
A Case Report of Hyperhemolytic Syndrome in Sickle Cell Disease, with a Special Focus on Avoiding the Use of Transfusions
 , ,
, ,
Abstract
:1. Introduction
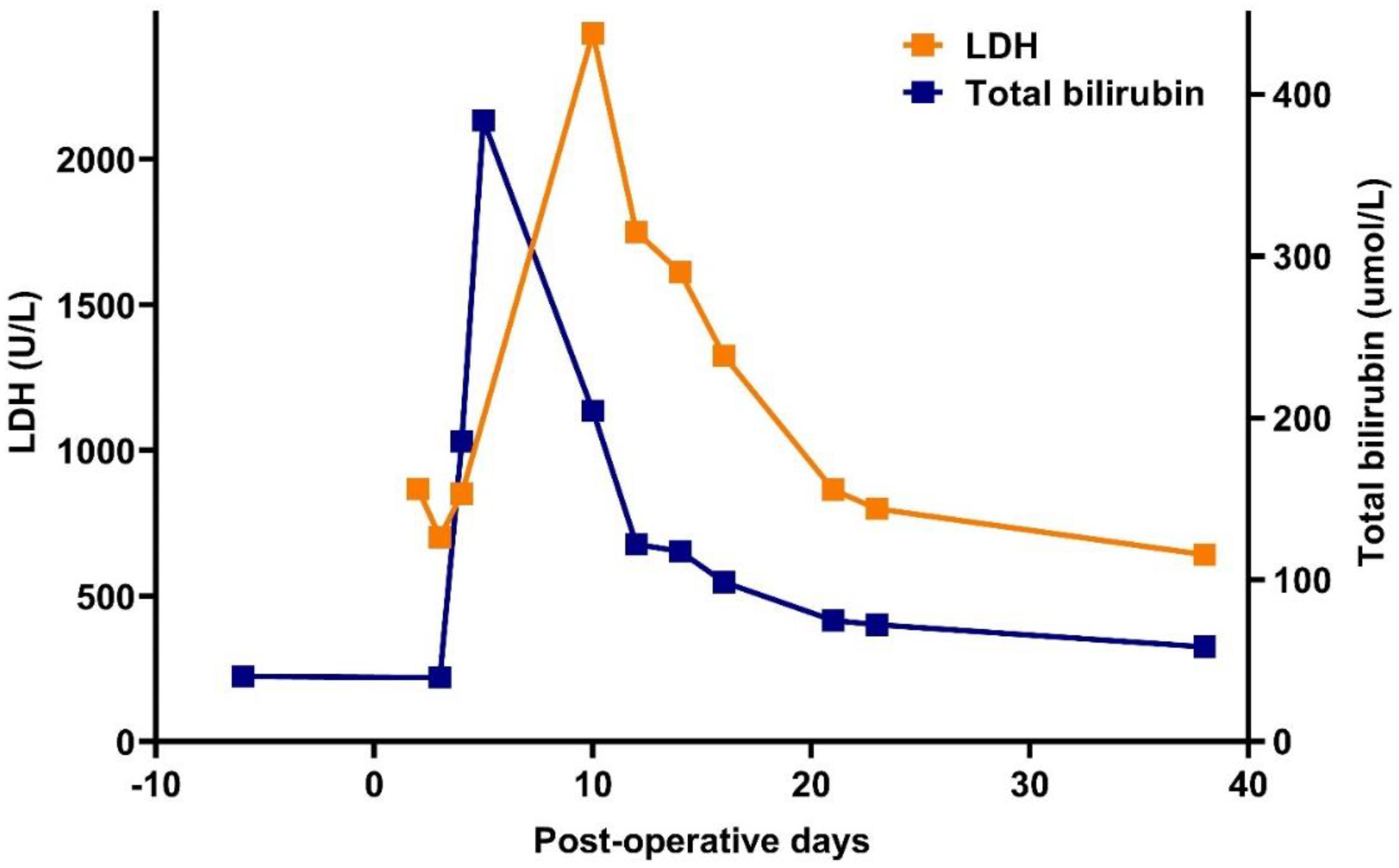
2. Case Report
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sundd, P.; Gladwin, M.T.; Novelli, E.M. Pathophysiology of sickle cell disease. Annu. Rev. Pathol. 2019, 14, 263–292. [Google Scholar] [CrossRef] [PubMed]
- Sparkenbaugh, E.; Pawlinski, R. Interplay between coagulation and vascular inflammation in sickle cell disease. Br. J. Hematol. 2013, 162, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Elendu, C.; Amaechi, D.C.; Alakwe-Ojimba, C.E.; Elendu, T.C.; Elendu, R.C.; Ayabazu, C.P.; Aina, T.O.; Aborisade, O.; Adenikinju, J.S. Understanding Sickle cell disease: Causes, symptoms, and treatment options. Medicine 2023, 102, e35237. [Google Scholar] [CrossRef] [PubMed]
- Rees, D.C.; Brousse, V.A.M.; Brewin, J.N. Determinants of severity in sickle cell disease. Blood Rev. 2022, 56, 100983. [Google Scholar] [CrossRef]
- Rees, D.C.; Robinson, S.; Howard, J. How I manage red cell transfusions in patients with sickle cell disease. Br. J. Haematol. 2018, 180, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Pirenne, F.; Yazdanbakhsh, K. How I safely transfuse patients with sickle-cell disease and manage delayed hemolytic transfusion reactions. Blood 2018, 131, 2773–2781. [Google Scholar] [CrossRef] [PubMed]
- Kelly, S.; Deng, X.; Hopper, C.; Style, L. A pilot randomized trial of red blood cell transfusion for acute treatment of vaso-occlusive pain episodes in sickle cell disease. Br. J. Haematol. 2015, 171, 288–290. [Google Scholar] [CrossRef]
- Adams Rj McKie, V.C.; Hsu, L.; Files, B.; Vichinsky, E.; Pegelow, C.; Abboud, M.; Gallagher, D.; Kutlar, A.; Nichols, F.T.; Bonds, D.R.; et al. Prevention of first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography. N. Engl. J. Med. 1998, 339, 5–11. [Google Scholar] [CrossRef]
- Ware, R.E.; Davis, B.R.; Schultz, W.H.; Brown, R.C.; Aygun, B.; Sarnaik, S.; Odame, I.; Fuh, B.; George, A.; Owen, W.; et al. Hydroxycarbamide versus chronic transfusion for maintenance of transcranial doppler flow velocities in children with sickle cell anemia (TWiTCH): A multicentric, open-label, phase 3, non-inferiority trial. Lancet 2016, 387, 661–670. [Google Scholar] [CrossRef]
- Narbey, D.; Habibi, A.; Chadebech, P.; Mekontso-Dessap, A.; Khellaf, M.; Lelièvre, J.D.; Godeau, B.; Michel, M.; Galactéros, F.; Djoudi, R.; et al. Incidence and predictive score for delayed hemolytic transfusion reaction in adult patients with sickle cell disease. Am. J. Hematol. 2017, 92, 1340–1348. [Google Scholar] [CrossRef]
- Pirenne, F.; Pondarré, C. Alloimmunization and hyperhemolysis in sickle cell disease. Hematology 2023, 2023, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Fasano, R.M.; Miller, M.J.; Chonat, S.; Stowell, S.R. Clinical presentation of delayed hemolytic transfusion reactions and hyperhemolysis in sickle cell disease. Transfus. Clin. Biol. 2019, 26, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Banks, M.; Shikle, J. Hyperhemolysis syndrome in patients with sickle cell disease. Arch. Pathol. Lab. Med. 2017, 142, 1425–1427. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.W.; Stephens, L.D.; Allen, E.S.; Binns, T.C.; Booth, G.S.; Hendrickson, J.E.; Karafin, M.S.; Tormey, C.A.; Woo, J.S.; Adkins, B.D. Epidemiological and clinical features, therapeutic strategies and outcomes in patients with hyperhaemolysis: A systematic review. Br. J. Haematol. 2023, 201, 1025–1032. [Google Scholar] [CrossRef]
- Shankar, K.; Shah, D.; Huffman, D.L.; Peterson, C.; Bhagavatula, R. Hyperhemolysis Syndrome in a Patient With Sickle Cell Disease and Acute Chest Syndrome. Cureus 2021, 13, e13017. [Google Scholar] [CrossRef]
- Win, N.; New, H.; Lee, E.; de la Fuente, J. Hyperhemolysis syndrome in sickle cell disease: A case report (recurrent episode) and literature review. Transfusion 2008, 48, 1231–1238. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, K.; Abbas, A.; Kazmi, R.; Shaaban, H.; Miller, R. Hyperhemolytic crisis following transfusion in sickle cell disease with acute hepatic crisis: A case report. Cureus 2022, 14, e27844. [Google Scholar] [CrossRef] [PubMed]
- Kalter, J.A.; Gupta, R.; Greenberg, M.R.; Miller, A.J.; Allen, J. Hyperhemolysis Syndrome in a Patient with Sickle Cell Disease: A Case Report. Clin. Pract. Cases Emerg. Med. 2021, 5, 101–104. [Google Scholar] [CrossRef]
- Win, N. Hyperhemolysis syndrome in sickle cell disease. Expert Rev. Hematol. 2009, 2, 111–115. [Google Scholar] [CrossRef]
- Roumenina, L.T.; Bartolucci, P.; Pirenne, F. The role of Complement in Post-Transfusion Hemolysis and Hyperhemolysis Reaction. Transfus. Med. Rev. 2019, 33, 225–230. [Google Scholar] [CrossRef]
- Madu, A.J.; Ugwu, A.O.; Efobi, C. Hyperhemolytic Syndrome in Sickle Cell Disease: Clearing the Cobwebs. Med. Princ. Pract. 2021, 30, 236–243. [Google Scholar] [CrossRef]
- Menakuru, S.R.; Priscu, A.; Dhillon, V.; Salih, A. Acute Hyperhemolysis Syndrome in a Patient with Known Sickle Cell Anemia Refractory to Steroids and IVIG Treated with Tocilizumab and Erythropoietin: A Case Report and Review of Literature. Hematol. Rep. 2022, 14, 235–239. [Google Scholar] [CrossRef]
- Dumas, G.; Habibi, A.; Onimus, T.; Merle, J.C.; Razazi, K.; Mekontso Dessap, A.; Galactéros, F.; Michel, M.; Frémeaux Bacchi, V.; Noizat Pirenne, F.; et al. Eculizumab salvage therapy for delayed hemolysis transfusion reaction in sickle cell disease patients. Blood 2016, 127, 1062–1064. [Google Scholar] [CrossRef]
- Vichinsky, E.; Hoppe, C.C.; Ataga, K.I.; Ware, R.E.; Nduba, V.; El-Beshlawy, A.; Hassab, H.; Achebe, M.M.; Alkindi, S.; Brown, R.C.; et al. A Phase 3 Randomized Trial of Voxelotor in Sickle Cell Disease. N. Engl. J. Med. 2019, 381, 509–519. [Google Scholar] [CrossRef]
- Kuriri, F.A. Hope on the Horizon: New and Future Therapies for sickle cell disease. J. Clin. Med. 2023, 12, 5692. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.T.; Alsawas, M.; Fasano, R.M.; Field, J.J.; Hendrickson, J.E.; Howard, J.; Kameka, M.; Kwiatkowski, J.L.; Pirenne, F.; Shi, P.A.; et al. American Society of Hematology 2020 guidelines for sickle cell disease: Transfusion support. Blood Adv. 2020, 4, 327–355. [Google Scholar] [CrossRef]
- Ballas, S.K.; Kuypers, F.A.; Gordeuk, V.R.; Hankins, J.S.; Thompson, A.A.; Vichinsky, E. Time to rethink haemoglobin threshold guidelines in sickle cell disease. Br. J. Haematol. 2021, 195, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Shaulov, A.; Rund, D.; Filon, D.; Nachmias, B.; Khalili, A.; Manny, N.; Zelig, O. Successful treatment with plasma exchange in life-threatening hyperhemolytic syndrome unrelated to sickle cell disease. Transfusion 2023, 63, 1100–1106. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Days | Baseline | P+ 1 | P+ 3 | P+ 9 | P+ 11 | P+ 16 | P+ 24 | P+ 38 |
|---|---|---|---|---|---|---|---|---|
| Hgb (g/L) | 97 | 65 | 52 | 39 | 35 | 67 | 77 | 81 |
| LDH (U/L) | 300 | 867 | 851 | 4221 | 2432 | 1329 | 799 | 642 |
| tBi (umol/L) | 39.8 | NA | 185.7 | 437.3 | 204 | 98.5 | 72.3 | 58.5 |
| diBi (umol/L) | 13.5 | NA | 86.1 | 249.8 | 170.9 | 82 | 60.1 | 41.3 |
| CRP (mg/L) | 4.4 | NA | NA | 168 | 104 | 52 | 5.98 | |
| Haptoglobin (g/L) | NA | NA | NA | <0.1 | <0.1 | <0.1 | <0.1 | <0.1 |
| Reticulocytes (G/L) | NA | NA | 154 | 247 | NA | 50 | 49 | 155 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Obajed Al-Ali, O.; Pfliegler, G.; Magyari, F.; Borics, F.; Pinczés, L.I.; Illés, Á.; Brúgós, B. A Case Report of Hyperhemolytic Syndrome in Sickle Cell Disease, with a Special Focus on Avoiding the Use of Transfusions. Thalass. Rep. 2024, 14, 18-25. https://doi.org/10.3390/thalassrep14010003
Obajed Al-Ali O, Pfliegler G, Magyari F, Borics F, Pinczés LI, Illés Á, Brúgós B. A Case Report of Hyperhemolytic Syndrome in Sickle Cell Disease, with a Special Focus on Avoiding the Use of Transfusions. Thalassemia Reports. 2024; 14(1):18-25. https://doi.org/10.3390/thalassrep14010003
Chicago/Turabian StyleObajed Al-Ali, Omar, György Pfliegler, Ferenc Magyari, Fanni Borics, László Imre Pinczés, Árpád Illés, and Boglárka Brúgós. 2024. "A Case Report of Hyperhemolytic Syndrome in Sickle Cell Disease, with a Special Focus on Avoiding the Use of Transfusions" Thalassemia Reports 14, no. 1: 18-25. https://doi.org/10.3390/thalassrep14010003