
Understanding the Intricacies of Iron Overload Associated with β-Thalassemia: A Comprehensive Review
 and
and
Abstract
:1. Introduction
2. Physiological Iron Metabolism and Its Regulation
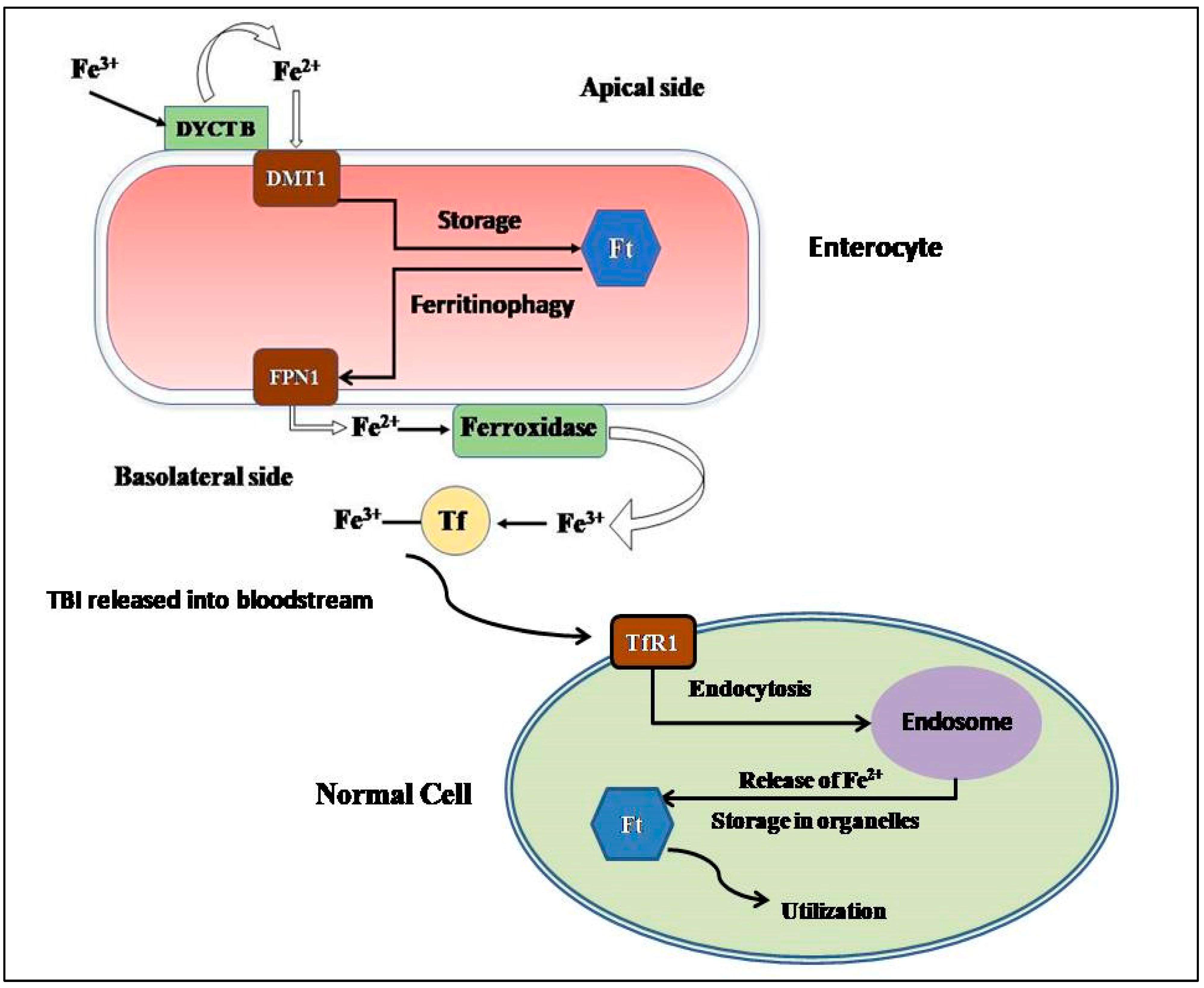
2.1. Absorption and Cellular Uptake
2.2. Storage
2.3. Consumption and Recycling
2.4. Regulation of Iron Metabolism
3. Iron Overload in Beta-Thalassemia
3.1. TDT vs. NTDT
3.2. Differential Expression of Different Proteins in Iron Overload Conditions
3.3. Detection of Iron Overload
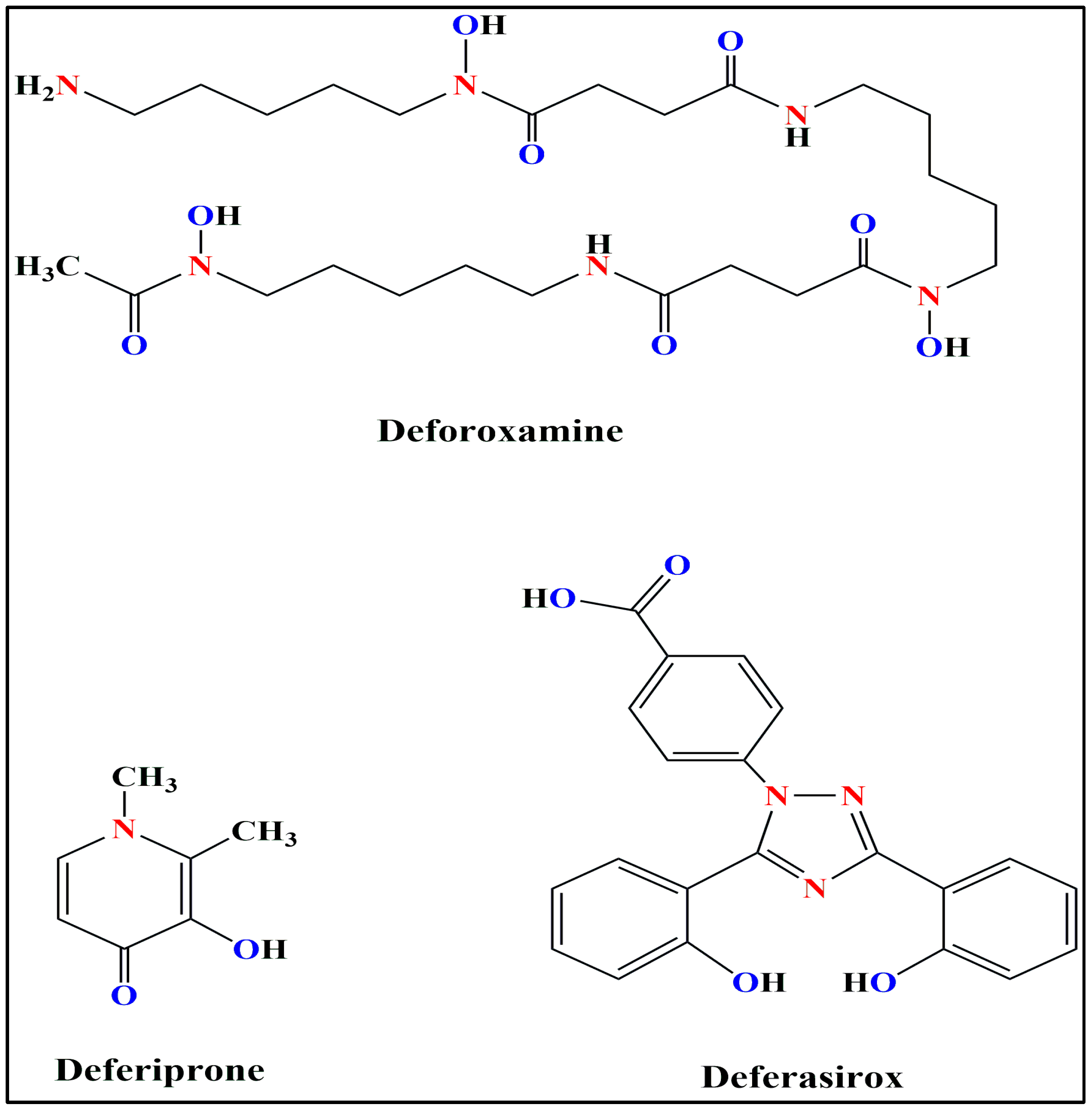
4. Iron Chelators
5. Guidelines for Usage of Iron Chelators in India
- DFO: continuous subcutaneous injection over 8–12 h or more with the help of an infusion pump, dispersed in water; dosage, 25–50 mg/kg/day;
- DFP: orally, in 2–3 divided dosages; dosage, 50–100 mg/kg/day;
- DFX: orally, dispersed in water or juice; dosage, 20–40 mg/kg/day;
- Combination therapy: when patients no longer respond to monotherapies, it is advisable to shift to combined regimens of DFX and DFO [125].
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marengo-Rowe, A.J. The thalassemias and related disorders. Bayl. Univ. Med. Cent. Proc. 2007, 20, 27–31. [Google Scholar] [CrossRef] [Green Version]
- Modell, B. Global epidemiology of haemoglobin disorders and derived service indicators. Bull. World Health Organ. 2008, 86, 480–487. [Google Scholar] [CrossRef]
- Galanello, R.; Origa, R. Beta-thalassemia. Genet. Med. 2010, 5, 11. [Google Scholar] [CrossRef] [Green Version]
- Giardine, B.; Borg, J.; Higgs, D.R.; Peterson, K.R.; Philipsen, S.; Maglott, D.; Singleton, B.K.; Anstee, D.J.; Basak, A.N.; Clark, B.; et al. Systematic documentation and analysis of human genetic variation in hemoglobinopathies using the microattribution approach. Nat. Genet. 2011, 43, 295–301. [Google Scholar] [CrossRef]
- Origa, R. β-Thalassemia. Genet. Med. 2017, 19, 609–619. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, R.; Prakash, A.; Aggarwal, M. Thalassemia: An overview. J. Sci. Soc. 2014, 41, 3–6. [Google Scholar] [CrossRef]
- Ministry of Health and Family Welfare, Government of India. National Health Mission—Guidelines on Hemoglobinopathies in India: Prevention and Control of Hemoglobinopathies in India; Ministry of Health and Family Welfare, Government of India: New Delhi, India, 2016; pp. 1–138. [Google Scholar]
- Thein, S.L. Pathophysiology of beta thalassemia—A guide to molecular therapies. Hematol. Am. Soc. Hematol. Educ. Program 2005, 2005, 31–37. [Google Scholar] [CrossRef]
- Bou-Fakhredin, R.; De Franceschi, L.; Motta, I.; Cappellini, M.D.; Taher, A.T. Pharmacological Induction of Fetal Hemoglobin in β-Thalassemia and Sickle Cell Disease: An Updated Perspective. Pharmaceuticals 2022, 15, 753. [Google Scholar] [CrossRef]
- Rivella, S. β-thalassemias: Paradigmatic diseases for scientific discoveries and development of innovative therapies. Haematologica 2015, 100, 418–430. [Google Scholar] [CrossRef] [Green Version]
- Musallam, K.M.; Rivella, S.; Vichinsky, E.; Rachmilewitz, E.A. Non-transfusion-dependent thalassemias. Haematologica 2013, 98, 833–844. [Google Scholar] [CrossRef] [Green Version]
- Beard, J.; Han, O. Systemic iron status. Biochim. Biophys. Acta 2008, 1790, 584–588. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yu, C.; Kang, R.; Tang, D. Iron Metabolism in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 590226. [Google Scholar] [CrossRef]
- Frey, P.A.; Reed, G.H. The ubiquity of iron. ACS Chem. Biol. 2012, 7, 1477–1481. [Google Scholar] [CrossRef] [PubMed]
- Mancardi, D.; Mezzanotte, M.; Arrigo, E.; Barinotti, A.; Roetto, A. Iron Overload, Oxidative Stress, and Ferroptosis in the Failing Heart and Liver. Antioxidants 2021, 10, 1864. [Google Scholar] [CrossRef]
- Lawen, A.; Lane, D.J.R. Mammalian iron homeostasis in health and disease: Uptake, storage, transport, and molecular mechanisms of action. Antioxid. Redox Signal. 2013, 18, 2473–2507. [Google Scholar] [CrossRef] [PubMed]
- Malcovati, L. Impact of transfusion dependency and secondary iron overload on the survival of patients with myelodysplastic syndromes. Leuk. Res. 2007, 31, S2–S6. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Musallam, K.M.; Taher, A.T.; Rivella, S. Ineffective Erythropoiesis: Anemia and Iron Overload. Hematol. Oncol. Clin. N. Am. 2018, 32, 213–221. [Google Scholar] [CrossRef]
- Oikonomidou, P.R.; Rivella, S. What can we learn from ineffective erythropoiesis in thalassemia? Blood Rev. 2018, 32, 130–143. [Google Scholar] [CrossRef]
- Melchiori, L.; Gardenghi, S.; Rivella, S. Beta-Thalassemia: HiJAKing Ineffective Erythropoiesis and Iron Overload. Adv. Hematol. 2010, 2010, 938640. [Google Scholar] [CrossRef] [Green Version]
- Gardenghi, S.; Grady, R.W.; Rivella, S. Anemia, ineffective erythropoiesis, and hepcidin: Interacting factors in abnormal iron metabolism leading to iron overload in β-thalassemia. Hematol. Oncol. Clin. N. Am. 2010, 24, 1089–1107. [Google Scholar] [CrossRef] [Green Version]
- Colah, R.; Gorakshakar, A.; Nadkarni, A. Global burden, distribution and prevention of β-thalassemias and hemoglobin E disorders. Expert Rev. Hematol. 2010, 3, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.A. Intestinal iron absorption: Regulation by dietary & systemic factors. Int. J. Vitam. Nutr. Res. 2010, 80, 231–242. [Google Scholar] [CrossRef]
- Silva, B.; Faustino, P. An overview of molecular basis of iron metabolism regulation and the associated pathologies. Biochim. Biophys. Acta 2015, 1852, 1347–1359. [Google Scholar] [CrossRef] [Green Version]
- Gunshin, H.; MacKenzie, B.; Berger, U.V.; Gunshin, Y.; Romero, M.F.; Boron, W.F.; Nussberger, S.; Gollan, J.L.; Hediger, M.A. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 1997, 388, 482–488. [Google Scholar] [CrossRef]
- Le, N.T.; Richardson, D.R. Ferroportin1: A new iron export molecule? Int. J. Biochem. Cell Biol. 2002, 34, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Yeh, K.-Y.; Yeh, M.; Mims, L.; Glass, J.; Gulec, S.; Anderson, G.J.; Collins, J.F.; Polk, P.; Hudson, D.M.; Curtis, S.B.; et al. Iron feeding induces ferroportin 1 and hephaestin migration and interaction in rat duodenal epithelium. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G55–G65. [Google Scholar] [CrossRef] [Green Version]
- Brittin, G.M.; Chee, Q.T. Relation of ferroxidase (ceruloplasmin) to iron absorption. J. Lab. Clin. Med. 1969, 74, 53–59. [Google Scholar]
- Anderson, G.J.; Frazer, D.M. Hepatic iron metabolism. Semin. Liver Dis. 2005, 25, 420–432. [Google Scholar] [CrossRef]
- Ponka, P.; Lok, C.N. The transferrin receptor: Role in health and disease. Int. J. Biochem. Cell Biol. 1999, 31, 1111–1137. [Google Scholar] [CrossRef]
- Herbison, C.E.; Thorstensen, K.; Chua, A.C.G.; Graham, R.M.; Leedman, P.; Olynyk, J.K.; Trinder, D.; Rapisarda, C.; Puppi, J.; Hughes, R.D.; et al. The role of transferrin receptor 1 and 2 in transferrin-bound iron uptake in human hepatoma cells. Am. J. Physiol. Physiol. 2009, 297, C1567–C1575. [Google Scholar] [CrossRef] [Green Version]
- Trinder, D.; Baker, E. Transferrin receptor 2: A new molecule in iron metabolism. Int. J. Biochem. Cell Biol. 2003, 35, 292–296. [Google Scholar] [CrossRef]
- Hershko, C.; Graham, G.; Bates, G.W.; Rachmilewitz, E.A. Non-Specific serum iron in thalassaemia: An abnormal serum iron fraction of potential toxicity. Br. J. Haematol. 1978, 40, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Grootveld, M.; Bell, J.D.; Halliwell, B.; I Aruoma, O.; Bomford, A.; Sadler, P.J. Non-transferrin-bound iron in plasma or serum from patients with idiopathic hemochromatosis. Characterization by high performance liquid chromatography and nuclear magnetic resonance spectroscopy. J. Biol. Chem. 1989, 264, 4417–4422. [Google Scholar] [CrossRef] [PubMed]
- Knutson, M.D. Non-transferrin-bound iron transporters. Free. Radic. Biol. Med. 2019, 133, 101–111. [Google Scholar] [CrossRef]
- Fleming, R.E.; Ponka, P. Iron overload in human disease. N. Engl. J. Med. 2012, 366, 348–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Theil, E.C. Ferritin as an iron concentrator and chelator target. Ann. N. Y. Acad. Sci. 2005, 1054, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Arosio, P.; Elia, L.; Poli, M. Ferritin, cellular iron storage and regulation. IUBMB Life 2017, 69, 414–422. [Google Scholar] [CrossRef] [Green Version]
- Dev, S.; Babitt, J.L. Overview of iron metabolism in health and disease. Hemodial. Int. 2017, 21 (Suppl. S1), S6–S20. [Google Scholar] [CrossRef] [Green Version]
- Levi, S.; Corsi, B.; Bosisio, M.; Invernizzi, R.; Volz, A.; Sanford, D.; Arosio, P.; Drysdale, J. A human mitochondrial ferritin encoded by an intronless gene. J. Biol. Chem. 2001, 276, 24437–24440. [Google Scholar] [CrossRef] [Green Version]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef] [Green Version]
- Fuhrmann, D.C.; Mondorf, A.; Beifuß, J.; Jung, M.; Brüne, B. Hypoxia inhibits ferritinophagy, increases mitochondrial ferritin, and protects from ferroptosis. Redox Biol. 2020, 36, 101670. [Google Scholar] [CrossRef]
- Senn, H.; Wüthrich, K. Amino acid sequence, haem-iron co-ordination geometry and functional properties of mitochondrial and bacterial c-type cytochromes. Q. Rev. Biophys. 1985, 18, 111–134. [Google Scholar] [CrossRef] [Green Version]
- Lill, R. Function and biogenesis of iron–sulphur proteins. Nature 2009, 460, 831–838. [Google Scholar] [CrossRef]
- Philpott, C.C.; Ryu, M.-S. Special delivery: Distributing iron in the cytosol of mammalian cells. Front. Pharmacol. 2014, 5, 173. [Google Scholar] [CrossRef] [PubMed]
- Koury, M.J.; Ponka, P. New insights into erythropoiesis: The roles of folate, vitamin B12, and iron. Annu. Rev. Nutr. 2004, 24, 105–131. [Google Scholar] [CrossRef] [PubMed]
- Philpott, C.C. The flux of iron through ferritin in erythrocyte development. Curr. Opin. Hematol. 2018, 25, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Hamdi, A.; Roshan, T.M.; Kahawita, T.M.; Mason, A.B.; Sheftel, A.D.; Ponka, P. Erythroid cell mitochondria receive endosomal iron by a “kiss-and-run” mechanism. Biochim. Biophys. Acta 2016, 1863, 2859–2867. [Google Scholar] [CrossRef] [PubMed]
- Paradkar, P.N.; Zumbrennen, K.B.; Paw, B.H.; Ward, D.M.; Kaplan, J. Regulation of Mitochondrial iron import through differential turnover of mitoferrin 1 and mitoferrin 2. Mol. Cell. Biol. 2009, 29, 1007–1016. [Google Scholar] [CrossRef] [Green Version]
- Sano, S.; Inoue, S.; Tanabe, Y.; Sumiya, C.; Koike, S. Significance of mitochondria for porphyrin and heme biosynthesis. Science 1959, 129, 275–276. [Google Scholar] [CrossRef]
- Kispal, G.; Csere, P.; Prohl, C.; Lill, R. The mitochondrial proteins Atm1p and Nfs1p are essential for biogenesis of cytosolic Fe/S proteins. EMBO J. 1999, 18, 3981–3989. [Google Scholar] [CrossRef] [Green Version]
- Low, P.S.; Waugh, S.M.; Zinke, K.; Drenckhahn, D. The role of hemoglobin denaturation and band 3 clustering in red blood cell aging. Science 1985, 227, 531–533. [Google Scholar] [CrossRef]
- Bratosin, D.; Mazurier, J.; Tissier, J.-P.; Slomianny, C.; Estaquier, J.; Russo-Marie, F.; Huart, J.-J.; Freyssinet, J.-M.; Aminoff, D.; Ameisen, J.-C.; et al. Molecular mechanisms of erythrophagocytosis. Characterization of the senescent erythrocytes that are phagocytized by macrophages. Comptes Rendus L’académie Sci.-Ser. III-Sci. Vie 1997, 320, 811–818. [Google Scholar] [CrossRef]
- Bosman, G.; Willekens, F.; Werre, J. Erythrocyte aging: A more than superficial resemblance to apoptosis? Cell. Physiol. Biochem. 2005, 16, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrons, J.V.; Casafont, L.B.; Frasnedo, E.F. Concise review: How do red blood cells born, live, and die? Ann. Hematol. 2021, 100, 2425–2433. [Google Scholar] [CrossRef]
- Yeh, K.-Y.; Yeh, M.; Glass, J. Hepcidin regulation of ferroportin 1 expression in the liver and intestine of the rat. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G385–G394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, C.H.; Valore, E.V.; Waring, A.J.; Ganz, T. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J. Biol. Chem. 2001, 276, 7806–7810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vokurka, M.; Krijt, J.; Šulc, K.; Nečas, E. Hepcidin mRNA levels in mouse liver respond to inhibition of erythropoiesis. Physiol. Res. 2006, 55, 667–674. [Google Scholar] [CrossRef]
- Nicolas, G.; Chauvet, C.; Viatte, L.; Danan, J.L.; Bigard, X.; Devaux, I.; Beaumont, C.; Kahn, A.; Vaulont, S. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J. Clin. Investig. 2002, 110, 1037–1044. [Google Scholar] [CrossRef]
- Schmidt, P.J.; Toran, P.T.; Giannetti, A.M.; Bjorkman, P.J.; Andrews, N.C. The transferrin receptor modulates hfe-dependent regulation of hepcidin expression. Cell Metab. 2008, 7, 205–214. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Valore, E.V.; Nemeth, E.; Goodnough, J.B.; Gabayan, V.; Ganz, T. Iron transferrin regulates hepcidin synthesis in primary hepatocyte culture through hemojuvelin and BMP2/4. Blood 2007, 110, 2182–2189. [Google Scholar] [CrossRef] [Green Version]
- Kanamori, Y.; Murakami, M.; Sugiyama, M.; Hashimoto, O.; Matsui, T.; Funaba, M. Hepcidin and IL-1β. Vitam. Horm. 2019, 110, 143–156. [Google Scholar] [CrossRef]
- Babitt, J.L.; Huang, F.W.; Wrighting, D.M.; Xia, Y.; Sidis, Y.; Samad, T.A.; Campagna, J.A.; Chung, R.T.; Schneyer, A.L.; Woolf, C.J.; et al. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat. Genet. 2006, 38, 531–539. [Google Scholar] [CrossRef]
- Silvestri, L.; Pagani, A.; Nai, A.; De Domenico, I.; Kaplan, J.; Camaschella, C. The serine protease matriptase-2 (TMPRSS6) inhibits hepcidin activation by cleaving membrane hemojuvelin. Cell Metab. 2008, 8, 502–511. [Google Scholar] [CrossRef] [Green Version]
- Anderson, C.P.; Shen, M.; Eisenstein, R.S.; Leibold, E.A. Mammalian iron metabolism and its control by iron regulatory proteins. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2012, 1823, 1468–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.D.; Tan, E.-K. Iron regulatory protein (IRP)-iron responsive element (IRE) signaling pathway in human neurodegenerative diseases. Mol. Neurodegener. 2017, 12, 75. [Google Scholar] [CrossRef] [PubMed]
- Casey, J.L.; Hentze, M.W.; Koeller, D.M.; Caughman, S.W.; Rouault, T.A.; Klausner, R.D.; Harford, J.B. Iron-responsive elements: Regulatory rna sequences that control mrna levels and translation. Science 1988, 240, 924–928. [Google Scholar] [CrossRef]
- Zhang, D.-L.; Hughes, R.M.; Ollivierre-Wilson, H.; Ghosh, M.C.; Rouault, T.A. A ferroportin transcript that lacks an iron-responsive element enables duodenal and erythroid precursor cells to evade translational repression. Cell Metab. 2009, 9, 461–473. [Google Scholar] [CrossRef] [Green Version]
- Tchernitchko, D.; Bourgeois, M.; Martin, M.-E.; Beaumont, C. Expression of the two mRNA isoforms of the iron transporter Nramp2/DMTI in mice and function of the iron responsive element. Biochem. J. 2002, 363, 449–455. [Google Scholar] [CrossRef]
- Anderson, G.J.; Frazer, D.M. Current understanding of iron homeostasis. Am. J. Clin. Nutr. 2017, 106 (Suppl. S6), 1559S–1566S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouault, T.A. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat. Chem. Biol. 2006, 2, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Sukhbaatar, N.; Weichhart, T. Iron Regulation: Macrophages in Control. Pharmaceuticals 2018, 11, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakhlon, O.; Cabantchik, Z. The labile iron pool: Characterization, measurement, and participation in cellular processes(1). Free. Radic. Biol. Med. 2002, 33, 1037–1046. [Google Scholar] [CrossRef]
- Melis, M.A.; Cau, M.; Deidda, F.; Barella, S.; Cao, A.; Galanello, R. H63D mutation in the HFE gene increases iron overload in beta-thalassemia carriers. Haematologica 2002, 87, 242–245. [Google Scholar] [PubMed]
- Zekavat, O.R.; Jahromi, M.Z.; Haghpanah, S.; Jahromi, Z.B.K.; Cohan, N. Association of HFE Gene Mutations With Serum Ferritin Level and Heart and Liver Iron Overload in Patients With Transfusion-dependent Beta-Thalassemia. J. Pediatr. Hematol. Oncol. 2021, 43, e26–e28. [Google Scholar] [CrossRef] [PubMed]
- Nienhuis, A.W.; Nathan, D.G. Pathophysiology and Clinical Manifestations of the β-Thalassemias. Cold Spring Harb. Perspect. Med. 2012, 2, a011726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leecharoenkiat, K.; Lithanatudom, P.; Sornjai, W.; Smith, D.R. Iron dysregulation in beta-thalassemia. Asian Pac. J. Trop. Med. 2016, 9, 1035–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coates, T.D. Physiology and pathophysiology of iron in hemoglobin-associated diseases. Free Radic. Biol. Med. 2014, 72, 23–40. [Google Scholar] [CrossRef] [Green Version]
- Kumfu, S.; Chattipakorn, S.; Fucharoen, S.; Chattipakorn, N. Ferric iron uptake into cardiomyocytes of β-thalassemic mice is not through calcium channels. Drug Chem. Toxicol. 2013, 36, 329–334. [Google Scholar] [CrossRef]
- Kumfu, S.; Chattipakorn, S.; Chattipakorn, N.; Cardiac Electrophysiology Research and Training Center. Silencing of lipocalin-2 and its receptor improved cardiomyocytes viability via decreasing iron uptake, mitochondrial fission, mitophagy and apoptosis under iron overload condition. Eur. Heart J. 2020, 41 (Suppl. S2), ehaa946-3392. [Google Scholar] [CrossRef]
- Russo, V.; Rago, A.; Papa, A.A.; Nigro, G. Electrocardiographic Presentation, Cardiac Arrhythmias, and Their Management in β-Thalassemia Major Patients. Ann. Noninvasive Electrocardiol. 2016, 21, 335–342. [Google Scholar] [CrossRef]
- Farmakis, D.; Porter, J.; Taher, A.; Cappellini, M.D.; Angastiniotis, M.; Eleftheriou, A. 2021 Thalassaemia International Federation Guidelines for the Management of Transfusion-dependent Thalassemia. HemaSphere 2022, 6, e732. [Google Scholar] [CrossRef] [PubMed]
- Tanno, T.; Miller, J.L. Iron Loading and Overloading due to Ineffective Erythropoiesis. Adv. Hematol. 2010, 2010, 358283. [Google Scholar] [CrossRef] [Green Version]
- Pootrakul, P.; Kitcharoen, K.; Yansukon, P.; Wasi, P.; Fucharoen, S.; Charoenlarp, P.; Brittenham, G.; Pippard, M.J.; Finch, C. The effect of erythroid hyperplasia on iron balance. Blood 1988, 71, 1124–1129. [Google Scholar] [CrossRef] [Green Version]
- Pigeon, C.; Ilyin, G.; Courselaud, B.; Leroyer, P.; Turlin, B.; Brissot, P.; Loréal, O. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J. Biol. Chem. 2001, 276, 7811–7819. [Google Scholar] [CrossRef] [Green Version]
- Ganz, T. Hepcidin and iron regulation, 10 years later. Blood 2011, 117, 4425–4433. [Google Scholar] [CrossRef] [Green Version]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.V.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Origa, R.; Galanello, R.; Ganz, T.; Giagu, N.; Maccioni, L.; Faa, G.; Nemeth, E. Liver iron concentrations and urinary hepcidin in beta-thalassemia. Haematologica 2007, 92, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Viprakasit, V.; Musallam, K.M.; Cappellini, M.D. Treating iron overload in patients with non-transfusion-dependent thalassemia. Am. J. Hematol. 2013, 88, 409–415. [Google Scholar] [CrossRef] [Green Version]
- Camaschella, C.; Nai, A. Ineffective erythropoiesis and regulation of iron status in iron loading anaemias. Br. J. Haematol. 2016, 172, 512–523. [Google Scholar] [CrossRef] [Green Version]
- Kautz, L.; Jung, G.; Du, X.; Gabayan, V.; Chapman, J.; Nasoff, M.; Nemeth, E.; Ganz, T. Erythroferrone contributes to hepcidin suppression and iron overload in a mouse model of β-thalassemia. Blood 2015, 126, 2031–2037. [Google Scholar] [CrossRef] [Green Version]
- Taher, A.T.; Musallam, K.M.; Wood, J.C.; Cappellini, M.D. Magnetic resonance evaluation of hepatic and myocardial iron deposition in transfusion-independent thalassemia intermedia compared to regularly transfused thalassemia major patients. Am. J. Hematol. 2010, 85, 288–290. [Google Scholar] [CrossRef]
- Taher, A.T.; Musallam, K.M.; Karimi, M.; El-Beshlawy, A.; Belhoul, K.; Daar, S.; Saned, M.-S.; El-Chafic, A.-H.; Fasulo, M.R.; Cappellini, M.D. Overview on practices in thalassemia intermedia management aiming for lowering complication rates across a region of endemicity: The OPTIMAL CARE study. Blood 2010, 115, 1886–1892. [Google Scholar] [CrossRef] [Green Version]
- Taher, A.T.; Saliba, A.N. Iron overload in thalassemia: Different organs at different rates. Hematol. Am. Soc. Hematol. Educ. Program 2017, 2017, 265–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canonne-Hergaux, F.; Levy, J.E.; Fleming, M.D.; Montross, L.K.; Andrews, N.C.; Gros, P. Expression of the DMT1 (NRAMP2/DCT1) iron transporter in mice with genetic iron overload disorders. Blood 2001, 97, 1138–1140. [Google Scholar] [CrossRef] [PubMed]
- Awadallah, S.M.; Nimer, N.A.; Atoum, M.F.; Saleh, S.A. Association of haptoglobin phenotypes with ceruloplasmin ferroxidase activity in β-thalassemia major. Clin. Chim. Acta 2011, 412, 975–979. [Google Scholar] [CrossRef]
- Mishra, A.K.; Tiwari, A. Iron overload in Beta thalassaemia major and intermedia patients. Maedica 2013, 8, 328–332. [Google Scholar]
- Hershko, C. Pathogenesis and management of iron toxicity in thalassemia. Ann. N. Y. Acad. Sci. 2010, 1202, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Borgna-Pignatti, C.; Rugolotto, S.; De Stefano, P.; Zhao, H.; Cappellini, M.D.; Del Vecchio, G.C.; Romeo, M.A.; Forni, G.L.; Gamberini, M.R.; Ghilardi, R.; et al. Survival and complications in patients with thalassemia major treated with transfusion and deferoxamine. Haematologica 2004, 89, 1187–1193. [Google Scholar]
- Taher, A.T.; Porter, J.; Viprakasit, V.; Kattamis, A.; Chuncharunee, S.; Sutcharitchan, P.; Siritanaratkul, N.; Galanello, R.; Karakas, Z.; Lawniczek, T.; et al. Deferasirox reduces iron overload significantly in nontransfusion-dependent thalassemia: 1-year results from a prospective, randomized, double-blind, placebo-controlled study. Blood 2012, 120, 970–977. [Google Scholar] [CrossRef] [Green Version]
- Telfer, P.T.; Prestcott, E.; Holden, S.; Walker, M.; Hoffbrand, A.V.; Wonke, B. Hepatic iron concentration combined with long-term monitoring of serum ferritin to predict complications of iron overload in thalassaemia major. Br. J. Haematol. 2000, 110, 971–977. [Google Scholar] [CrossRef]
- Borgna-Pignatti, C.; Vergine, G.; Lombardo, T.; Cappellini, M.D.; Cianciulli, P.; Maggio, A.; Renda, D.; Lai, M.E.; Mandas, A.; Forni, G.; et al. Hepatocellular carcinoma in the thalassaemia syndromes. Br. J. Haematol. 2004, 124, 114–117. [Google Scholar] [CrossRef] [PubMed]
- Ansah, D.; Husain, N.; Ruh, A.; Berhane, H.; Smith, A.; Thompson, A.; De Freitas, A.; Rigsby, C.K.; Robinson, J.D. Cardiac Magnetic Resonance Strain in Beta Thalassemia Major Correlates with Cardiac Iron Overload. Children 2023, 10, 271. [Google Scholar] [CrossRef]
- Anderson, L.; Holden, S.; Davis, B.; Prescott, E.; Charrier, C.; Bunce, N.; Firmin, D.; Wonke, B.; Porter, J.; Walker, J.; et al. Cardiovascular T2-star (T2*) magnetic resonance for the early diagnosis of myocardial iron overload. Eur. Heart J. 2001, 22, 2171–2179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liguori, C.; Pitocco, F.; Di Giampietro, I.; de Vivo, A.E.; Schena, E.; Cianciulli, P.; Zobel, B.B. Relationship between myocardial T2* values and cardiac volumetric and functional parameters in β-thalassemia patients evaluated by cardiac magnetic resonance in association with serum ferritin levels. Eur. J. Radiol. 2013, 82, e441–e447. [Google Scholar] [CrossRef] [PubMed]
- Gammella, E.; Recalcati, S.; Rybinska, I.; Buratti, P.; Cairo, G. Iron-Induced Damage in Cardiomyopathy: Oxidative-Dependent and Independent Mechanisms. Oxidative Med. Cell. Longev. 2015, 2015, 230182. [Google Scholar] [CrossRef] [Green Version]
- Cui, H.-J.; He, H.-Y.; Yang, A.-L.; Zhou, H.-J.; Wang, C.; Luo, J.-K.; Lin, Y.; Tang, T. Efficacy of Deferoxamine in Animal Models of Intracerebral Hemorrhage: A Systematic Review and Stratified Meta-Analysis. PLoS ONE 2015, 10, e0127256. [Google Scholar] [CrossRef]
- Uygun, V.; Kurtoglu, E. Iron-chelation therapy with oral chelators in patients with thalassemia major. Hematology 2013, 18, 50–55. [Google Scholar] [CrossRef]
- Kuo, K.H.; Mrkobrada, M. A systematic review and meta-analysis of deferiprone monotherapy and in combination with deferoxamine for reduction of iron overload in chronically transfused patients with β-thalassemia. Hemoglobin 2014, 38, 409–421. [Google Scholar] [CrossRef]
- Kontoghiorghes, G.J.; Aldouri, M.; Hoffbrand, A.V.; Barr, J.; Wonke, B.; Kourouclaris, T.; Sheppard, L. Effective chelation of iron in beta thalassaemia with the oral chelator 1,2-dimethyl-3-hydroxypyrid-4-one. BMJ 1987, 295, 1509–1512. [Google Scholar] [CrossRef] [Green Version]
- Binding, A.; Ward, R.; Tomlinson, G.; Kuo, K.H.M. Deferiprone exerts a dose-dependent reduction of liver iron in adults with iron overload. Eur. J. Haematol. 2019, 103, 80–87. [Google Scholar] [CrossRef]
- Waheed, N.; Ali, S.; Butt, M.A. Comparison of deferiprone and deferrioxamine for the treatment of transfusional iron overload in children with beta thalassemia major. J. Ayub. Med. Coll. Abbottabad 2014, 26, 297–300. [Google Scholar]
- Nisbet-Brown, E.; Olivieri, N.F.; Giardina, P.J.; Grady, R.W.; Neufeld, E.J.; Séchaud, R.; Krebs-Brown, A.J.; Anderson, J.R.; Alberti, D.; Sizer, K.C.; et al. Effectiveness and safety of ICL670 in iron-loaded patients with thalassaemia: A randomised, double-blind, placebo-controlled, dose-escalation trial. Lancet 2003, 361, 1597–1602. [Google Scholar] [CrossRef]
- Nick, H.; Acklin, P.; Lattmann, R.; Buehlmayer, P.; Hauffe, S.; Schupp, J.; Alberti, D. Development of tridentate iron chelators: From desferrithiocin to ICL670. Curr. Med. Chem. 2003, 10, 1065–1076. [Google Scholar] [CrossRef]
- Entezari, S.; Haghi, S.M.; Norouzkhani, N.; Sahebnazar, B.; Vosoughian, F.; Akbarzadeh, D.; Islampanah, M.; Naghsh, N.; Abbasalizadeh, M.; Deravi, N. Iron chelators in treatment of iron overload. J. Toxicol. 2022, 2022, 4911205. [Google Scholar] [CrossRef] [PubMed]
- Hershko, C.; Weatherall, D.J.; Finch, C. Iron-Chelating Therapy. CRC Crit. Rev. Clin. Lab. Sci. 1988, 26, 303–345. [Google Scholar] [CrossRef] [PubMed]
- Al-Refaie, F.N.; Sheppard, L.N.; Nortey, P.; Wonke, B.; Hoffbrand, A.V. Pharmacokinetics of the oral iron chelator deferiprone (L1) in patients with iron overload. Br. J. Haematol. 1995, 89, 403–408. [Google Scholar] [CrossRef] [PubMed]
- Jain, M.; Jitani, A.; Dolai, T. Current practices in the management of Beta-hemoglobinopathies. In Fetal Hemoglobin; Nova Science Publishers Inc.: Hauppauge, NY, USA, 2020; p. 11. [Google Scholar]
- Hider, R.C.; Kong, X.; Abbate, V.; Harland, R.; Conlon, K.; Luker, T. Deferitazole, a new orally active iron chelator. Dalton Trans. 2015, 44, 5197–5204. [Google Scholar] [CrossRef] [PubMed]
- Taher, A.T.; Saliba, A.N.; Kuo, K.H.; Giardina, P.J.; Cohen, A.R.; Neufeld, E.J.; Aydinok, Y.; Kwiatkowski, J.L.; Jeglinski, B.I.; Pietropaolo, K.; et al. Safety and pharmacokinetics of the oral iron chelator SP-420 in β-thalassemia. Am. J. Hematol. 2017, 92, 1356–1361. [Google Scholar] [CrossRef]
- Soliman, Y.; Abdelaziz, A.; Mouffokes, A.; Amer, B.E.; Goudy, Y.M.; Abdelwahab, O.A.; Badawy, M.M.; Diab, R.A.; Elsharkawy, A. Efficacy and Safety of Calcium Channel Blockers in Preventing Cardiac Siderosis in Thalassemia Patients: An Updated Meta-Analysis with Trial Sequential Analysis. Eur. J. Haematol. 2023, 110, 414–425. [Google Scholar] [CrossRef]
- Porter, J.; Cappellini, M.D.; Coates, T.; Hermine, O.; Viprakasit, V.; Voskaridou, E.; Liew, H.K.; Perrotta, S.; Khelif, A.; Kattamis, A.; et al. Effects of Luspatercept on Iron Overload and Impact on Responders to Luspatercept: Results from the BELIEVE Trial. Blood 2019, 134, 2245. [Google Scholar] [CrossRef]
- Hermine, O.; Cappellini, M.D.D.; Taher, A.T.; Coates, T.D.; Viprakasit, V.; Kattamis, A.; Shetty, J.K.; Weisskopf, M.B.; Holot, N.; Vodala, S.; et al. Effect of Luspatercept on Red Blood Cell (RBC) Transfusion Burden, Iron Chelation Therapy (ICT), and Iron Overload in Adults with Transfusion-Dependent β-Thalassemia (TDT) from the BELIEVE Trial: A Long-Term Analysis. Blood 2022, 140, 8215–8217. [Google Scholar] [CrossRef]
- Coates, T.D. Iron overload in transfusion-dependent patients. Hematology 2019, 2019, 337–344. [Google Scholar] [CrossRef]
- Government of India; Ministry of Health and Family Welfare. Prevention and control of hemoglobinopathies in india-thalassemia, sickle cell disease and other variant hemoglobins. In Guidelines on Hemoglobinopathies in India; Ministry of Health & Family Welfare Government of India: New Delhi, India, 2016; p. 146. Available online: https://nhm.gov.in/images/pdf/programmes/RBSK/Resource_Documents/Guidelines_on_Hemoglobinopathies_in%20India.pdf (accessed on 27 June 2023).
- Bhattacharyya, D.; Chowdhury, R.; Choudhuri, S.; Ghosh, P.; Bhattacharyya, M. Efficacy and Safety of Deferasirox in Chelation Naïve HbEβ Thalassemia Patients: Initial Experience from a Tertiary Centre of Eastern India. Blood 2017, 130 (Suppl. S1), 4760. [Google Scholar] [CrossRef]
- Sidhu, S.; Kakkar, S.; Dewan, P.; Bansal, N.; Sobti, P.C. Adherence to Iron Chelation Therapy and Its Determinants. Int. J. Hematol. Stem Cell Res. 2021, 15, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Tripathy, I.; Panja, A.; Dolai, T.K.; Mallick, A.K. Comparative Efficacy and Safety Between Deferiprone and Deferasirox with Special Reference to Serum Ferritin Level and Cardiac Function in Bengali β-Thalassemia Major Children. Hemoglobin 2021, 45, 296–302. [Google Scholar] [CrossRef]
- DivakarJose, R.R.; Delhikumar, C.G.; Kumar, G.R. Efficacy and Safety of Combined Oral Chelation with Deferiprone and Deferasirox on Iron Overload in Transfusion Dependent Children with Thalassemia—A Prospective Observational Study. Indian J. Pediatr. 2021, 88, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Chandra, J.; Parakh, N.; Sidharth Singh, N.; Sharma, S.; Goel, M.; Pemde, H. Efficacy and Safety of Thalidomide in Patients With Transfusion-Dependent Thalassemia. Indian Pediatr. 2021, 58, 611–616. [Google Scholar] [CrossRef]
- Naithani, R.; Chandra, J.; Sharma, S. Safety of oral iron chelator deferiprone in young thalassaemics. Eur. J. Haematol. 2005, 74, 217–220. [Google Scholar] [CrossRef]
- Mohamed, R.; Rahman, A.H.A.; Masra, F.; Latiff, Z.A. Barriers to adherence to iron chelation therapy among adolescent with transfusion dependent thalassemia. Front. Pediatr. 2022, 10, 951947. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Characteristics | Deferoxamine | Deferiprone | Deferasirox |
|---|---|---|---|
| Structure | Hexadentate | Bidentate | Tridentate |
| Route of Administration | Subcutaneous or Intravenous Injections | Oral | Oral |
| Mechanism of Action | Chelates NTBI, Ft-bound iron; promotes ferritinophagy [115] | Chelates labile iron in cytosol [115] | Chelates labile iron in cytosol; increases hepcidin levels [115] |
| Route of Excretion | Biliary and Urinary [116] | Urinary [117] | Fecal |
| Adverse Effects | Hearing disorders, Growth Retardation, Lung/Renal Toxicity, Bone Abnormalities, Visual disorders, Pain at site of injection [118]. | Severe Agranulocytosis, Gastrointestinal problems, Arthiritis [118]. | Rash, Renal disorders, Gastrointestinal problems [118] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Basu, S.; Rahaman, M.; Dolai, T.K.; Shukla, P.C.; Chakravorty, N. Understanding the Intricacies of Iron Overload Associated with β-Thalassemia: A Comprehensive Review. Thalass. Rep. 2023, 13, 179-194. https://doi.org/10.3390/thalassrep13030017
Basu S, Rahaman M, Dolai TK, Shukla PC, Chakravorty N. Understanding the Intricacies of Iron Overload Associated with β-Thalassemia: A Comprehensive Review. Thalassemia Reports. 2023; 13(3):179-194. https://doi.org/10.3390/thalassrep13030017
Chicago/Turabian StyleBasu, Subhangi, Motiur Rahaman, Tuphan Kanti Dolai, Praphulla Chandra Shukla, and Nishant Chakravorty. 2023. "Understanding the Intricacies of Iron Overload Associated with β-Thalassemia: A Comprehensive Review" Thalassemia Reports 13, no. 3: 179-194. https://doi.org/10.3390/thalassrep13030017