
Leg Ulcers in Sickle Cell Disease: A Multifactorial Analysis Highlights the Hemolytic Profile

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Clinical Data
2.3. Laboratory Determinations
2.4. Statistical Analyses
3. Results
3.1. Clinical Characterization of SLUs
3.2. Laboratory Parameters in SLU− and SLU+ Patients
3.3. Genetic Parameters in SLU− and SLU+ Patients
3.4. Hemolytic Index in SLU Occurrence
3.5. Hemolytic Biomarkers and HbF Levels in SLU Recurrence
3.6. Laboratory Biomarkers and Clinical Characteristics of SLU
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sundd, P.; Gladwin, M.T.; Novelli, E.M. Pathophysiology of Sickle Cell Disease. Annu. Rev. Pathol. Mech. Dis. 2019, 14, 263–292. [Google Scholar] [CrossRef]
- Ingram, V.M. Gene Mutations in Human Hæmoglobin: The Chemical Difference Between Normal and Sickle Cell Hæmoglobin. Nature 1957, 180, 326–328. [Google Scholar] [CrossRef]
- Rees, D.C.; Williams, T.N.; Gladwin, M.T. Sickle-cell disease. Lancet 2010, 376, 2018–2031. [Google Scholar] [CrossRef] [PubMed]
- Serjeant, G.R.; Vichinsky, E. Variability of homozygous sickle cell disease: The role of alpha and beta globin chain variation and other factors. Blood Cells Mol. Dis. 2018, 70, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Hunt, J.A.; Ingram, V.M. Allelomorphism and the chemical differences of the human hæmoglobins A, S and C. Nature 1958, 181, 1062–1063. [Google Scholar] [CrossRef] [PubMed]
- da Guarda, C.C.; Yahouédéhou, S.C.M.A.; Santiago, R.P.; Neres, J.S.D.S.; Fernandes, C.F.D.L.; Aleluia, M.M.; Figueiredo, C.V.B.; Fiuza, L.M.; Carvalho, S.P.; Oliveira, R.M.D.; et al. Sickle cell disease: A distinction of two most frequent genotypes (HbSS and HbSC). PLoS ONE 2020, 15, e0228399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abboud, M.R. Standard management of sickle cell disease complications. Hematol. Stem Cell Ther. 2020, 13, 85–90. [Google Scholar] [CrossRef]
- Minniti, C.P.; Kato, G.J. Critical Reviews: How we treat sickle cell patients with leg ulcers. Am. J. Hematol. 2015, 91, 22–30. [Google Scholar] [CrossRef]
- Minniti, C.P.; Eckman, J.; Sebastiani, P.; Steinberg, M.; Ballas, S.K. Leg ulcers in sickle cell disease. Am. J. Hematol. 2010, 85, 831–833. [Google Scholar] [CrossRef] [Green Version]
- AlDallal, S.M. Mini review: Leg ulcers—A secondary complication of sickle cell disease. Int. J. Gen. Med. 2019, 12, 279–282. [Google Scholar] [CrossRef]
- Ministry of Health; Secretary of Health Care; Department of Specialized Care. Sickle Cell Disease: Ulcers: Prevention and Treatment, 1st ed.; Ministry of Health: Brasília, Brazil, 2012.
- Senet, P.; Blas-Chatelain, C.; Levy, P.; Manea, E.; Peschanski, M.; Mirault, T.; Stankovic-Stojanovic, K.; Debure, C.; Debbache, K.; Girot, R.; et al. Factors predictive of leg-ulcer healing in sickle cell disease: A multicentre, prospective cohort study. Br. J. Dermatol. 2017, 177, 206–211. [Google Scholar] [CrossRef] [PubMed]
- Antwi-Boasiako, C.; Andemariam, B.; Colombatti, R.; Asare, E.V.; Strunk, C.; Piccone, C.M.; Manwani, D.; Boruchov, D.; Farooq, F.; Urbonya, R.; et al. A study of the geographic distribution and associated risk factors of leg ulcers within an international cohort of sickle cell disease patients: The CASiRe group analysis. Ann. Hematol. 2020, 99, 2073–2079. [Google Scholar] [CrossRef] [PubMed]
- Yahouédéhou, S.C.M.A.; da Guarda, C.C.; Figueiredo, C.V.B.; Santiago, R.P.; Carvalho, S.P.; Fiuza, L.M.; Ndidi, U.S.; Oliveira, R.M.; Carvalho, M.O.S.; Nascimento, V.M.L.; et al. Hydroxyurea alters hematological, biochemical and inflammatory biomarkers in Brazilian children with SCA: Investigating associations with βS haplotype and α-thalassemia. PLoS ONE 2019, 14, e0218040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, B.; Delgadinho, M.; Ferreira, J.; Germano, I.; Miranda, A.; Arez, A.P.; Faustino, P.; Brito, M. Co-Inheritance of alpha-thalassemia and sickle cell disease in a cohort of Angolan pediatric patients. Mol. Biol. Rep. 2020, 47, 5397–5402. [Google Scholar] [CrossRef]
- Bryan, N.S.; Grisham, M.B. Methods to detect nitric oxide and its metabolites in biological samples. Free Radic. Biol. Med. 2007, 43, 645–657. [Google Scholar] [CrossRef] [Green Version]
- Chong, S.S.; Boehm, C.D.; Higgs, D.R.; Cutting, G.R. Single-tube multiplex-PCR screen for common deletional determinants of α-thalassemia. Blood 2000, 95, 360–362. [Google Scholar] [CrossRef]
- Genser, B.; Cooper, P.J.; Yazdanbakhsh, M.; Barreto, M.L.; Rodrigues, L.C. A guide to modern statistical analysis of immunological data. BMC Immunol. 2007, 8, 27. [Google Scholar] [CrossRef] [Green Version]
- Cumming, V.; King, L.; Fraser, R.; Serjeant, G.; Reid, M. Venous incompetence, poverty and lactate dehydrogenase in Jamaica are important predictors of leg ulceration in sickle cell anaemia. Br. J. Haematol. 2008, 142, 119–125. [Google Scholar] [CrossRef]
- Koshy, M.; Entsuah, R.; Koranda, A.; Kraus, A.; Johnson, R.; Bellvue, R.; Flournoy-Gill, Z.; Levy, P. Leg ulcers in patients with sickle cell disease. Blood 1989, 74, 1403–1408. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.P.; Minniti, C.P. Leg Ulceration in Sickle Cell Disease: An Early and Visible Sign of End-Organ Disease. In Sickle Cell Disease—Pain and Common Chronic Complications; InTech: London, UK, 2016; pp. 171–202. [Google Scholar] [CrossRef]
- Olatunya, O.S.; Albuquerque, D.M.; Adekile, A.D.; Costa, F.F. Evaluation of sociodemographic, clinical, and laboratory markers of sickle leg ulcers among young nigerians at a tertiary health institution. Niger. J. Clin. Pract. 2018, 21, 882–887. [Google Scholar]
- Vi, J.G.T.; Nolan, V.G.; Mendelsohn, L.; Kato, G.J.; Gladwin, M.T.; Steinberg, M.H. Chronic Hyper-Hemolysis in Sickle Cell Anemia: Association of Vascular Complications and Mortality with Less Frequent Vasoocclusive Pain. PLoS ONE 2008, 3, e2095. [Google Scholar] [CrossRef] [Green Version]
- Kato, G.; Steinberg, M.H.; Gladwin, M.T. Intravascular hemolysis and the pathophysiology of sickle cell disease. J. Clin. Investig. 2017, 127, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Minniti, C.P.; Vi, J.G.T.; Hildesheim, M.; O’Neal, P.; Wilson, J.; Castro, O.; Gordeuk, V.R.; Kato, G. Laboratory and echocardiography markers in sickle cell patients with leg ulcers. Am. J. Hematol. 2011, 86, 705–708. [Google Scholar] [CrossRef]
- Minniti, C.P.; Delaney, K.-M.H.; Gorbach, A.M.; Xu, D.; Lee, C.-C.; Malik, N.; Koroulakis, A.; Antalek, M.; Maivelett, J.; Peters-Lawrence, M.; et al. Vasculopathy, inflammation, and blood flow in leg ulcers of patients with sickle cell anemia. Am. J. Hematol. 2013, 89, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubert, M.; Elion, J.; Tolo, A.; Diallo, D.A.; Diop, S.; Diagne, I.; Sanogo, I.; Belinga, S.; Guifo, O.; Wamba, G.; et al. Degree of anemia, indirect markers of hemolysis, and vascular complications of sickle cell disease in Africa. Blood 2017, 130, 2215–2223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minniti, C.P.; Sable, C.; Campbell, A.; Rana, S.; Ensing, G.; Dham, N.; Onyekwere, O.; Nouraie, M.; Kato, G.; Gladwin, M.T.; et al. Elevated tricuspid regurgitant jet velocity in children and adolescents with sickle cell disease: Association with hemolysis and hemoglobin oxygen desaturation. Haematologica 2009, 94, 340–347. [Google Scholar] [CrossRef]
- Adôrno, E.V.; Zanette, Â.; Lyra, I.; Seixas, M.O.; Reis, M.G.; Gonçalves, M.S.; Ii, I. Clinical and molecular characteristics of sickle cell anemia in the northeast of Brazil. Genet. Mol. Biol. 2008, 31, 621–625. [Google Scholar] [CrossRef] [Green Version]
- Aslan, M.; Ryan, T.M.; Adler, B.; Townes, T.M.; Parks, D.A.; Thompson, J.A.; Tousson, A.; Gladwin, M.T.; Patel, R.P.; Tarpey, M.M.; et al. Oxygen radical inhibition of nitric oxide-dependent vascular function in sickle cell disease. Proc. Natl. Acad. Sci. USA 2001, 98, 15215–15220. [Google Scholar] [CrossRef] [Green Version]
- Theocharidou, E.; Suddle, A.R. The Liver in Sickle Cell Disease. Clin. Liver Dis. 2019, 23, 177–189. [Google Scholar] [CrossRef]
- Ballas, S.K. Sickle cell anemia with few painful crises is characterized by decreased red cell deformability and increased number of dense cells. Am. J. Hematol. 1991, 36, 122–130. [Google Scholar] [CrossRef]
- Donaldson, A.; Thomas, P.; Serjeant, B.E.; Serjeant, G.R. Fetal hemoglobin in homozygous sickle cell disease: A study of patients with low HBF levels. Clin. Lab. Haematol. 2001, 23, 285–289. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Leg ulcer age at onset (years) | N (%) | Leg ulcer location | N (%) | Origin | N (%) |
| 13 to 17 | 7 (41.2) | Right malleolus | 6 (35.3) | Traumatic | 10 (58.8) |
| 18 to 30 | 6 (35.2) | Left malleolus | 5 (29.4) | Spontaneous | 7 (41.2) |
| 31 to 46 | 4 (23.6) | Right and left malleolus | 4 (23.5) | ||
| Atypical | 2 (11.8) | ||||
| Leg ulcer duration (months) | N (%) | Leg ulcer recurrence (episodes) | N (%) | Wound bed tissue * | N (%) |
| 1 to 3 | 3 (17.6) | 1 to 3 | 14 (82.4) | Necrotic tissue | 7 (53.8) |
| 4 to 12 | 4 (23.5) | 4 to 7 | 3 (17.6) | Sloughy tissue | 13 (100) |
| 13 to 24 | 4 (23.5) | Granulation tissue | 6 (46.1) | ||
| 25 to 36 | 2 (11.9) | Epithelial tissue | 1 (7.6) | ||
| >37 | 4 (23.5) | ||||
| Exudate characteristics * | N (%) | Lesion edges * | N (%) | Perilesional skin * | N (%) |
| Serous | 13 (100) | Maceration | 4 (30.7) | Hyperpigmentation | 10 (77.0) |
| Bloody | 1 (7.6) | Dehydration | 5 (38.4) | Dryness | 5 (38.4) |
| Purulence | 7 (53.8) | Healthy | 6 (46.1) | Edema | 5 (38.4) |
| Healthy | 3 (23.0) |
| Laboratory Parameters | SLU− (N = 51) | SLU+ (N = 17) | p-Value |
|---|---|---|---|
| Mean ± SD | Mean ± SD | ||
| Hb pattern | |||
| HbS, % | 62.61 ± 17.52 | 76.55 ± 15.73 | 0.005 |
| HbF, % | 4.46 ± 5.70 | 9.52 ± 7.96 | 0.024 |
| Erythrocytes | |||
| RBC, ×106/μL | 3.71 ± 1.03 | 2.62 ± 0.92 | <0.001 |
| Hemoglobin, g/dL | 10.59 ± 2.89 | 9.19 ± 2.51 | 0.037 * |
| Hematocrit, % | 32.21 ± 7.36 | 27.75 ± 7.38 | 0.010 |
| MCV, fL | 92.20 ± 13.43 | 109.07 ± 17.08 | <0.001 |
| MCH, pg | 30.47 ± 4.64 | 36.07 ± 5.75 | <0.001 |
| MCHC, g/dL | 33.04 ± 0.94 | 33.07 ± 0.75 | 0.896 |
| RDW, % | 17.53 ± 2.76 | 17.25 ± 3.05 | 0.733 |
| Reticulocyte count, % | 3.57 ± 1.41 | 2.95 ± 1.22 | 0.110 |
| Reticulocyte count, /mL | 137,078 ± 72,073 | 81,537 ± 51,981 | 0.005 |
| Leukocytes | |||
| WBC, /μL | 9226 ± 3369.9 | 8458 ± 3786.8 | 0.433 |
| Segment count, /μL | 4639 ± 2329.42 | 3184 ± 1621.44 | 0.020 |
| Eosinophil count, /μL | 352 ± 441.83 | 327 ± 337.30 | 0.832 |
| Basophils count, /μL | 28.24 ± 55.58 | 24.24 ± 50.75 | 0.794 |
| Lymphocyte count, /μL | 3375 ± 1394.17 | 4021 ± 2094.30 | 0.249 |
| Monocyte count, /μL | 831 ± 372.78 | 901 ± 519.47 | 0.546 |
| Platelets | |||
| Platelet count, ×103/μL | 331.55 ± 137.34 | 426.12 ± 184.54 | 0.028 |
| MPV, fL | 9.87 ± 1.23 | 9.41 ± 0.99 | 0.165 |
| Laboratory Parameters | SLU− (N = 48) | SLU+ (N = 16) | p-Value |
|---|---|---|---|
| Mean ± SD | Mean ± SD | ||
| Hemolysis markers | |||
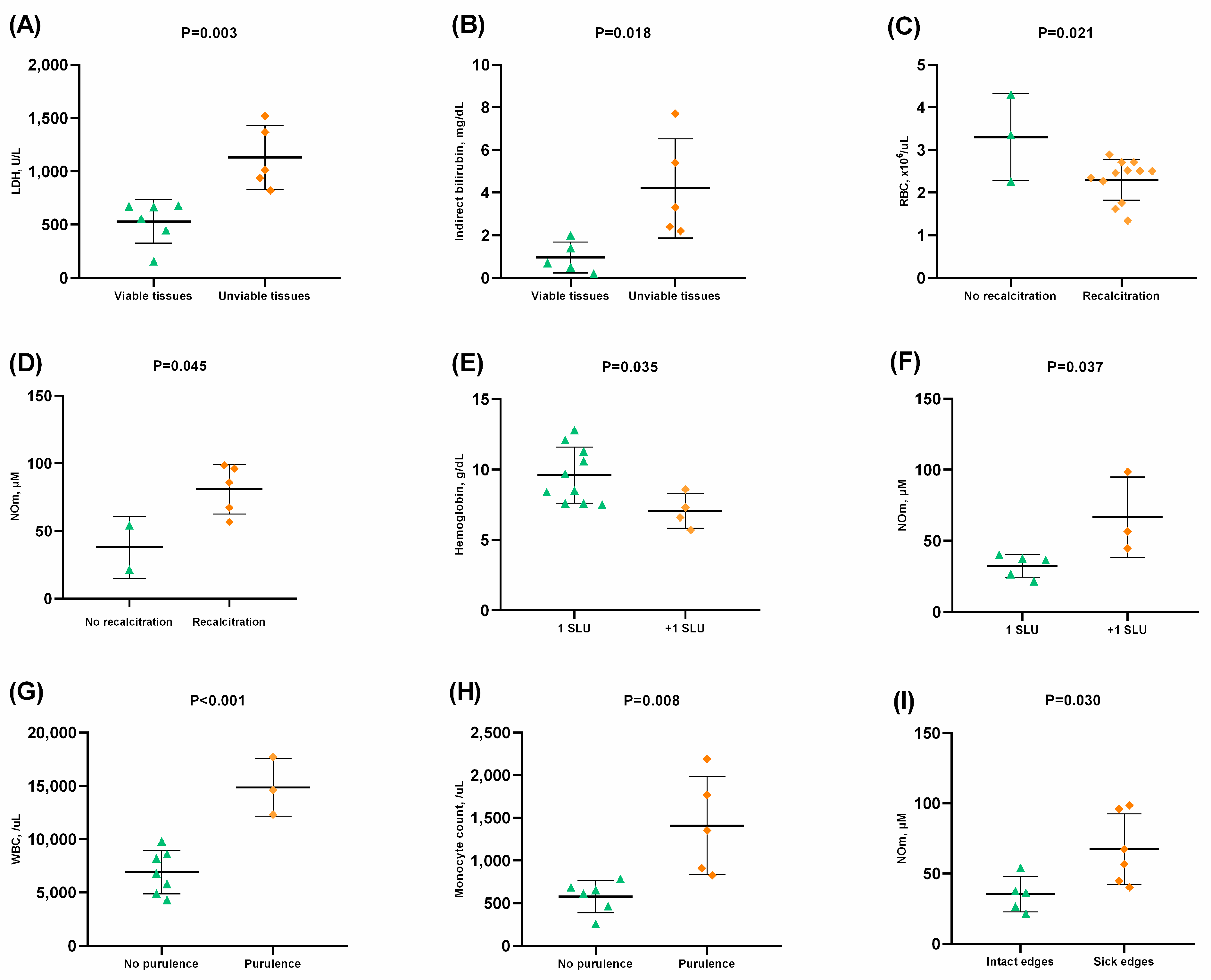
| LDH, U/L | 579.33 ± 326.14 | 763.19 ± 347.95 | 0.021 * |
| Total bilirubin, mg/dL | 1.73 ± 2.07 | 2.88 ± 2.20 | 0.024 * |
| Direct bilirubin, mg/dL | 0.39 ± 0.91 | 0.31 ± 0.16 | 0.119 * |
| Indirect bilirubin, mg/dL | 1.33 ± 1.41 | 2.56 ± 2.16 | 0.022 * |
| Hepatic profile | |||
| AST, U/L | 34.56 ± 28.95 | 55.81 ± 39.88 | 0.025 |
| ALT, U/L | 22.60 ± 15.54 | 23.56 ± 16.10 | 0.833 |
| GGT, U/L | 46.59 ± 33.11 | 88.19 ± 67.71 | 0.030 |
| Renal profile | |||
| Urea, mg/dL | 23.63 ± 9.23 | 26.38 ± 19.57 | 0.450 |
| Creatinine, mg/dL | 0.75 ± 0.19 | 0.78 ± 0.38 | 0.757 |
| Uric acid, mg/dL | 5.22 ± 1.84 | 6.45 ± 2.68 | 0.044 |
| Iron metabolism | |||
| Iron, mcg/dL | 103.58 ± 43.059 | 146.81 ± 68.059 | 0.027 |
| Ferritin, ng/mL | 411.74 ± 488.91 | 867.39 ± 2360.13 | 0.455 |
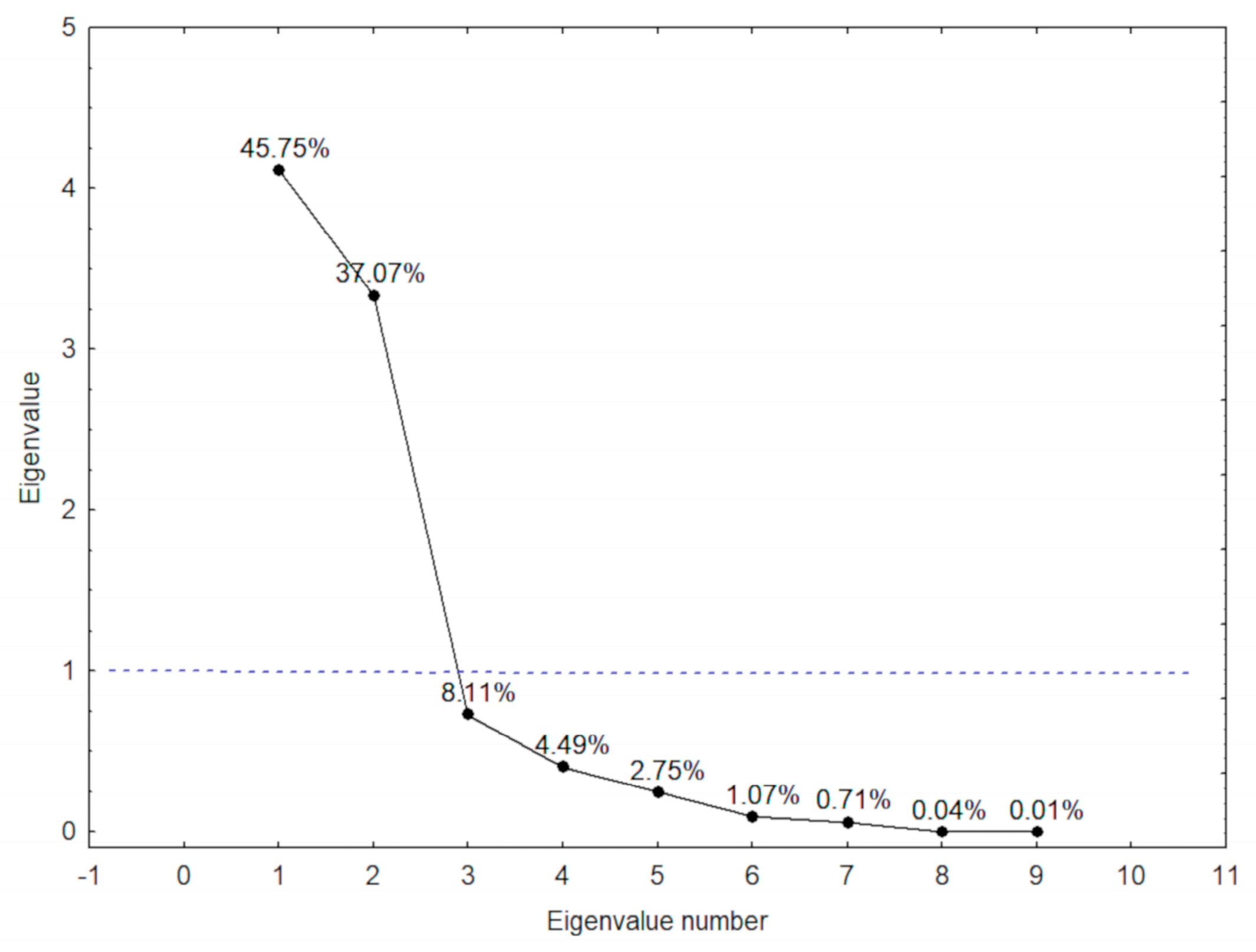
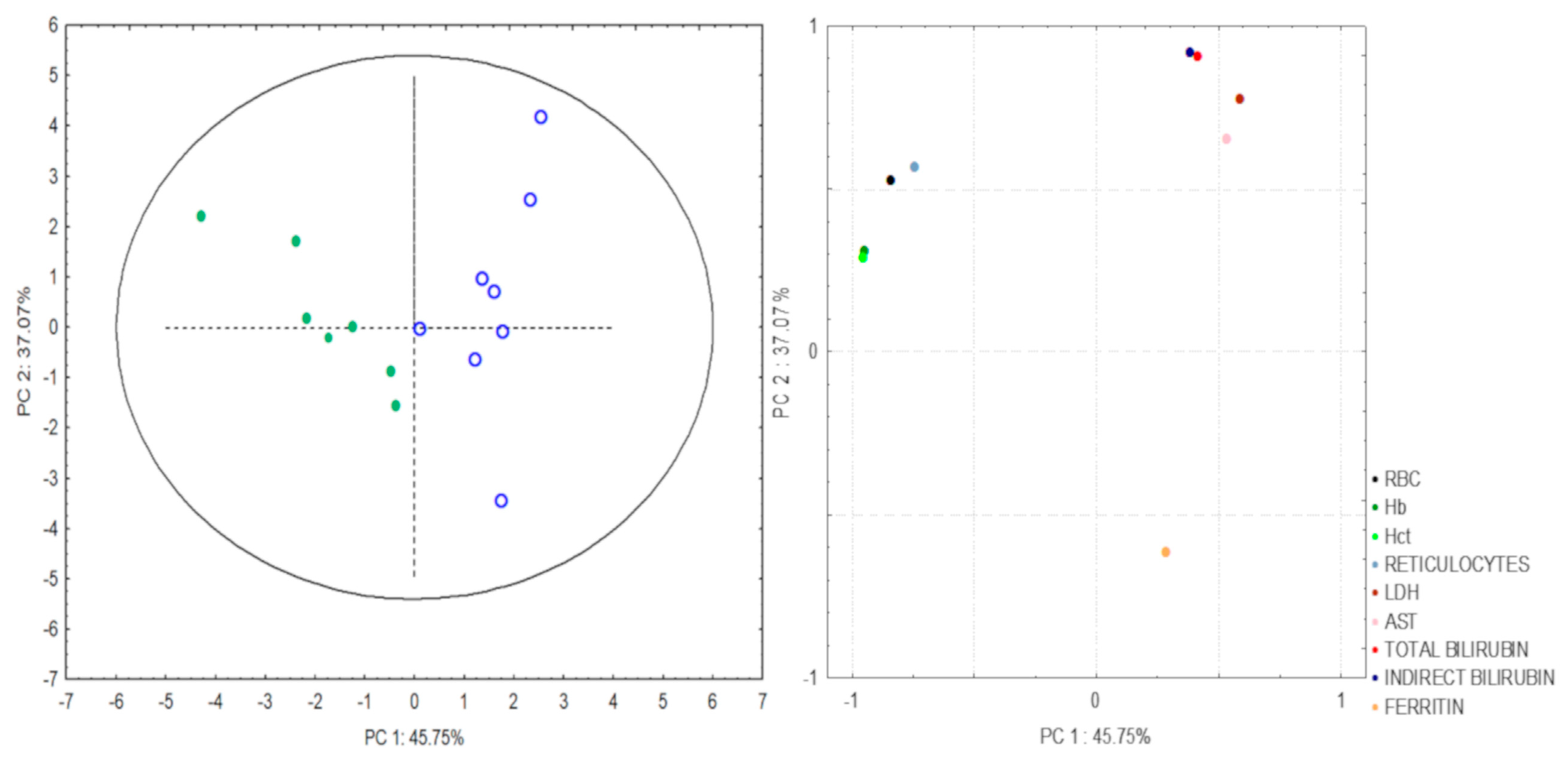
| PC | Variables | r |
|---|---|---|
| 1 | RBC | −0.84 |
| Hb | −0.95 | |
| Hct | −0.96 | |
| Reticulocytes | −0.74 | |
| 2 | LDH | 0.71 |
| AST | 0.59 | |
| Total Bilirubin | 0.84 | |
| Indirect Bilirubin | 0.85 | |
| Ferritin | −0.68 |
| Independent Variables | Dependent Variable | p-Value | β | R2 | p-Value of the Model |
|---|---|---|---|---|---|
| RBC, ×106/mL | Recurrence of SLU | 0.226 | 0.647 | 0.981 | 0.013 |
| Hemoglobin, g/dL | 0.004 | 10.691 | |||
| Hematocrit, % | 0.002 | −12.284 | |||
| LDH, U/L | 0.005 | 1.216 | |||
| Indirect bilirubin, mg/dL | 0.002 | −2.270 | |||
| Ferritin, ng/dL | 0.014 | −0.746 | |||
| HbF, % | 0.011 | −0.911 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santos, E.d.C.; Santana, P.V.B.; Jesus, L.L.S.d.; Melo, G.I.V.; Yahouédéhou, S.C.M.A.; Guarda, C.C.d.; Santiago, R.P.; Fiuza, L.M.; Carvalho, S.P.; Santos, L.O.d.; et al. Leg Ulcers in Sickle Cell Disease: A Multifactorial Analysis Highlights the Hemolytic Profile. Hematol. Rep. 2023, 15, 119-129. https://doi.org/10.3390/hematolrep15010013
Santos EdC, Santana PVB, Jesus LLSd, Melo GIV, Yahouédéhou SCMA, Guarda CCd, Santiago RP, Fiuza LM, Carvalho SP, Santos LOd, et al. Leg Ulcers in Sickle Cell Disease: A Multifactorial Analysis Highlights the Hemolytic Profile. Hematology Reports. 2023; 15(1):119-129. https://doi.org/10.3390/hematolrep15010013
Chicago/Turabian StyleSantos, Edvan do Carmo, Paulo Vinícius Bispo Santana, Laíne Lopes Silva de Jesus, Gabriela Imbassahy Valentim Melo, Sètondji Cocou Modeste Alexandre Yahouédéhou, Caroline Conceição da Guarda, Rayra Pereira Santiago, Luciana Magalhães Fiuza, Suéllen Pinheiro Carvalho, Liz Oliveira dos Santos, and et al. 2023. "Leg Ulcers in Sickle Cell Disease: A Multifactorial Analysis Highlights the Hemolytic Profile" Hematology Reports 15, no. 1: 119-129. https://doi.org/10.3390/hematolrep15010013
{kind=link}