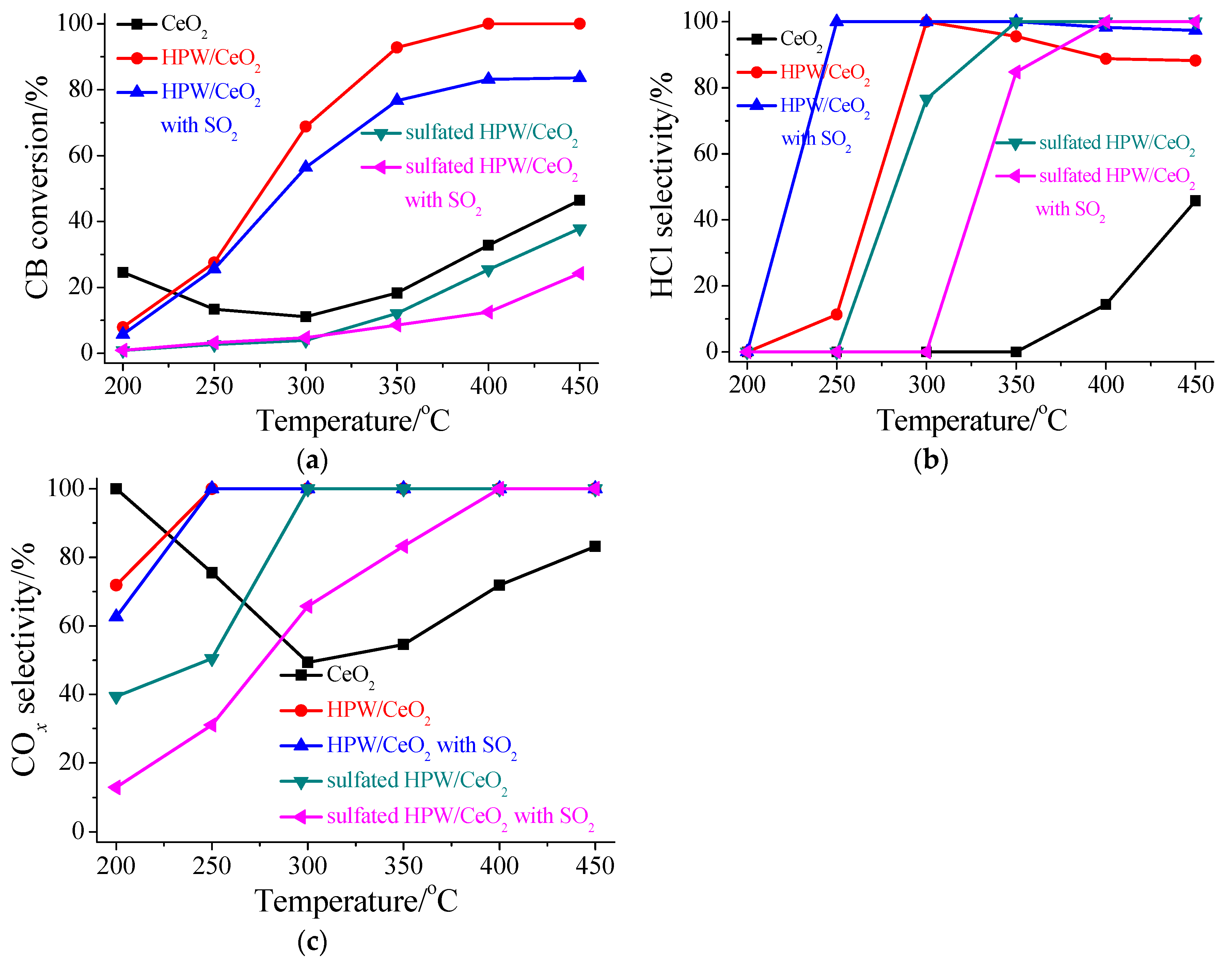
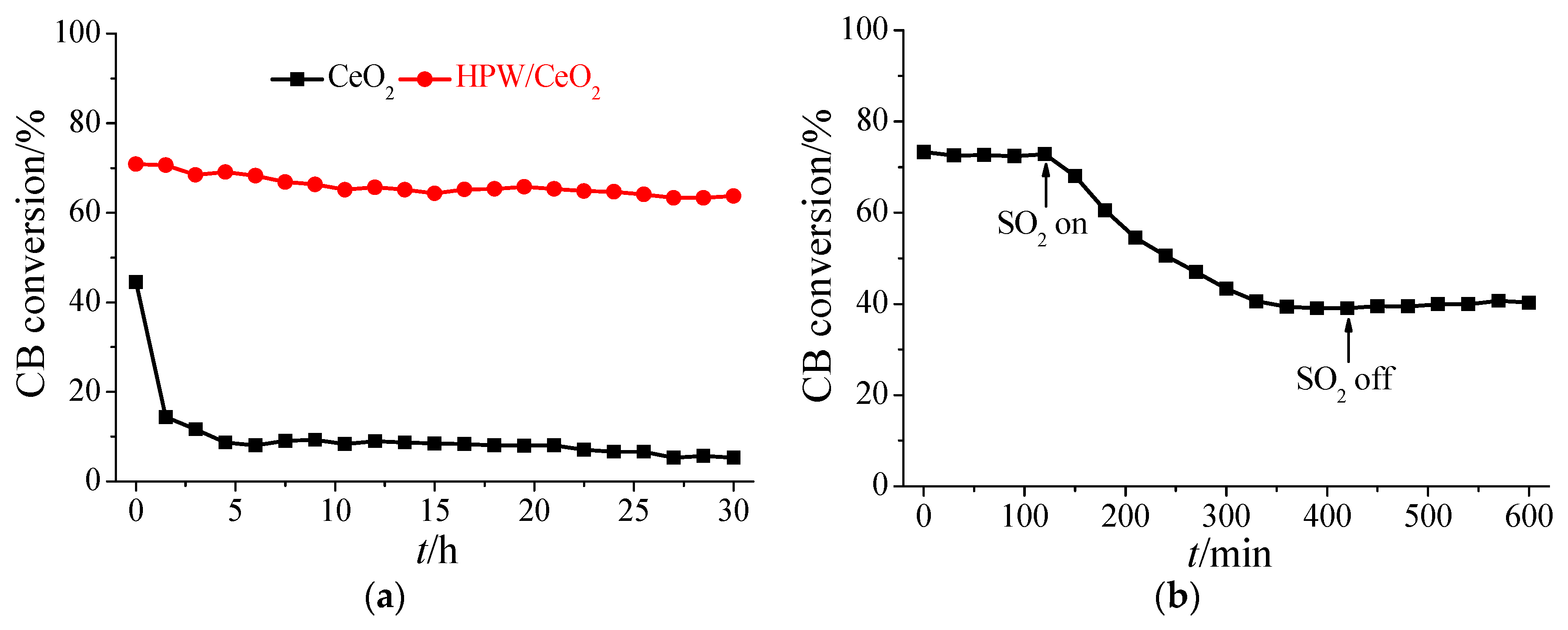
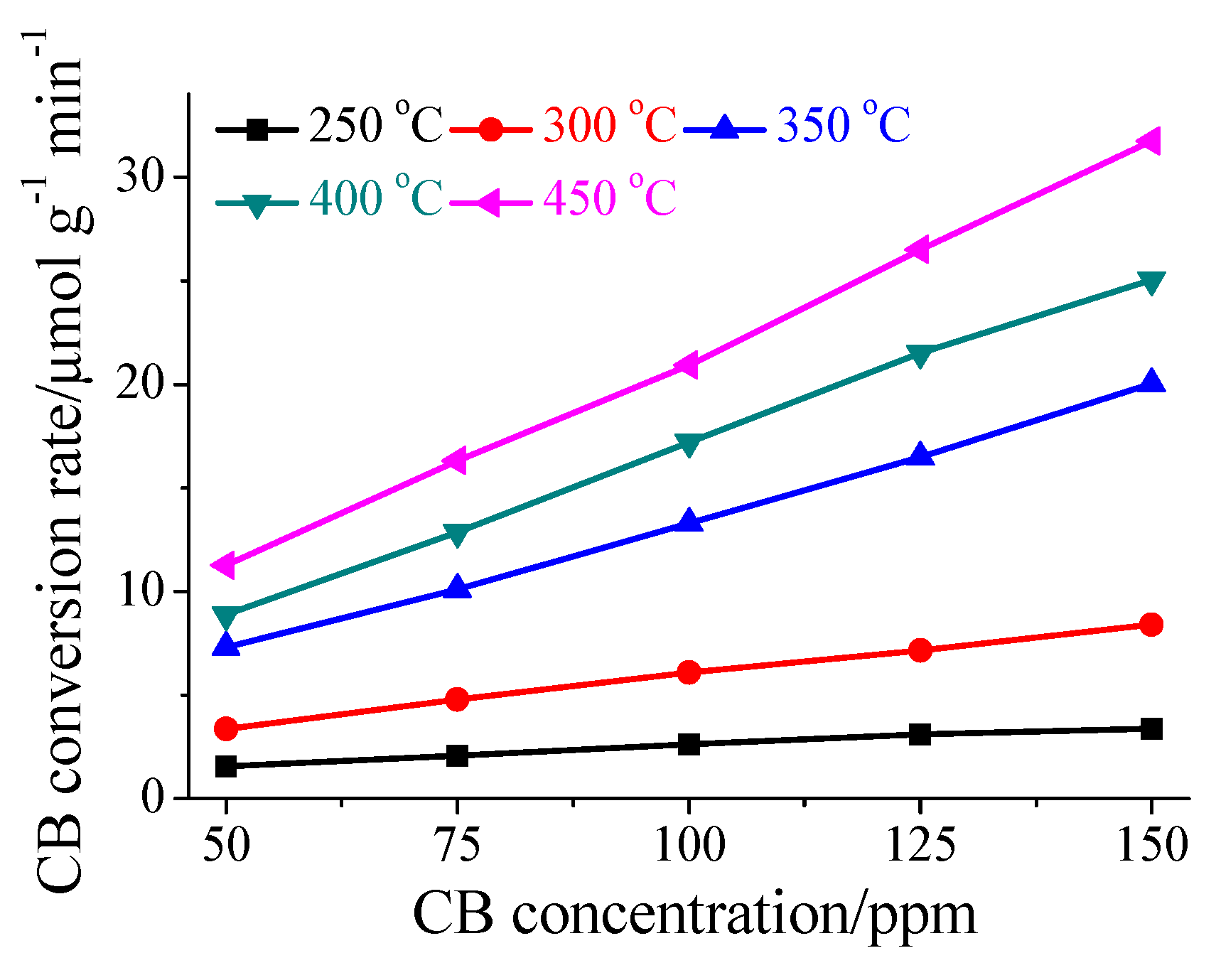
3.1. Performance for CB Oxidation
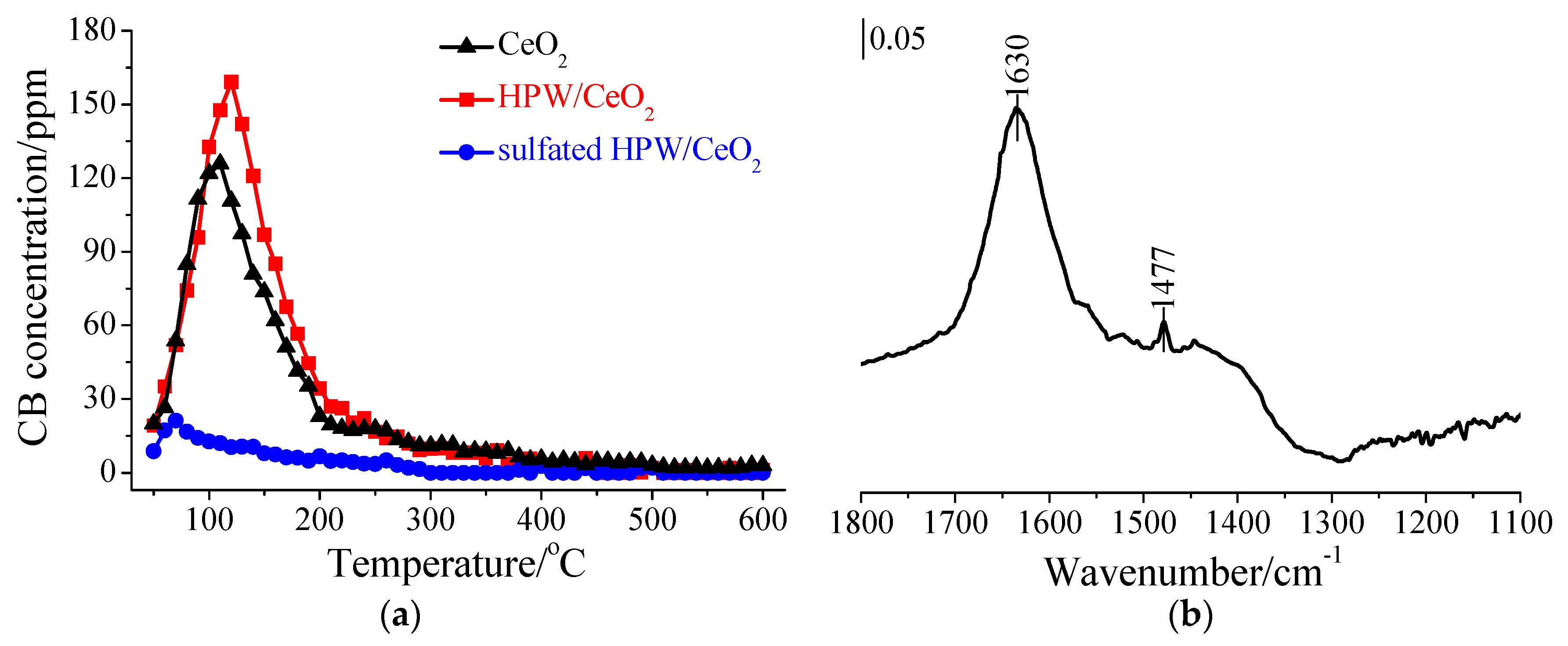
3.2. Influence of SO2 in the Flue Gas
3.3. Characterization
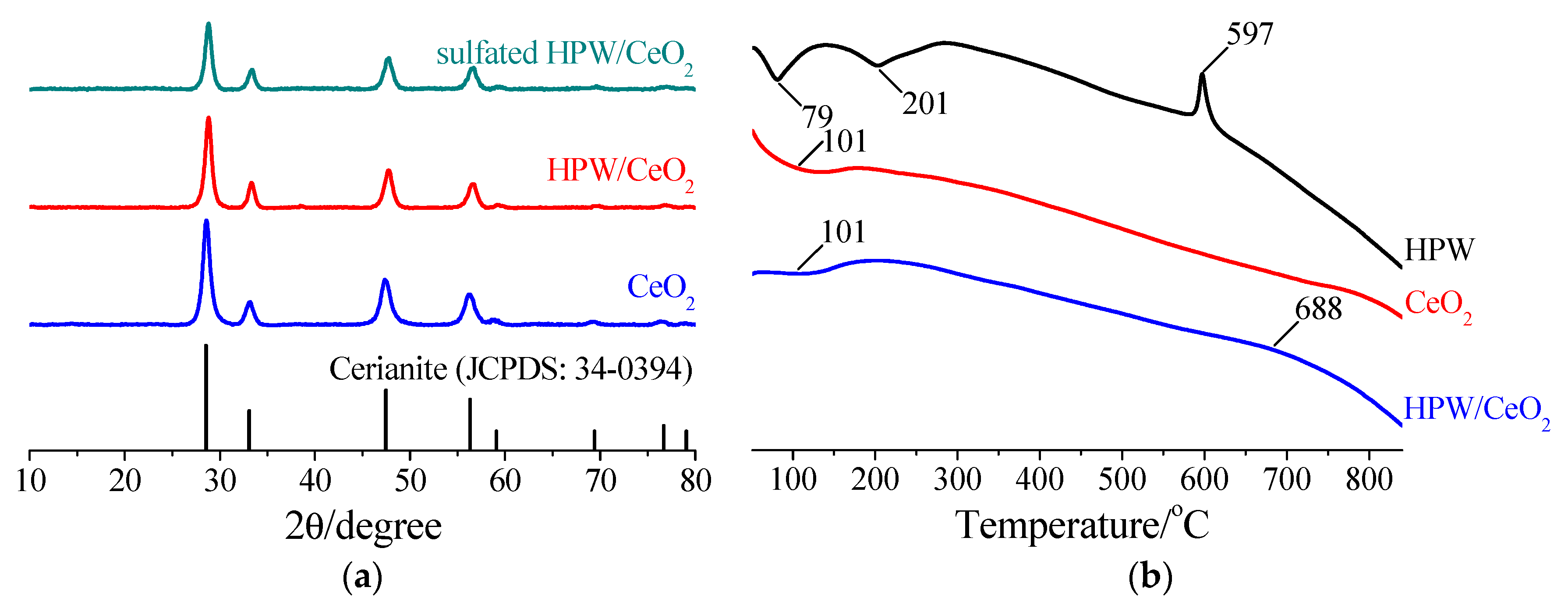
3.3.1. XRD and BET Surface Area
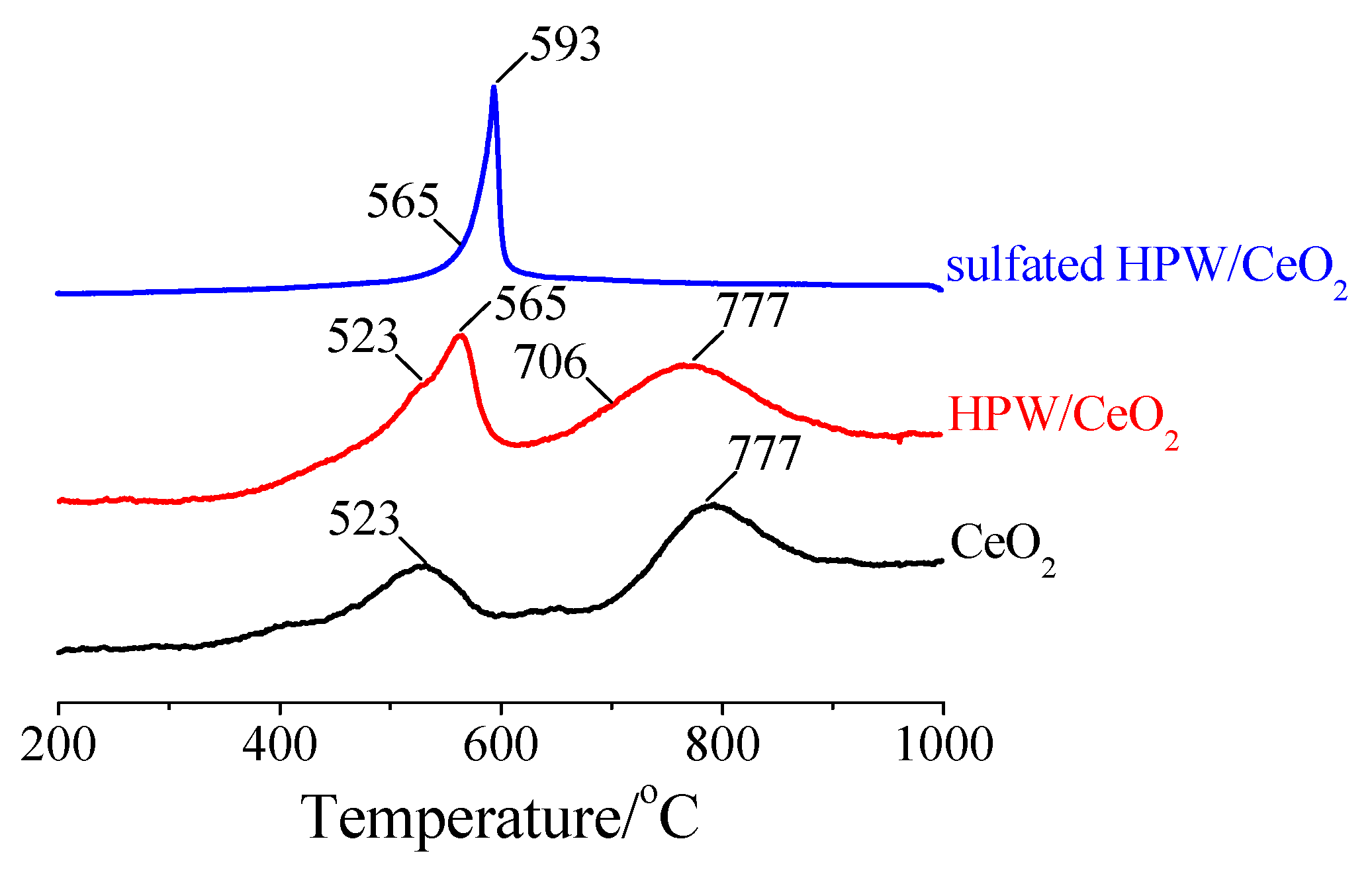
3.3.2. DTA
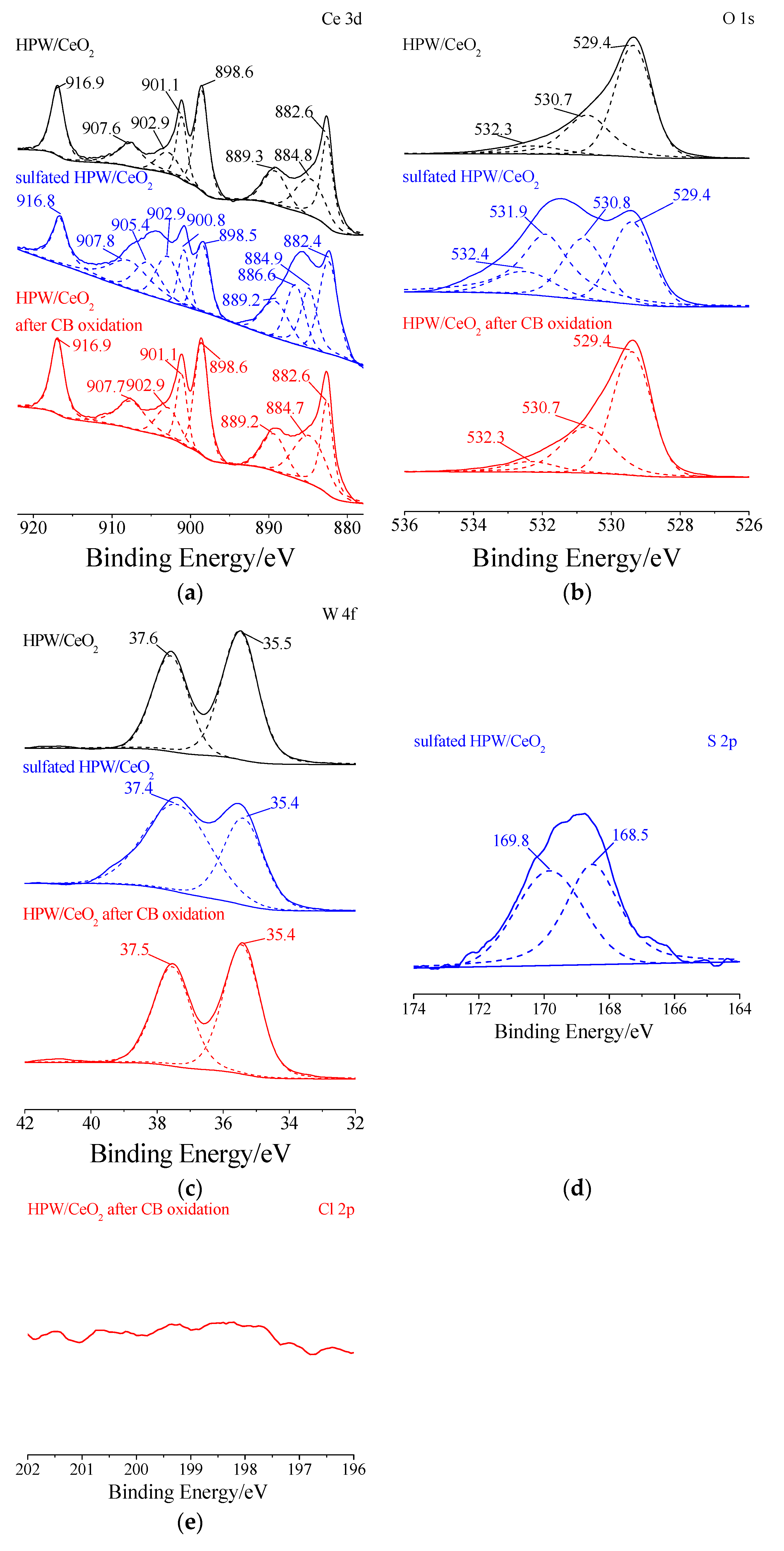
3.3.3. XPS
3.3.4. H2–TPR
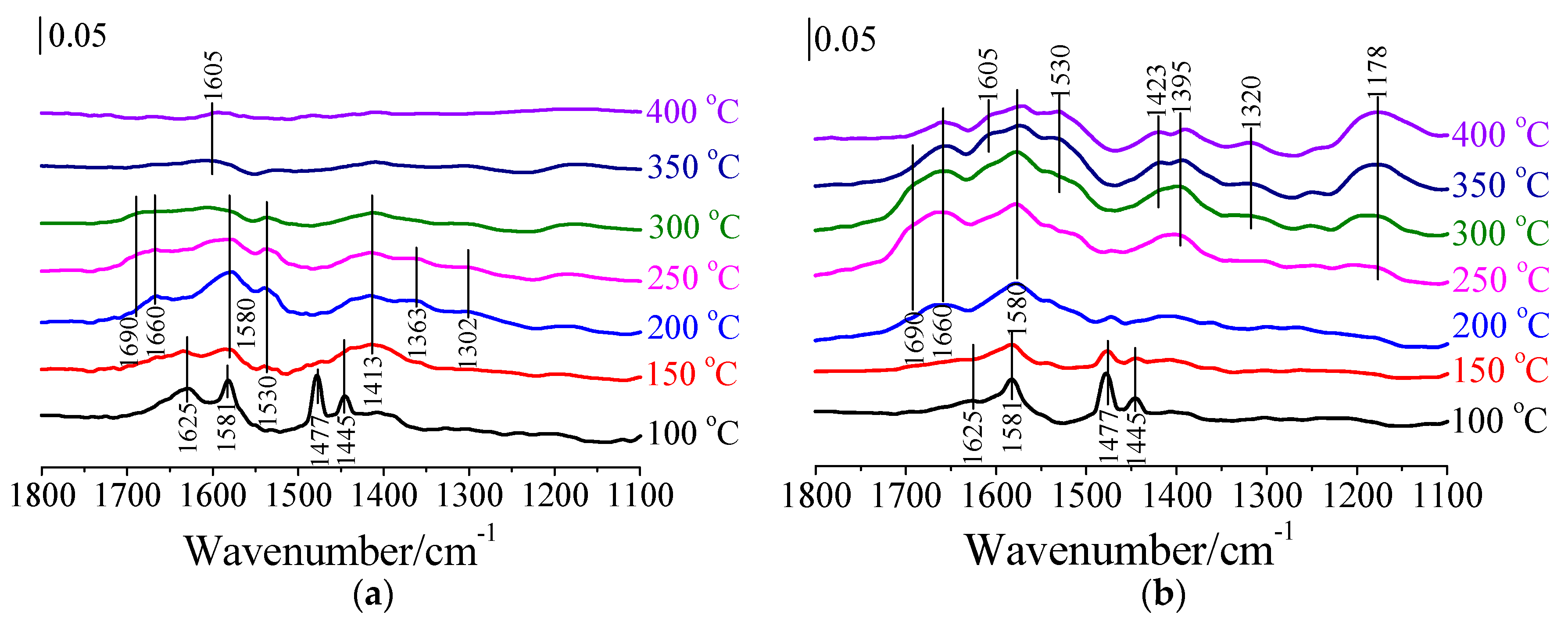
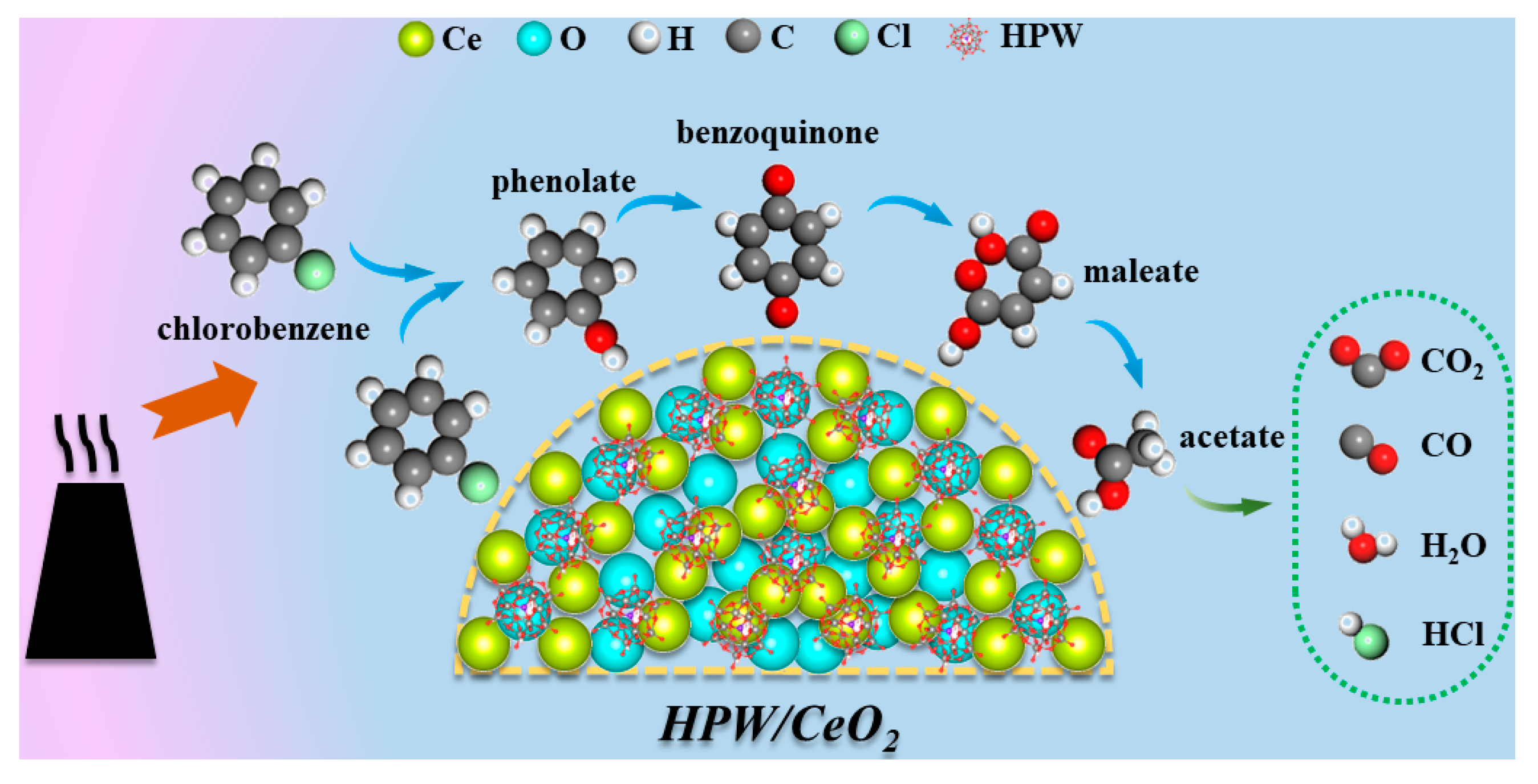
3.4. Mechanism of CB Oxidation
3.5. Synergistic Effect of HPW and CeO2 on CB Oxidation
3.6. Inhibition Mechanism of SO2 on CB Oxidation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Huang, B.B.; Lei, C.; Wei, C.H.; Zeng, G.M. Chlorinated volatile organic compounds (Cl-VOCs) in environment: Sources, potential human health impacts, and current remediation technologies. Environ. Int. 2014, 71, 118–138. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.W.; Zhang, Z.M.; Li, N.; Yan, B.B.; He, C.; Hao, Z.P.; Chen, G.Y. How to achieve complete elimination of Cl-VOCs: A critical review on byproducts formation and inhibition strategies during catalytic oxidation. Chem. Eng. J. 2021, 404, 126534. [Google Scholar] [CrossRef]
- Lin, F.W.; Xiang, L.; Zhang, Z.M.; Li, N.; Yan, B.B.; He, C.; Hao, Z.P.; Chen, G.Y. Comprehensive review on catalytic degradation of Cl-VOCs under the practical application conditions. Crit. Rev. Environ. Sci. Technol. 2022, 52, 311–355. [Google Scholar] [CrossRef]
- Dai, Q.G.; Shen, K.; Deng, W.; Cai, Y.P.; Yan, J.R.; Wu, J.Y.; Guo, L.M.; Liu, R.; Wang, X.Y.; Zhan, W.C. HCl-tolerant HxPO4/RuOx-CeO2 catalysts for extremely efficient catalytic elimination of chlorinated VOCs. Environ. Sci. Technol. 2021, 55, 4007–4016. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Cheng, J.; Zhang, X.; Douthwaite, M.; Pattisson, S.; Hao, Z.P. Recent advances in the catalytic oxidation of volatile organic compounds: A review based on pollutant sorts and sources. Chem. Rev. 2019, 119, 4471–4568. [Google Scholar] [CrossRef] [PubMed]
- Puértolas, B.; Smith, A.; Vázquez, I.; Dejoz, A.; Moragues, A.; Garcia, T.; Solsona, B. The different catalytic behaviour in the propane total oxidation of cobalt and manganese oxides prepared by a wet combustion procedure. Chem. Eng. J. 2013, 229, 547–558. [Google Scholar] [CrossRef]
- Li, W.B.; Wang, J.X.; Gong, H. Catalytic combustion of VOCs on non-noble metal catalysts. Catal. Today 2009, 148, 81–87. [Google Scholar] [CrossRef]
- Kan, J.W.; Deng, L.; Li, B.; Huang, Q.; Zhu, S.M.; Shen, S.B.; Chen, Y.W. Performance of co-doped Mn-Ce catalysts supported on cordierite for low concentration chlorobenzene oxidation. Appl. Catal. A Gen. 2017, 530, 21–29. [Google Scholar] [CrossRef]
- Chen, J.; Wang, C.Q.; Lv, X.L.; Huang, G.X.; Xu, W.J.; Li, X.L.; Jia, H.P. Pt/CeO2 coated with polyoxometallate chainmail to regulate oxidation of chlorobenzene without hazardous by-products. J. Hazard. Mater. 2023, 441, 129925. [Google Scholar] [CrossRef]
- Rachapudi, R.; Chintawar, P.S.; Greene, H.L. Aging and structure/activity characteristics of CR-ZSM-5 catalysts during exposure to chlorinated VOCs. J. Catal. 1999, 185, 58–72. [Google Scholar] [CrossRef]
- Bertinchamps, F.; Attianese, A.; Mestdagh, M.M.; Gaigneaux, E.M. Catalysts for chlorinated VOCs abatement: Multiple effects of water on the activity of VOx based catalysts for the combustion of chlorobenzene. Catal. Today 2006, 112, 165–168. [Google Scholar] [CrossRef]
- Geng, Y.; Xiong, S.C.; Li, B.; Liao, Y.; Xiao, X.; Yang, S.J. H3PW12O40 grafted on CeO2: A high-performance catalyst for the selective catalytic reduction of NOx with NH3. Ind. Eng. Chem. Res. 2018, 57, 856–866. [Google Scholar] [CrossRef]
- Wei, X.J.; Rang, X.D.; Zhu, W.H.; Xiang, M.; Deng, Y.Y.; Jiang, F.H.; Mao, R.; Zhang, Z.W.; Kong, X.Q.; Wang, F. Morphology effect of CeO2 on Ni/CeO2 catalysts for selective hydrogenation of cinnamaldehyde. Chem. Phys. 2021, 542, 111079. [Google Scholar] [CrossRef]
- Dai, Q.G.; Zhang, Z.Y.; Yan, J.R.; Wu, J.Y.; Johnson, G.; Sun, W.; Wang, X.Y.; Zhang, S.; Zhan, W.C. Phosphate-functionalized CeO2 nanosheets for efficient catalytic oxidation of dichloromethane. Environ. Sci. Technol. 2018, 52, 13430–13437. [Google Scholar] [CrossRef]
- Yang, P.; Yang, S.S.; Shi, Z.N.; Meng, Z.H.; Zhou, R.X. Deep oxidation of chlorinated VOCs over CeO2-based transition metal mixed oxide catalysts. Appl. Catal. B Environ. 2015, 162, 227–235. [Google Scholar] [CrossRef]
- Rivas, B.D.; Sampedro, C.; García-Real, M.; López-Fonseca, R.; Gutiérrez-Ortiz, J.I. Promoted activity of sulphated Ce/Zr mixed oxides for chlorinated VOC oxidative abatement. Appl. Catal. B Environ. 2013, 129, 225–235. [Google Scholar] [CrossRef]
- Jin, D.F.; Hou, Z.Y.; Luo, Y.M.; Zheng, X.M. Synthesis of di(methylphenyl)methane over heteropolyacids (H3PW12O40) catalysts. Catal. Lett. 2005, 102, 109–113. [Google Scholar] [CrossRef]
- Qiu, B.; Yi, X.D.; Lin, L.; Fang, W.P.; Wan, H.L. The hydrocracking of n-decane over bifunctional Ni-H3PW12O40/SiO2 catalysts. Catal. Today 2008, 131, 464–471. [Google Scholar] [CrossRef]
- Li, G.B.; Shen, K.; Wu, P.; Zhang, Y.P.; Hu, Y.Q.; Xiao, R.; Wang, B.; Zhang, S.L. SO2 poisoning mechanism of the multi-active center catalyst for chlorobenzene and NOx synergistic degradation at dry and humid environments. Environ. Sci. Technol. 2021, 55, 13186–13197. [Google Scholar]
- Gan, L.N.; Li, K.Z.; Yang, W.N.; Chen, J.J.; Peng, Y.; Li, J.H. Core-shell-like structured α-MnO2@CeO2 catalyst for selective catalytic reduction of NO: Promoted activity and SO2 tolerance. Chem. Eng. J. 2020, 391, 123473. [Google Scholar] [CrossRef]
- Mulik, N.; Bokade, V. Immobilization of HPW on UiO-66-NH2 MOF as efficient catalyst for synthesis of furfuryl ether and alkyl levulinate as biofuel. Mol. Catal. 2022, 531, 112689. [Google Scholar] [CrossRef]
- Li, B.S.; Liu, Z.X.; Liu, J.J.; Zhou, Z.Y.; Gao, X.H.; Pang, X.M.; Sheng, H.T. Preparation, characterization and application in deep catalytic ODS of the mesoporous silica pillared clay incorporated with phosphotungstic acid. J. Colloid Interface Sci. 2011, 362, 450–456. [Google Scholar] [CrossRef]
- Su, Z.; Li, X.; Si, W. Probing the actual role and activity of oxygen vacancies in toluene catalytic oxidation: Evidence from in situ XPS/NEXAFS and DFT+ U calculation. ACS Catal. 2023, 13, 3444–3455. [Google Scholar] [CrossRef]
- Su, Z.A.; Si, W.Z.; Liu, H.; Xiong, S.C.; Chu, X.F.; Yang, W.H.; Peng, Y.; Chen, J.J.; Cao, X.Z.; Li, J.H. Boosting the catalytic performance of CeO2 in toluene combustion via the Ce-Ce homogeneous interface. Environ. Sci. Technol. 2021, 55, 12630–12639. [Google Scholar] [CrossRef]
- Geng, Y.; Jin, K.; Mei, J.; Su, G.Y.; Ma, L.; Yang, S.J. CeO2 grafted with different heteropoly acids for selective catalytic reduction of NOx with NH3. J. Hazard. Mater. 2020, 382, 121032. [Google Scholar] [CrossRef]
- Yan, H.; Yao, S.; Wang, J.Y.; Zhao, S.M.; Sun, Y.H.; Liu, M.Y.; Zhou, X.; Zhang, G.Y.; Jin, X.; Feng, X.; et al. Engineering Pt-Mn2O3 interface to boost selective oxidation of ethylene glycol to glycolic acid. Appl. Catal. B Environ. 2021, 284, 119803. [Google Scholar] [CrossRef]
- Freitas, E.F.; Araújo, A.A.L.; Paiva, M.F.; Dias, S.C.L.; Dias, J.A. Comparative acidity of BEA and Y zeolite composites with 12-tungstophosphoric and 12-tungstosilicic acids. Mol. Catal. 2018, 458, 152–160. [Google Scholar] [CrossRef]
- Costa, N.L.D.; Pereira, L.G.; Resende, J.V.M.; Mendoza, C.A.D.; Ferreira, K.K.; Detoni, C.; Souza, M.; Gomes, F. Phosphotungstic acid on activated carbon: A remarkable catalyst for 5-hydroxymethylfurfural production. Mol. Catal. 2021, 500, 111334. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Jirsak, T.; Freitag, A.; Hanson, J.C.; Larese, J.Z.; Chaturvedi, S. Interaction of SO2 with CeO2 and Cu/CeO2 catalysts: Photoemission, XANES and TPD studies. Catal. Lett. 1999, 62, 113–119. [Google Scholar] [CrossRef]
- Wang, S.; Meng, X.; Liu, N.; Shi, L. Effective catalyst for removing trace olefins from reforming aromatics: Sulfated zirconia with high sulfur content synthesized by sulfonation with chlorosulfonic acid. Ind. Eng. Chem. Res. 2022, 61, 5103–5116. [Google Scholar] [CrossRef]
- Luo, M.F.; Zhong, Y.J.; Yuan, X.X.; Zheng, X.M. TPR and TPD studies of CuO/CeO2 catalysts for low temperature CO oxidation. Appl. Catal. A Gen. 1997, 162, 121–131. [Google Scholar] [CrossRef]
- Yang, S.J.; Guo, Y.F.; Chang, H.Z.; Ma, L.; Peng, Y.; Qu, Z.; Yan, N.Q.; Wang, C.Z.; Li, J.H. Novel effect of SO2 on the SCR reaction over CeO2: Mechanism and significance. Appl. Catal. B Environ. 2013, 136, 19–28. [Google Scholar] [CrossRef]
- Sun, Y.K.; Xu, S.; Bai, B.; Li, L.; Kang, Y.; Hu, X.Q.; Liao, Z.H.; He, C. Biotemplate fabrication of hollow tubular CexSr1-xTiO3 with regulable surface acidity and oxygen mobility for efficient destruction of chlorobenzene: Intrinsic synergy effect and reaction mechanism. Environ. Sci. Technol. 2022, 56, 5796–5807. [Google Scholar] [CrossRef]
- Su, Y.; Fu, K.X.; Pang, C.H.; Zheng, Y.F.; Song, C.F.; Ji, N.; Ma, D.G.; Lu, X.B.; Liu, C.X.; Han, R.; et al. Recent advances of chlorinated volatile organic compounds’ oxidation catalyzed by multiple catalysts: Reasonable adjustment of acidity and redox properties. Environ. Sci. Technol. 2022, 56, 9854–9871. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Gu, Y.F.; Zhao, J.; Wang, X.Y. Catalytic combustion of chlorobenzene over VOx/CeO2 catalysts. J. Catal. 2015, 326, 54–68. [Google Scholar] [CrossRef]
- Weng, X.L.; Sun, P.F.; Long, Y.; Meng, Q.J.; Wu, Z.B. Catalytic oxidation of chlorobenzene over MnxCe1-xO2/HZSM-5 catalysts: A study with practical implications. Environ. Sci. Technol. 2017, 51, 8057–8066. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, W.C.; Guan, Y.J.; Ying, P.L.; Li, C. FT-IR spectroscopic study of the oxidation of chlorobenzene over Mn-based catalyst. Langmuir 2002, 18, 6229–6232. [Google Scholar] [CrossRef]
- Wang, J.; Wang, X.; Liu, X.L.; Zhu, T.Y.; Guo, Y.Y.; Qi, H. Catalytic oxidation of chlorinated benzenes over V2O5/TiO2 catalysts: The effects of chlorine substituents. Cataly. Today 2015, 241, 92–99. [Google Scholar] [CrossRef]
- Lomnicki, S.; Lichtenberger, J.; Xu, Z.T.; Waters, M.; Kosman, J.; Amiridis, M.D. Catalytic oxidation of 2,4,6-trichlorophenol over vanadia/titania-based catalysts. Appl. Catal. B Environ. 2003, 46, 105–119. [Google Scholar] [CrossRef]
- Liu, X.L.; Zeng, J.L.; Shi, W.B.; Wang, J.; Zhu, T.Y.; Chen, Y.F. Catalytic oxidation of benzene over ruthenium-cobalt bimetallic catalysts and study of its mechanism. Catal. Sci. Technol. 2017, 7, 213–221. [Google Scholar] [CrossRef]
- Hauchecorne, B.; Terrens, D.; Verbruggen, S.; Martens, J.A.; Van Langenhove, H.; Demeestere, K.; Lenaerts, S. Elucidating the photocatalytic degradation pathway of acetaldehyde: An FTIR in situ study under atmospheric conditions. Appl. Catal. B Environ. 2011, 106, 630–638. [Google Scholar] [CrossRef]
- Zhang, K.Y.; Dai, L.Y.; Liu, Y.X.; Deng, J.G.; Jing, L.; Zhang, K.F.; Hou, Z.Q.; Zhang, X.; Wang, J.; Feng, Y.; et al. Insights into the active sites of chlorine-resistant Pt-based bimetallic catalysts for benzene oxidation. Appl. Catal. B Environ. 2020, 279, 119372. [Google Scholar] [CrossRef]
- Hu, Z.Y.; Chen, J.; Yan, D.X.; Li, Y.; Jia, H.P.; Lu, C.Z. Enhanced catalytic activities of MnOx/Co3O4 nanocomposites prepared via MOFs-templated approach for chlorobenzene oxidation. Appl. Surf. Sci. 2021, 551, 149453. [Google Scholar] [CrossRef]
- Lichtenberger, J.; Amiridis, M.D. Catalytic oxidation of chlorinated benzenes over V2O5/TiO2 catalysts. J. Catal. 2004, 223, 296–308. [Google Scholar] [CrossRef]
- Liu, X.L.; Chen, L.; Zhu, T.Y.; Ning, R.L. Catalytic oxidation of chlorobenzene over noble metals (Pd, Pt, Ru, Rh) and the distributions of polychlorinated by-products. J. Hazard. Mater. 2019, 363, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Dong, F.; Han, W.G.; Zhang, J.Y.; Tang, Z.C. Significant enhanced SO2 resistance of Pt/SiO2 catalysts by building the ultrathin metal oxide shell for benzene catalytic combustion. ACS Appl. Mater. Interfaces 2023, 15, 42541–42556. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.J.; Peng, Y.; Zhao, X.G.; Liu, H.; Gao, C.; Si, W.Z.; Li, J.H. Roles of Ru on the V2O5-WO3/TiO2 catalyst for the simultaneous purification of NOx and chlorobenzene: A dechlorination promoter and a redox inductor. ACS Catal. 2022, 12, 11505–11517. [Google Scholar] [CrossRef]
- Huang, X.; Liu, Z.; Wang, D.; Peng, Y.; Li, J.H. The effect of additives and intermediates on vanadia-based catalyst for multi-pollutant control. Catal. Sci. Technol. 2020, 10, 323–326. [Google Scholar] [CrossRef]
- Yu, S.X.; Niu, X.W.; Song, Z.J.; Huang, X.; Peng, Y.; Li, J.H. Improvement of Al2O3 on the multi-pollutant control performance of NOx and chlorobenzene in vanadia-based catalysts. Chemosphere 2022, 289, 133156. [Google Scholar] [CrossRef] [PubMed]
- Hua, Z.S.; Song, H.; Zhou, C.; Xin, Q.; Zhou, F.Y.; Fan, W.T.; Liu, S.J.; Zhang, X.; Zheng, C.H.; Yang, Y.; et al. A promising catalyst for catalytic oxidation of chlorobenzene and slipped ammonia in SCR exhaust gas: Investigating the simultaneous removal mechanism. Chem. Eng. J. 2023, 473, 145106. [Google Scholar] [CrossRef]
- Zhu, X.; Yuan, X.; Song, Z.J.; Peng, Y.; Li, J.H. A dual-balance strategy via phosphate modification on MnO2-CeO2 for NOx and chlorobenzene synergistic catalytic control. Appl. Catal. B Environ. Energy 2024, 342, 123364. [Google Scholar] [CrossRef]
- Yan, X.; Zhao, L.K.; Huang, Y.; Zhang, J.F.; Jiang, S. Three-dimensional porous CuO-modified CeO2-Al2O3 catalysts with chlorine resistance for simultaneous catalytic oxidation of chlorobenzene and mercury: Cu-Ce interaction and structure. J. Hazard. Mater. 2023, 455, 131585. [Google Scholar] [CrossRef] [PubMed]
- Yin, R.Q.; Chen, J.J.; Mi, J.X.; Liu, H.Y.; Yan, T.; Shan, L.; Lang, J.Y.; Li, J.H. Breaking the activity-selectivity trade-off for simultaneous catalytic elimination of nitric oxide and chlorobenzene via FeVO4-Fe2O3 interfacial charge transfer. ACS Catal. 2022, 12, 3797–3806. [Google Scholar] [CrossRef]
- Song, Z.J.; Yu, S.X.; Liu, H.; Wang, Y.; Gao, C.Y.; Wang, Z.S.; Qin, Y.M.; Peng, Y.; Li, J.H. Carbon/chlorinate deposition on MnOx-CeO2 catalyst in chlorobenzene combustion: The effect of SCR flue gas. Chem. Eng. J. 2022, 433, 133552. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Temperature/°C | /μmol g−1 min−1 | |||
|---|---|---|---|---|
| kE–R | kMvK | R2 | ||
| HPW/CeO2 | 250 | 0.00939 | 1.71 | 0.999 |
| 300 | 0.0347 | 2.81 | 0.999 | |
| 350 | 0.0961 | 3.56 | 0.998 | |
| 400 | 0.157 | 4.91 | 0.996 | |
| 450 | 0.213 | 4.16 | 0.999 | |
| Ce Species | O Species | ||||||
|---|---|---|---|---|---|---|---|
| Ce3+–O | Ce4+–O | Ce4+–SO42− | Olat | Oad | HPW | SO42− | |
| HPW/CeO2 | 4.0 | 16.8 | - | 46.1 | 24.2 | 0.13 | - |
| sulfated HPW/CeO2 | 4.4 | 7.9 | 3.1 | 21.0 | 17.1 | 0.22 | 7.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, K.; Dong, L.; Shen, Q.; Wu, W.; Wu, X.; Mei, J.; Yang, S. Chlorobenzene Oxidation over Phosphotungstic-Acid-Coated Cerium Oxide: Synergistic Effect of Phosphotungstic and Cerium Oxide and Inhibition Mechanism of Sulfur Dioxide. Sustainability 2024, 16, 2245. https://doi.org/10.3390/su16062245
Jiang K, Dong L, Shen Q, Wu W, Wu X, Mei J, Yang S. Chlorobenzene Oxidation over Phosphotungstic-Acid-Coated Cerium Oxide: Synergistic Effect of Phosphotungstic and Cerium Oxide and Inhibition Mechanism of Sulfur Dioxide. Sustainability. 2024; 16(6):2245. https://doi.org/10.3390/su16062245
Chicago/Turabian StyleJiang, Keyu, Leyuan Dong, Qi Shen, Wei Wu, Xue Wu, Jian Mei, and Shijian Yang. 2024. "Chlorobenzene Oxidation over Phosphotungstic-Acid-Coated Cerium Oxide: Synergistic Effect of Phosphotungstic and Cerium Oxide and Inhibition Mechanism of Sulfur Dioxide" Sustainability 16, no. 6: 2245. https://doi.org/10.3390/su16062245