
Spectrum of Thalassemia and Hemoglobinopathy Using Capillary Zone Electrophoresis: A Facility-Based Single Centred Study at icddr,b in Bangladesh
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Ethical Consideration
2.3. Sample Preparation and Analysis
2.4. Sickling Test to Exclude Hb D-Punjab
2.5. Red Cell Indices and Parameter of Anemia
2.6. Data Management
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Hossain, M.S.; Raheem, E.; Sultana, T.A.; Ferdous, S.; Nahar, N.; Islam, S.; Arifuzzaman, M.; Razzaque, M.A.; Alam, R.; Aziz, S.; et al. Thalassemias in South Asia: Clinical lessons learnt from Bangladesh. Orphanet J. Rare Dis. 2017, 12, 93. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, T.; Chakravarty, A.; Chakravarty, S. Population screening and prevention strategies for thalassemias and other hemoglobinopathies of Eastern India: Experience of 18,166 cases. Hemoglobin 2015, 39, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Galanello, R.; Origa, R. Beta-thalassemia. Orphanet J. Rare Dis. 2010, 5, 11. [Google Scholar] [CrossRef] [PubMed]
- Garfunkel, L.C.; Kaczorowski, J.; Christy, C. Pediatric Clinical Advisor E-Book: Instant Diagnosis and Treatment, 2nd ed.; Elsevier Health Sciences: Amsterdam, The Netherlands, 2007. [Google Scholar]
- Huisman, T.H.; Carver, M.F.; Efremov, G.D. A Syllabus of Human Hemoglobin Variants; Sickle Cell Anemia Foundation Augusta, GAUSA: Atlanta, IL, USA, 1998; Available online: https://globin.bx.psu.edu/html/huisman/variants/ (accessed on 20 November 2022).
- Alsaeed, E.S.; Farhat, G.N.; Assiri, A.M.; Memish, Z.; Ahmed, E.M.; Saeedi, M.Y.; Al-Dossary, M.F.; Bashawri, H. Distribution of hemoglobinopathy disorders in Saudi Arabia based on data from the premarital screening and genetic counseling program, 2011–2015. J. Epidemiol. Glob. Health 2018, 7, S41–S47. [Google Scholar] [CrossRef]
- Silberstein, P. Thalassemia, in xPharm: The Comprehensive Pharmacology Reference; Elsevier Inc.: Amsterdam, The Netherlands, 2008; pp. 1–6. [Google Scholar]
- Chandrashekar, V.; Soni, M. Hemoglobin disorders in south India. Int. Sch. Res. Not. 2011, 2011, 748939. [Google Scholar] [CrossRef]
- Hoffbrand, A.V.; Steensma, D.P. Hoffbrand’s Essential Haematology; John Wiley & Sons: Hoboken, NJ, USA, 2019. [Google Scholar]
- Henderson, S.; Timbs, A.; McCarthy, J.; Gallienne, A.; Van Mourik, M.; Masters, G.; May, A.; Khalil, M.S.M.; Schuh, A.; Old, J. Incidence of haemoglobinopathies in various populations—The impact of immigration. Clin. Biochem. 2009, 42, 1745–1756. [Google Scholar] [CrossRef]
- Eleftheriou, A.; Angastiniotis, M. Global Thalassaemia Review; Thalassaemia International Federation: Strovolos, Cyprus, 2021. [Google Scholar]
- Shannon, K.L.; Ahmed, S.; Rahman, H.; Prue, C.S.; Khyang, J.; Ram, M.; Haq, M.Z.; Chowdhury, A.; Akter, J.; Glass, G.E.; et al. Hemoglobin E and glucose-6-phosphate dehydrogenase deficiency and Plasmodium falciparum malaria in the Chittagong Hill Districts of Bangladesh. Am. J. Trop. Med. Hyg. 2015, 93, 281–286. [Google Scholar] [CrossRef]
- Weatherall, D.J. The challenge of haemoglobinopathies in resource-poor countries. Br. J. Haematol. 2011, 154, 736–744. [Google Scholar] [CrossRef]
- Ramprakash, S.; Agarwal, R.; Dhanya, R.; Marwah, P.; Soni, R.; Yaqub, N.; Fatima, I.; Zhara, T.; Gooneratne, L.; Williams, S.; et al. Low-cost matched sibling bone marrow transplant for standard-risk thalassemia in a limited-resource setting. Pediatr. Hematol. Oncol. J. 2017, 2, 107–113. [Google Scholar] [CrossRef]
- Panyasai, S.; Sakkhachornphop, S.; Pornprasert, S. Diagnosis of Compound Heterozygous Hb Tak/β-Thalassemia and HbD-Punjab/β-Thalassemia by HbA 2 Levels on Capillary Electrophoresis. Indian J. Hematol. Blood Transfus. 2018, 34, 110–114. [Google Scholar] [CrossRef]
- Borbely, N.; Phelan, L.; Szydlo, R.; Bain, B. Capillary zone electrophoresis for haemoglobinopathy diagnosis. J. Clin. Pathol. 2013, 66, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Dai, Q.; Zhang, X.; Zhou, W.; Gao, J.; Zhang, G. Comparison between capillary zone electrophoresis and capillary isoelectric focusing for thalassemia screening in southern China. J. Clin. Lab. Anal. 2018, 32, e22567. [Google Scholar] [CrossRef] [PubMed]
- Pauline, N.; Cabral, B.N.P.; Anatole, P.C.; Jocelyne, A.M.V.; Bruno, M.; Jeanne, N.Y. The in vitro antisickling and antioxidant effects of aqueous extracts Zanthoxyllum heitzii on sickle cell disorder. BMC Complement. Altern. Med. 2013, 13, 162. [Google Scholar] [CrossRef] [PubMed]
- Perumbeti, A. Hemoglobinopathies and Thalassemia Syndromes; Elsevier: Amsterdam, The Netherlands, 2014; pp. 1506–1531. [Google Scholar] [CrossRef]
- Sarma, P.R. Red cell indices. In Clinical Methods: The History, Physical, and Laboratory Examinations, 3rd ed.; Butterworths: Boston, MA, USA, 1990. Available online: https://www.ncbi.nlm.nih.gov/books/NBK260/ (accessed on 31 March 2023).
- Pagana, K.; Pagana, T.; Pagana, T. Mosby’s Diagnostic & Laboratory Test Reference, 14th ed.; Elsevier: St. Louis, MO, USA, 2019. [Google Scholar]
- DeLoughery, T.G. Microcytic anemia. N. Engl. J. Med. 2014, 371, 1324–1331. [Google Scholar] [CrossRef] [PubMed]
- Hoffbrand, V.; Provan, D. ABC of clinical haematology: Macrocytic anaemias. BMJ 1997, 314, 430–433. [Google Scholar] [CrossRef]
- Munkongdee, T.; Chen, P.; Winichagoon, P.; Fucharoen, S.; Paiboonsukwong, K. Update in laboratory diagnosis of thalassemia. Front. Mol. Biosci. 2020, 7, 74. [Google Scholar] [CrossRef]
- McPherson, R.A. Henry’s Clinical Diagnosis and Management by Laboratory Methods: First South Asia Edition_e-Book; Elsevier India: New Delhi, India, 2017. [Google Scholar]
- Kundrapu, S.; Noguez, J. Laboratory assessment of anemia. Adv. Clin. Chem. 2018, 83, 197–225. [Google Scholar] [CrossRef]
- WHO. Haemoglobin Concentrations for the Diagnosis of Anaemia and Assessment of Severity; World Health Organization: Geneva, Switzerland, 2011; pp. 1–6. Available online: http://www.who.int/vmnis/indicators/haemoglobin.pdf (accessed on 20 November 2022).
- Stevens, G.A.; Finucane, M.M.; De-Regil, L.M.; Paciorek, C.J.; Flaxman, S.R.; Branca, F.; Peña-Rosas, J.P.; Bhutta, Z.A.; Ezzati, M.; Nutrition Impact Model Study Group (Anaemia). Global, regional, and national trends in haemoglobin concentration and prevalence of total and severe anaemia in children and pregnant and non-pregnant women for 1995–2011: A systematic analysis of population-representative data. Lancet Glob. Health 2013, 1, e16–e25. [Google Scholar] [CrossRef]
- Korenromp, E.L.; Armstrong-Schellenberg, J.R.M.; Williams, B.G.; Nahlen, B.L.; Snow, R.W. Impact of malaria control on childhood anaemia in Africa—A quantitative review. Trop. Med. Int. Health 2004, 9, 1050–1065. [Google Scholar] [CrossRef]
- Chulilla, J.A.M.; Colás, M.S.R.; Martín, M.G. Classification of anemia for gastroenterologists. World J. Gastroenterol. 2009, 15, 4627. [Google Scholar] [CrossRef]
- Wayne, W.; Daniel, D.; Chad, L. Biostatistics. Basic Concepts and Methodology for the Health Sciences, 9th ed.; John Wiley & Sons, Inc.: New Delhi, India, 2010. [Google Scholar]
- Pradhan, S.; Chauhan, S.; Samal, P. Incidental detection of a rare hemoglobin variant (Hemoglobin N Seattle) leading to undetectable levels of HbA1c in a diabetic female: A case report. Thalass. Rep. 2017, 7, 5860. [Google Scholar] [CrossRef]
- Steinberg, M.H.; Rodgers, G.P. HbA2: Biology, clinical relevance and a possible target for ameliorating sickle cell disease. Br. J. Haematol. 2015, 170, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Torres, L.d.S.; Okumura, J.V.; da Silva, D.G.H.; Bonini-Domingos, C.R. Hemoglobin D-Punjab: Origin, distribution and laboratory diagnosis. Rev. Bras. Hematol. Hemoter. 2015, 37, 120–126. [Google Scholar] [CrossRef]
- Denic, S.; Souid, A.-K. Hemoglobin D-Punjab homozygotes and double heterozygotes in premarital screening: Case presentations and minireview. Eur. J. Med. Health Sci. 2021, 3, 90–94. [Google Scholar] [CrossRef]
- Adekile, A.D.; Kazanetz, E.G.; Leonova, J.Y.; Marouf, R.; Khmis, A.; Huisman, T.H.J. Co-inheritance of Hb D-Punjab (codon 121; GAA→CAA) and beta (0)-thalassemia (IVS-II-1; G→A). J. Pediatr. Hematol./Oncol. 1996, 18, 151–153. [Google Scholar] [CrossRef] [PubMed]
- Pornprasert, S.; Panyasai, S.; Rahad, S. Coinheritance of hemoglobin D-Punjab and β0-thalassemia 3.4 kb deletion in a Thai girl. Asian J. Transfus. Sci. 2017, 11, 199–202. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Mashon, R.S. Coinheritance of Hb D-Punjab and β-thalassemia: Diagnosis and implications in prenatal diagnosis. Hemoglobin 2015, 39, 138–140. [Google Scholar] [CrossRef] [PubMed]
- Giordano, P. Strategies for basic laboratory diagnostics of the hemoglobinopathies in multi-ethnic societies: Interpretation of results and pitfalls. Int. J. Lab. Hematol. 2013, 35, 465–479. [Google Scholar] [CrossRef]
- Bain, B.J. Haemoglobinopathy Diagnosis; John Wiley & Sons: Hoboken, NJ, USA, 2020. [Google Scholar]
- Thame, M.; Grandison, Y.; Mason, K.; Thompson, M.; Higgs, D.; Morris, J.; Serjeant, B.; Serjeant, G. The red cell distribution width in sickle cell disease—Is it of clinical value? Clin. Lab. Haematol. 1991, 13, 229–237. [Google Scholar] [CrossRef]
- Masiello, D.; Heeney, M.M.; Adewoye, A.H.; Eung, S.H.; Luo, H.-Y.; Steinberg, M.H.; Chui, D.H. Hemoglobin SE disease—A concise review. Am. J. Hematol. 2007, 82, 643–649. [Google Scholar] [CrossRef]
- Angastiniotis, M.; Eleftheriou, A.; Galanello, R.; Harteveld, C.L.; Petrou, M.; Traeger-Synodinos, J.; Giordano, P.; Jauniaux, E.; Modell, B.; Serour, G. Prevention of Thalassaemias and Other Haemoglobin Disorders: Volume 1: Principles; Thalassaemia International Federation: Strovolos, Cyprus, 2014. [Google Scholar]
- Avery, M.E. Avery’s Diseases of the Newborn, 8th ed.; Elsevier Health Sciences: Amsterdam, The Netherlands, 2005. [Google Scholar]
- Stephens, A.D.; Angastiniotis, M.; Baysal, E.; Chan, V.; Fucharoen, S.; Giordano, P.C.; Hoyer, J.D.; Mosca, A.; Wild, B.; The International Council for the Standardisation of Haematology (ICSH). ICSH recommendations for the measurement of haemoglobin A2. Int. J. Lab. Hematol. 2012, 34, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wahed, A.; Risin, S. Issues with Immunology and Serology Testing, in Accurate Results in the Clinical Laboratory; Elsevier: Amsterdam, The Netherlands, 2013; pp. 295–304. [Google Scholar] [CrossRef]
- Hossain, M.R.; Sarmin, M.; Rahman, H.; Shahrin, L.; Nyma, Z.; Ahmed, T.; Chisti, M.J. SARS-CoV-2 and dengue virus coinfection in an adult with beta-thalassemia (trait): A case report from Bangladesh with literature review. Heliyon 2021, 7, e08229. [Google Scholar] [CrossRef] [PubMed]
- Noor, F.A.; Sultana, N.; Bhuyan, G.S.; Islam, T.; Hossain, M.; Sarker, S.K.; Islam, K.; Khan, W.A.; Rahman, M.; Qadri, S.K.; et al. Nationwide carrier detection and molecular characterization of β-thalassemia and hemoglobin E variants in Bangladeshi population. Orphanet J. Rare Dis. 2020, 15, 15. [Google Scholar] [CrossRef] [PubMed]
- Fucharoen, S.; Weatherall, D.J. The hemoglobin E thalassemias. Cold Spring Harb. Perspect. Med. 2012, 2, a011734. [Google Scholar] [CrossRef]
- Gerald, P.S.; Diamond, L.K. A new hereditary hemoglobinopathy (the Lepore trait) and its interaction with thalassemia trait. Blood 1958, 13, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Fucharoen, S.; Viprakasit, V. Hb H disease: Clinical course and disease modifiers. ASH Educ. Program Book 2009, 2009, 26–34. [Google Scholar] [CrossRef]
- Riou, J.; Szuberski, J.; Godart, C.; Wajcman, H.; Oliveira, J.L.; Hoyer, J.D.; Bardakdjian-Michau, J. Precision of CAPILLARYS 2 for the detection of hemoglobin variants based on their migration positions. Am. J. Clin. Pathol. 2018, 149, 172–180. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Inherited Hb Disorders | Frequency, n (%) | Hb A, % (Mean ± SD) | Hb A2, % (Mean ± SD) | Hb F, % (Mean ± SD) | Altered Hb Variants, % (Mean ± SD) | ||
|---|---|---|---|---|---|---|---|
| Heterozygote Condition | |||||||
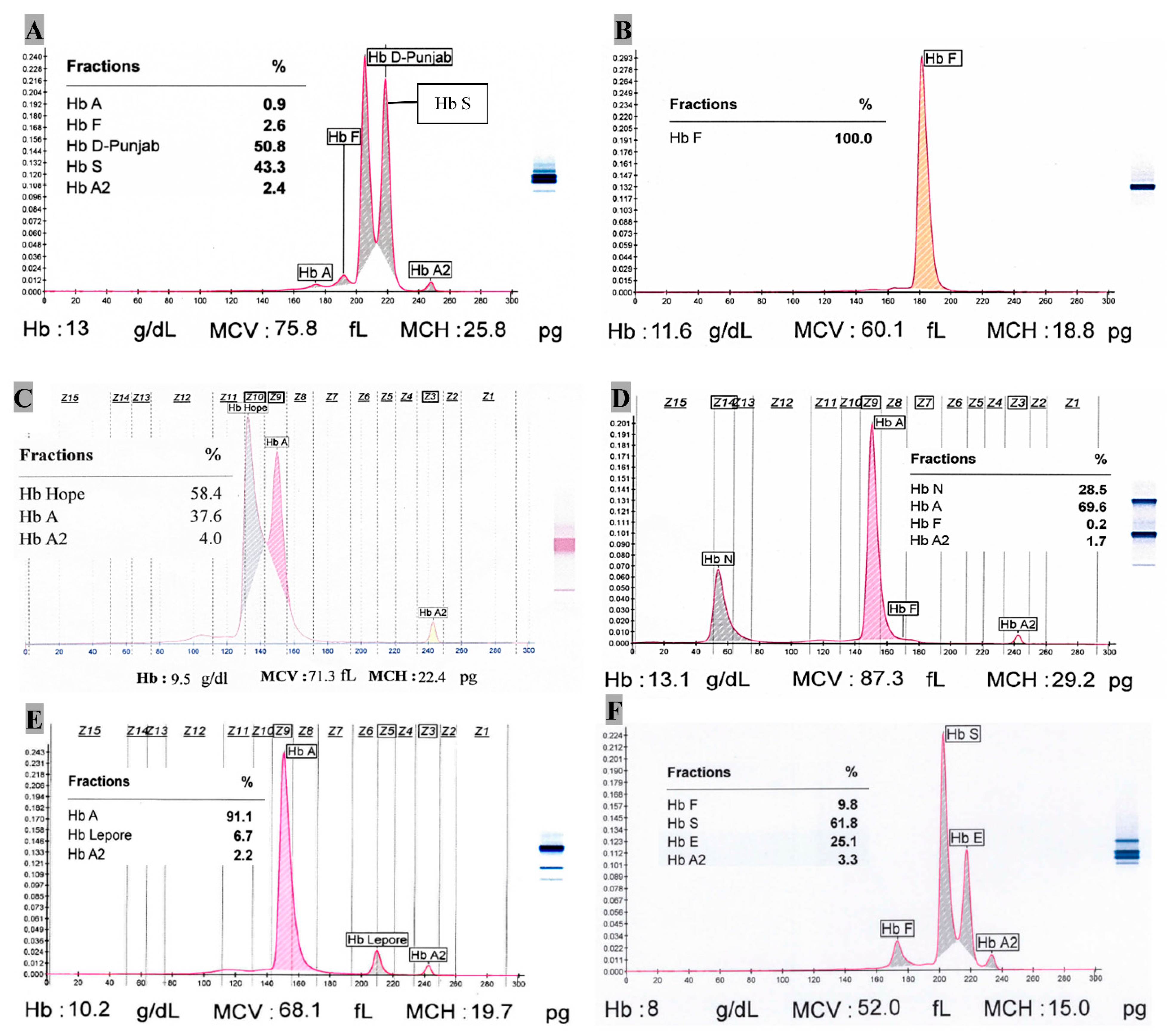
| Hb E | 907 (10.67) | 71.4 ± 2.3 | 3.4 ± 0.4 | 1.9 ± 2.3 | Hb E 23.8 ± 1.9 | - | |
| β-thalassemia | 714 (8.40) | 93.8 ± 1.6 | 5.2 ± 0.6 | 1.6 ± 1.6 | - | - | |
| Hb D-Punjab | 16 (0.19) | 58.7 ± 4.0 | 2.9 ± 0.4 | 1.0 ± 0.9 | Hb D-Punjab 37.2 ± 4.0 | - | |
| HPFH * | 13 (0.15) | 80.9 ± 3.3 | 2.5 ± 0.7 | 16.6 ± 3.1 | - | - | |
| Hb S | 9 (0.11) | 58.1 ± 2.0 | 2.7 ± 0.2 | 0.9 ± 0.5 | Hb S 38.6 ± 1.7 | - | |
| Double heterozygote condition | |||||||
| Hb E/β-thalassemia | Hb E/β+ | 101 (1.19) | 6.0 ± 2.6 | 5.1 ± 1.6 | 37.7 ± 11.8 | Hb E 50.9 ± 10.1 | - |
| Hb E/β0 | 33 (0.39) | - | 4.8 ± 1.6 | 45.3 ± 18.5 | Hb E 50.0 ± 17.4 | - | |
| Hb E/Hb S | 5 (<0.1) | - | 3.0 ± 0.7 | 21.6 ± 13.3 | Hb E 23.6 ± 2.7 | Hb S 51.8 ± 11.6 | |
| Hb S/β+-thalassemia | 1 (<0.1) | 7.0 | 4.8 | 22.1 | Hb S 66.1 | - | |
| Hb D-Punjab/β+-thalassemia | 1 (<0.1) | 4.1 | 5.2 | 1.4 | Hb D-Punjab 89.3 | - | |
| Hb S/Hb D-Punjab | 1 (<0.1) | 0.9 | 2.4 | 2.6 | Hb D-Punjab 50.8 | Hb S 43.3 | |
| Homozygote condition | |||||||
| Hb E | 135 (1.59) | 2.4 ± 2.1 | 4.6 ± 0.8 | 2.5 ± 3.0 | Hb E 91.9 ± 2.9 | - | |
| β-thalassemia | 9 (0.11) | 3.7 ± 2.7 | 2.4 ± 1.3 | 93.9 ± 3.9 | - | - | |
| Hb D-Punjab | 1 (<0.1) | - | 3.4 | 1.7 | Hb D-Punjab 94.9 | - | |
| HPFH * | 1 (<0.1) | - | - | 100 | - | - | |
| Other Hb variants | |||||||
| Hb Lepore | 10 (0.12) | 84.9 ± 5.5 | 2.5 ± 0.8 | 4.6 ± 5.5 | Hb Lepore 8.7 ± 2.1 | - | |
| Hb J variant | 7 (<0.1) | 70.3 ± 8.1 | 2.3 ± 0.7 | 0.8 ± 0.4 | Hb J 27.1 ± 7.9 | - | |
| Hb H (--/-α) | 3 (<0.1) | 94.2 ± 0.6 | 1.3 ± 0.5 | 0.4 | Hb H 4.1 ± 1.3 | - | |
| Hb N-Seattle | 2 (<0.1) | 69.3 | 1.7 | 0.2 | Hb N 28.8 | - | |
| Hb Hope | 1 (<0.1) | 37.6 | 4.0 | - | Hb Hope 58.4 | - | |
| Hb C | 1 (<0.1) | 53.7 | 5.3 | 0.9 | Hb C 40.1 | - | |
| Total | 1971 (23.18) | ||||||
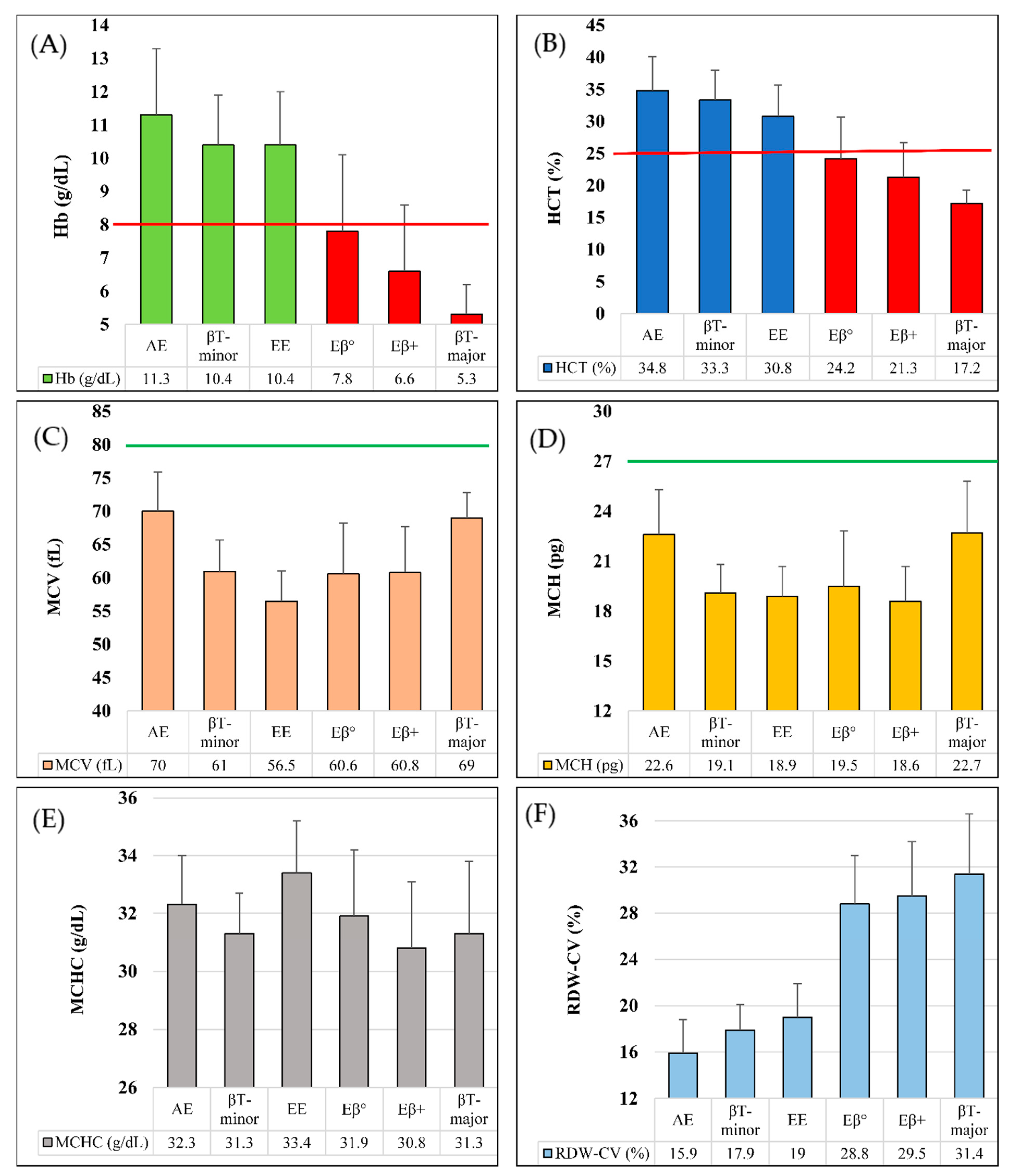
| Inherited Hb Disorders | Age * (Years), Median (IQR) | RBC (106/μL) Mean ± SD | Hb (g/dL) Mean ± SD | HCT (%) Mean ± SD | MCV (fL) Mean ± SD | MCH (pg) Mean ± SD | MCHC (g/dL) Mean ± SD | RDWCV (%) Mean ± SD |
|---|---|---|---|---|---|---|---|---|
| Hb E (Heterozygote) (n = 907) | 24 (5.5–36) | 5.0 ± 0.6 | 11.3 ± 2.0 | 34.8 ± 5.3 | 70.0 ± 5.9 | 22.6 ± 2.7 | 32.3 ± 1.7 | 15.9 ± 2.9 |
| β-thalassemia (Heterozygote) (n = 714) | 27.5 (9–40) | 5.5 ± 0.8 | 10.4 ± 1.5 | 33.3 ± 4.7 | 61.0 ± 4.7 | 19.1 ± 1.7 | 31.3 ± 1.4 | 17.9 ± 2.2 |
| Hb E (Homozygote) (n = 135) | 26 (12–40) | 5.5 ± 0.9 | 10.4 ± 1.6 | 30.8 ± 4.9 | 56.5 ± 4.6 | 18.9 ± 1.8 | 33.4 ± 1.8 | 19.0 ± 2.9 |
| Hb E/β+-thalassemia (n = 101) | 7 (2.5–22) | 3.6 ± 1.0 | 6.6 ± 2.0 | 21.3 ± 5.4 | 60.8 ± 6.9 | 18.6 ± 2.1 | 30.8 ± 2.3 | 29.5 ± 4.7 |
| Hb E/β0-thalassemia (n = 33) | 5 (1.25–17) | 3.9 ± 0.9 | 7.8 ± 2.3 | 24.2 ± 6.5 | 60.6 ± 7.6 | 19.5 ± 3.3 | 31.9 ± 2.3 | 28.8 ± 4.2 |
| β-thalassemia (Homozygote) (n = 9) | 0.7 (0.5–1) | 2.4 ± 0.6 | 5.3 ± 0.9 | 17.2 ± 2.1 | 69.0 ± 3.8 | 22.7 ± 3.1 | 32.0 ± 2.5 | 31.4 ± 5.2 |
| Hb D-Punjab (Heterozygote) (n = 16) | 6.5 (2–24.5) | 4.7 ± 0.8 | 10.2 ± 2.3 | 31.1 ± 5.2 | 66.8 ± 9.1 | 21.8 ± 4.2 | 32.5 ± 2.5 | 18.9 ± 4.2 |
| HPFH (Heterozygote) (n = 13) | 28 (22–41) | 4.9 ± 1.1 | 11.0 ± 2.4 | 35.9 ± 7.2 | 74.2 ± 10.1 | 22.8 ± 2.9 | 30.8 ± 1.1 | 19.5 ± 3.4 |
| Hb S (Heterozygote) (n = 9) | 24 (4–28) | 5.1 ± 0.4 | 12.2 ± 2.8 | 36.9 ± 7.3 | 73.0 ± 12.6 | 24.1 ± 4.7 | 32.9 ± 1.9 | 16.1 ± 4.1 |
| Hb Lepore (Heterozygote) (n = 10) | 24 (5.5–26) | 5.4 ± 1.0 | 11.0 ± 2.7 | 35.1 ± 7.8 | 65.4 ± 5.2 | 19.9 ± 1.8 | 30.8 ± 1.5 | 20.2 ± 3.7 |
| Hb E/Hb S (n = 5) | 65 (6–66) | 3.1 ± 1.2 | 9.5 ± 1.5 | 28.0 ± 4.6 | 83.0 ± 22.3 | 28.1 ± 9.1 | 34.5 ± 0.9 | 20.4 ± 7.7 |
| Hb H (--/-α) (n = 3) | 20 (1–45) | 4.5 ± 0.3 | 8.6 ± 0.2 | 29.9 ± 1.0 | 66.8 ± 1.9 | 19.2 ± 1.5 | 28.7 ± 1.5 | 22.6 ± 4.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasan, A.; Ahmed, J.; Chanda, B.C.; Aniqua, M.; Akther, R.; Dhar, P.K.; Hasan, K.A.B.; Siddique, A.R.; Islam, M.Z.; Urmee, S.Z.; et al. Spectrum of Thalassemia and Hemoglobinopathy Using Capillary Zone Electrophoresis: A Facility-Based Single Centred Study at icddr,b in Bangladesh. Thalass. Rep. 2023, 13, 131-143. https://doi.org/10.3390/thalassrep13020012
Hasan A, Ahmed J, Chanda BC, Aniqua M, Akther R, Dhar PK, Hasan KAB, Siddique AR, Islam MZ, Urmee SZ, et al. Spectrum of Thalassemia and Hemoglobinopathy Using Capillary Zone Electrophoresis: A Facility-Based Single Centred Study at icddr,b in Bangladesh. Thalassemia Reports. 2023; 13(2):131-143. https://doi.org/10.3390/thalassrep13020012
Chicago/Turabian StyleHasan, Anamul, Jigishu Ahmed, Bikash Chandra Chanda, Maisha Aniqua, Raisa Akther, Palash Kanti Dhar, Kazi Afrin Binta Hasan, Abdur Rouf Siddique, Md. Zahidul Islam, Sharmine Zaman Urmee, and et al. 2023. "Spectrum of Thalassemia and Hemoglobinopathy Using Capillary Zone Electrophoresis: A Facility-Based Single Centred Study at icddr,b in Bangladesh" Thalassemia Reports 13, no. 2: 131-143. https://doi.org/10.3390/thalassrep13020012