
Co-Inheritance of Heterozygous β0-Thalassemia with Single Functional α-Globin Gene: Challenges of Carrier Detection in Pre-Marital Screening Program for Thalassemia

Abstract
:1. Introduction
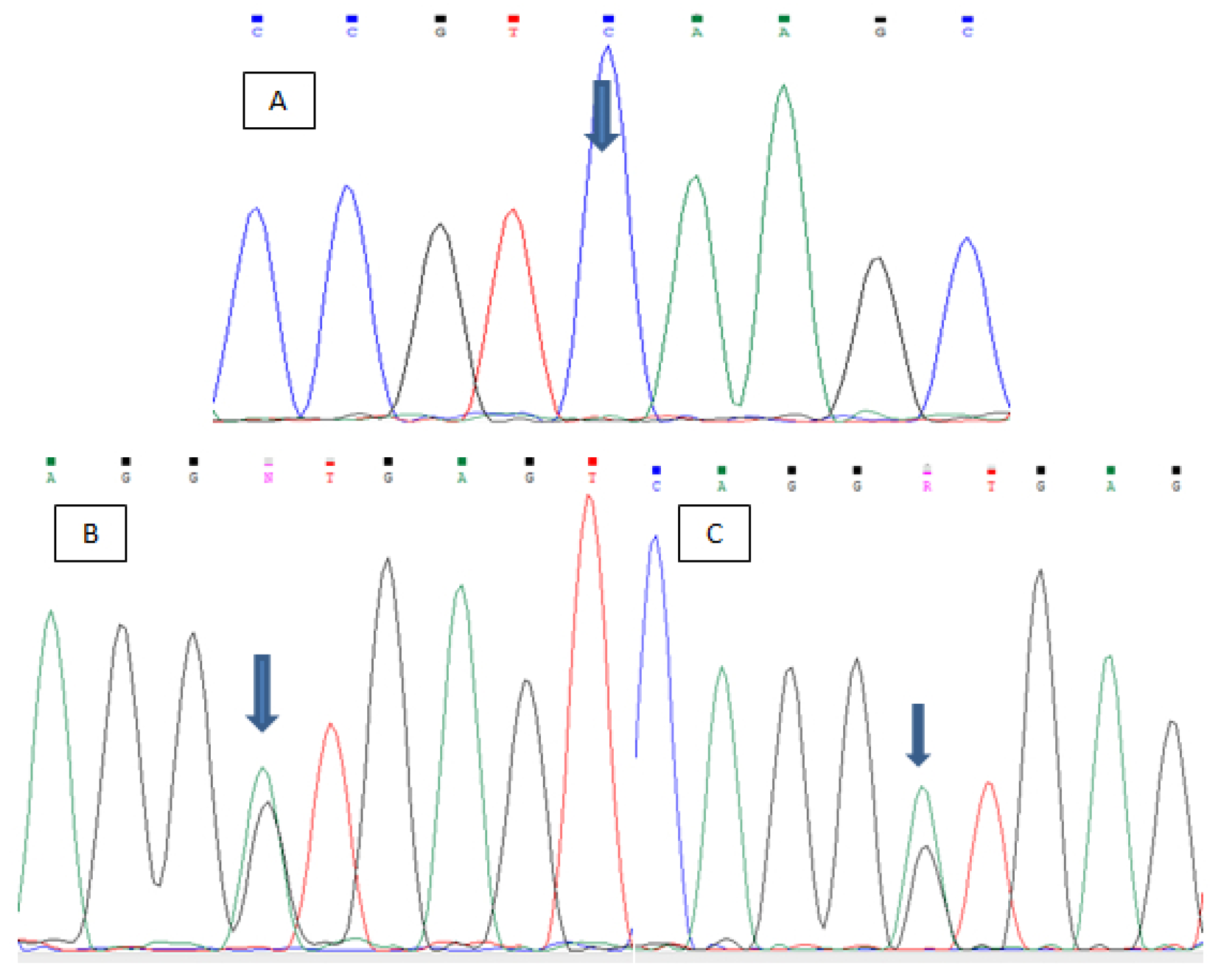
2. Case Presentation
3. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weatherall, D.J. The definition and epidemiology of non-transfusion-dependent thalassemia. Blood Rev. 2012, 26, S3–S6. [Google Scholar] [CrossRef]
- Mettananda, S.; Gibbons, R.J.; Higgs, D.R. α-Globin as a molecular target in the treatment of β-thalassemia. Blood J. Am. Soc. Hematol. 2015, 125, 3694–3701. [Google Scholar] [CrossRef]
- Abolghasemi, H.; Amid, A.; Zeinali, S.; Radfar, M.H.; Eshghi, P.; Rahiminejad, M.S.; Ehsani, M.A.; Najmabadi, H.; Akbari, M.T.; Afrasiabi, A. Thalassemia in Iran: Epidemiology, prevention, and management. J. Pediatric Hematol./Oncol. 2007, 29, 233–238. [Google Scholar] [CrossRef]
- Jalali, H.; Mahdavi, M.R.; Roshan, P.; Kosaryan, M.; Karami, H.; Mahdavi, M. Alpha thalassemia gene mutations in neonates from Mazandaran, Iran, 2012. Hematology 2012, 19, 192–195. [Google Scholar] [CrossRef] [PubMed]
- Mahdavi, M.R.; Kowsarian, M.; Karami, H.; Mohseni, A.; Vahidshahi, K.; Roshan, P.; Hojjati, M.T.; Ebrahimzadeh, M.A. Prevalence of hemoglobin alpha-chain gene deletion in neonates in North of Iran. Eur. Rev. Med. Pharmacol. Sci. 2010, 14, 871–875. [Google Scholar] [PubMed]
- Samavat, A.; Modell, B. Iranian national thalassaemia screening programme. BMJ 2004, 329, 1134–1137. [Google Scholar] [CrossRef] [PubMed]
- Moghaddam, Z.K.; Bayat, N.; Valaei, A.; Kordafshari, A.; Zarbakhsh, B.; Zeinali, S.; Karimipoor, M. Co-inheritance of α-and β-thalassemia: Challenges in prenatal diagnosis of thalassemia. Iran J. Blood Canc 2012, 2, 81–84. [Google Scholar]
- Zhong, L.; Gan, X.; Xu, L.; Liang, C.; Xie, Y.; Lin, W.; Chen, P.; Liu, M. The phenomena of balanced effect between α-globin gene and of β-globin gene. BMC Med. Genet. 2018, 19, 145. [Google Scholar] [CrossRef] [PubMed]
- Chong, S.S.; Boehm, C.D.; Higgs, D.R.; Cutting, G.R. Single-tube multiplex-PCR screen for common deletional determinants of α-thalassemia. Blood 2000, 95, 360–362. [Google Scholar] [CrossRef] [PubMed]
- Habibzadeh, F.; Yadollahi, M.; Merat, A.; Haghshenas, M. Thalassemia in Iran: Overviwe. Arch. Iran. Med. 1998, 1, 27–33. [Google Scholar]
- Eshghi, P.; Sanei Moghaddam, E.; Hasheini, S. Prevalence of hemoglobinopathies in Sistan and Balouchistan province in the southeast of Iran. Sci. J. Iran. Blood Transfus. Organ. 2013, 9, 406–413. [Google Scholar]
- Bazi, A.; Miri-Moghaddam, E. Spectrum of β-thalassemia Mutations in Iran, an Update. Iran. J. Pediatric Hematol. Oncol. 2016, 6, 190–202. [Google Scholar]
- Miri-Moghaddam, E.; Naderi, M.; Izadi, S.; Mashhadi, M. Causes of new cases of major thalassemia in sistan and balouchistan province in South-East of iran. Iran. J. Public Health 2012, 41, 67. [Google Scholar] [PubMed]
- Mahdavi, M.R.; Karami, H.; Akbari, M.T.; Jalali, H.; Roshan, P. Detection of rare beta globin gene mutation [+ 22 5UTR (G> A)] in an infant, despite prenatal screening. Case Rep. Hematol. 2013, 2013, 906292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.-Z.; Liao, C.; Zhou, J.-Y.; Xie, X.-M.; Li, J. Mild hemoglobin H-constant spring disease with β-thalassemia a case report. Ann. Hematol. 2011, 90, 123–124. [Google Scholar] [CrossRef] [PubMed]
- Cao, A.; Galanello, R. Beta-thalassemia. Genet. Med. 2010, 12, 61–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Age (y) | RBC (×106/µL) | Hb (g/dL) | MCV (fl) | MCH (pg) | Hb-A (%) | Hb-A2 (%) | Hb-F (%) | Other Hb (%) | |
|---|---|---|---|---|---|---|---|---|---|
| Male | 27 | 6.1 | 12 | 63.1 | 19.4 | 94.5 | 5.1 | >0.5 | - |
| Female | 23 | 6.8 | 10.3 | 46.9 | 15.1 | 93.8 | 3.2 | 2.2 | 0.8 (HbCS) |
| Reference Range | - | 4.5–6.3 | 12–16 | 80–95 | 27–32 | 94.5–98 | <3.5 | <2 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jalali, H.; Karami, H.; Mahdavi, M.R.; Mahdavi, M. Co-Inheritance of Heterozygous β0-Thalassemia with Single Functional α-Globin Gene: Challenges of Carrier Detection in Pre-Marital Screening Program for Thalassemia. Thalass. Rep. 2022, 12, 101-104. https://doi.org/10.3390/thalassrep12030015
Jalali H, Karami H, Mahdavi MR, Mahdavi M. Co-Inheritance of Heterozygous β0-Thalassemia with Single Functional α-Globin Gene: Challenges of Carrier Detection in Pre-Marital Screening Program for Thalassemia. Thalassemia Reports. 2022; 12(3):101-104. https://doi.org/10.3390/thalassrep12030015
Chicago/Turabian StyleJalali, Hossein, Hossein Karami, Mohammad Reza Mahdavi, and Mehrad Mahdavi. 2022. "Co-Inheritance of Heterozygous β0-Thalassemia with Single Functional α-Globin Gene: Challenges of Carrier Detection in Pre-Marital Screening Program for Thalassemia" Thalassemia Reports 12, no. 3: 101-104. https://doi.org/10.3390/thalassrep12030015