

A Case Report of a 5-Year-Old Girl with Self-Limited Epilepsy with Autonomic Seizures


Abstract
:1. Introduction
2. Case Report
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Panayiotopoulos, C.P.; Aicardi, J. Benign Nocturnal Childhood Occipital Epilepsy: A New Syndrome with Nocturnal Seizures, Tonic Deviation of the Eyes, and Vomiting. J. Child Neurol. 1989, 4, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Covanis, A. Panayiotopoulos Syndrome: A Benign Childhood Autonomic Epilepsy Frequently Imitating Encephalitis, Syncope, Migraine, Sleep Disorder, or Gastroenteritis. Pediatrics 2006, 118, e1237-43. [Google Scholar] [CrossRef]
- Ferrie, C.D.; Caraballo, R.; Covanis, A.; Demirbilek, V.; Dervent, A.; Fejerman, N.; Fusco, L.; Grünewald, R.A.; Kanazawa, O.; Koutroumanidis, M.; et al. Autonomic Status Epilepticus in Panayiotopoulos Syndrome and Other Childhood and Adult Epilepsies: A Consensus View. Epilepsia 2007, 48, 1165–1172. [Google Scholar] [CrossRef]
- Caraballo, R.; Koutroumanidis, M.; Panayiotopoulos, C.P.; Fejerman, N. Idiopathic Childhood Occipital Epilepsy of Gastaut: A Review and Differentiation From Migraine and Other Epilepsies. J. Child Neurol. 2009, 24, 1536–1542. [Google Scholar] [CrossRef] [PubMed]
- Specchio, N.; Wirrell, E.C.; Scheffer, I.E.; Nabbout, R.; Riney, K.; Samia, P.; Guerreiro, M.; Gwer, S.; Zuberi, S.M.; Wilmshurst, J.M.; et al. International League Against Epilepsy Classification and Definition of Epilepsy Syndromes with Onset in Childhood: Position Paper by the ILAE Task Force on Nosology and Definitions. Epilepsia 2022, 63, 1398–1442. [Google Scholar] [CrossRef] [PubMed]
- Covanis, A.; Ferrie, C.D.; Koutroumanidis, M.; Oguni, H.; Panayiotopoulos, C.P. Panayiotopoulos Syndrome and Gastaut Type Idiopathic Childhood Occipital Epilepsy. In Epileptic Syndromes in Infancy, Childhood and Adolescence; Roger, J., Bureau, M., Dravet, C., Genton, P., Tassinari, C.A., Wolf, P., Eds.; John Libbey Eurotext: Montrouge, France, 2005; pp. 227–253. [Google Scholar]
- Panayiotopoulos, C.P. Panayiotopoulos Syndrome: A Common and Benign Childhood Epileptic Syndrome; John Libbey & Company: London, UK, 2002. [Google Scholar]
- Kawakami, S.; Kubota, M.; Terashima, H.; Nagata, C.; Ishiguro, A. Differentiating Early Clinical Features of Panayiotopoulos Syndrome from Acute Encephalopathy. Brain Dev. 2022, 44, 386–390. [Google Scholar] [CrossRef]
- Panayiotopoulos, C.P. Autonomic Seizures and Autonomic Status Epilepticus Peculiar to Childhood: Diagnosis and Management. Epilepsy Behav. 2004, 5, 286–295. [Google Scholar] [CrossRef]
- Panayiotopoulos, C.P. Vomiting as an Ictal Manifestation of Epileptic Seizures and Syndromes. J. Neurol. Neurosurg. Psychiatry 1988, 51, 1448–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panayiotopoulos, C.P. Extraoccipital Benign Childhood Partial Seizures with Ictal Vomiting and Excellent Prognosis. J. Neurol. Neurosurg. Psychiatry 1999, 66, 82–85. [Google Scholar] [CrossRef]
- Rubin, D.I.; Patterson, M.C.; Westmoreland, B.F.; Klass, D.W. Angelman’s Syndrome: Clinical and Electroencephalographic Findings. Electroencephalogr. Clin. Neurophysiol. 1997, 102, 299–302. [Google Scholar] [CrossRef]
- Viani, F.; Romeo, A.; Viri, M.; Mastrangelo, M.; Lalatta, F.; Briscioli, V.; Gobbi, G.; Lanzi, G.; Bettio, D. Seizure and EEG Patterns in Angelman’s Syndrome. J. Child Neurol. 1995, 10, 467–471. [Google Scholar] [CrossRef]
- Wilson, S.J. Rethinking Neurobehavioral Comorbidity in Panayiotopoulos Syndrome. Dev. Med. Child Neurol. 2020, 62, 893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kivity, S.; Ephraim, T.; Weitz, R.; Tamir, A. Childhood Epilepsy with Occipital Paroxysms: Clinical Variants in 134 Patients. Epilepsia 2000, 41, 1522–1533. [Google Scholar] [CrossRef] [PubMed]
- Sanders, S.; Rowlinson, S.; Manidakis, I.; Ferrie, C.D.; Koutroumanidis, M. The Contribution of the EEG Technologists in the Diagnosis of Panayiotopoulos Syndrome (Susceptibility to Early Onset Benign Childhood Autonomic Seizures). Seizure 2004, 13, 565–573. [Google Scholar] [CrossRef] [Green Version]
- Parisi, P.; Ferri, R.; Pagani, J.; Cecili, M.; Montemitro, E.; Villa, M.P. Ictal Video-Polysomnography and EEG Spectral Analysis in a Child with Severe Panayiotopoulos Syndrome. Epileptic Disord. 2005, 7, 333–339. [Google Scholar]
- Panayiotopoulos, C.P. Benign Childhood Focal Seizures and Related Epileptic Syndromes. In Clinical Guide to Epileptic Syndromes and Their Treatment; Springer: London, UK, 2007; pp. 285–318. [Google Scholar]
- Dalla Bernardina, B.; Sgro, V.; Fejerman, N. Epilepsy with Centrotemporal Spikes and Related Syndromes. In Epileptic Syndromes in Infancy, Childhood and Adolescence; John Libbey Eurotext: Paris, France, 2005; pp. 203–225. [Google Scholar]
- Marini, C.; Mei, D.; Temudo, T.; Ferrari, A.R.; Buti, D.; Dravet, C.; Dias, A.I.; Moreira, A.; Calado, E.; Seri, S.; et al. Idiopathic Epilepsies with Seizures Precipitated by Fever and SCN1A Abnormalities. Epilepsia 2007, 48, 1678–1685. [Google Scholar] [CrossRef] [Green Version]
- Mulley, J.C.; Scheffer, I.E.; Petrou, S.; Dibbens, L.M.; Berkovic, S.F.; Harkin, L.A. SCN1A Mutations and Epilepsy. Hum. Mutat. 2005, 25, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Livingston, J.H.; Cross, J.H.; Mclellan, A.; Birch, R.; Zuberi, S.M. A Novel Inherited Mutation in the Voltage Sensor Region of SCN1A Is Associated with Panayiotopoulos Syndrome in Siblings and Generalized Epilepsy with Febrile Seizures Plus. J. Child Neurol. 2009, 24, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Miller, I.O.; Sotero de Menezes, M.A. SCN1A Seizure Disorders. University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Kivity, S.; Oliver, K.L.; Afawi, Z.; Damiano, J.A.; Arsov, T.; Bahlo, M.; Berkovic, S.F. SCN1A Clinical Spectrum Includes the Self-Limited Focal Epilepsies of Childhood. Epilepsy Res. 2017, 131, 9–14. [Google Scholar] [CrossRef]
- Grosso, S.; Orrico, A.; Galli, L.; Di Bartolo, R.; Sorrentino, V.; Balestri, P. SCN1A Mutation Associated with Atypical Panayiotopoulos Syndrome. Neurology 2007, 69, 609–611. [Google Scholar] [CrossRef]
- Panayiotopoulos, C.P.; Michael, M.; Sanders, S.; Valeta, T.; Koutroumanidis, M. Benign Childhood Focal Epilepsies: Assessment of Established and Newly Recognized Syndromes. Brain 2008, 131, 2264–2286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Specchio, N.; Trivisano, M.; Di Ciommo, V.; Cappelletti, S.; Masciarelli, G.; Volkov, J.; Fusco, L.; Vigevano, F. Panayiotopoulos Syndrome: A Clinical, EEG, and Neuropsychological Study of 93 Consecutive Patients. Epilepsia 2010, 51, 2098–2107. [Google Scholar] [CrossRef] [PubMed]
- Valeta, T. Parental Attitude, Reaction and Education in Benign Childhood Focal Seizures. In The Epilepsies: Seizures, Syndromes and Management; Panayiotopoulos, C.P., Ed.; Bladon Medical Publishing: Oxford, UK, 2005; pp. 258–261. [Google Scholar]
- Ferrie, C.; Caraballo, R.; Covanis, A.; Demirbilek, V.; Dervent, A.; Kivity, S.; Koutroumanidis, M.; Martinovic, Z.; Oguni, H.; Verrotti, A.; et al. Panayiotopoulos Syndrome: A Consensus View. Dev. Med. Child Neurol. 2006, 48, 236–240. [Google Scholar] [CrossRef] [Green Version]
- Beardsley, S.J.; Dostal, I.; Cole, J.; Gutierrez, A.; Robson, J. Valproate Use in Women Aged 15–44 Years: An Observational Study in General Practice. BJGP Open 2021, 5, BJGPO.2020.0104. [Google Scholar] [CrossRef]
- Kikumoto, K.; Yoshinaga, H.; Oka, M.; Ito, M.; Endoh, F.; Akiyama, T.; Ohtsuka, Y. EEG and Seizure Exacerbation Induced by Carbamazepine in Panayiotopoulos Syndrome. Epileptic Disord. 2006, 8, 53–56. [Google Scholar]
- Mujawar, Q.M.; Sen, S.; Ali, M.D.; Balakrishnan, P.; Patil, S. Panayiotopoulos Syndrome Presenting with Status Epilepticus and Cardiorespiratory Arrest: A Case Report. Pediatr. Emerg. Care 2011, 27, 754–757. [Google Scholar] [CrossRef]
- García, C.; Rubio, G. Efficacy and Safety of Levetiracetam in the Treatment of Panayiotopoulos Syndrome. Epilepsy Res. 2009, 85, 318–320. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, F.U. Febrile Seizures: Treatment and Prognosis. Epilepsia 2000, 41, 2–9. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Symptoms and Manifestations of SeLEAS | Characteristics and Frequency of Symptoms and Manifestations of SeLEAS | Symptoms and Manifestations of Our Patient 1 |
|---|---|---|
| Age of onset (years) | 1–14 (Mean: 4–5) | 5 years old |
| Duration of seizures | ||
| Less than 4 min | Rare | <5 min duration of seizures |
| ≥5 min | As a rule | |
| Seizures during sleep | >2/3 of cases | Seizures during sleep |
| Features of seizures | ||
| Ictal vomiting | Frequent | Ictal vomiting |
| Deviation of the eyes | Frequent | Deviation of the eyes |
| Impairment of consciousness | Frequent | Drowsiness |
| Visual hallucinations | Rare | |
| Loss of vision | Rare | |
| Autonomic semiology | As a rule | Urinary and faecal incontinence |
| Postictal migraine-like headache | Rare | |
| Seizures evolving into autonomic status epilepticus | Frequent | |
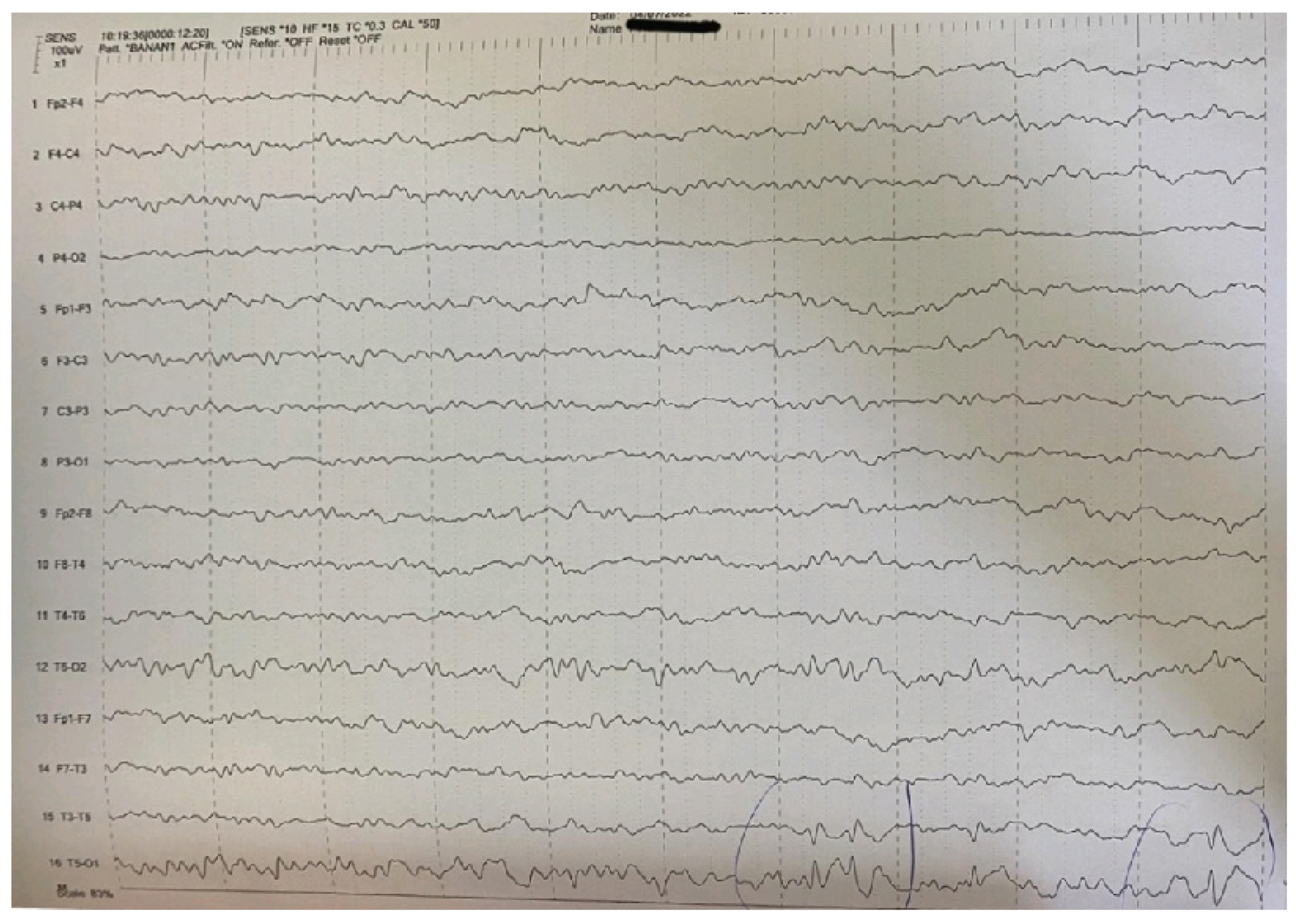
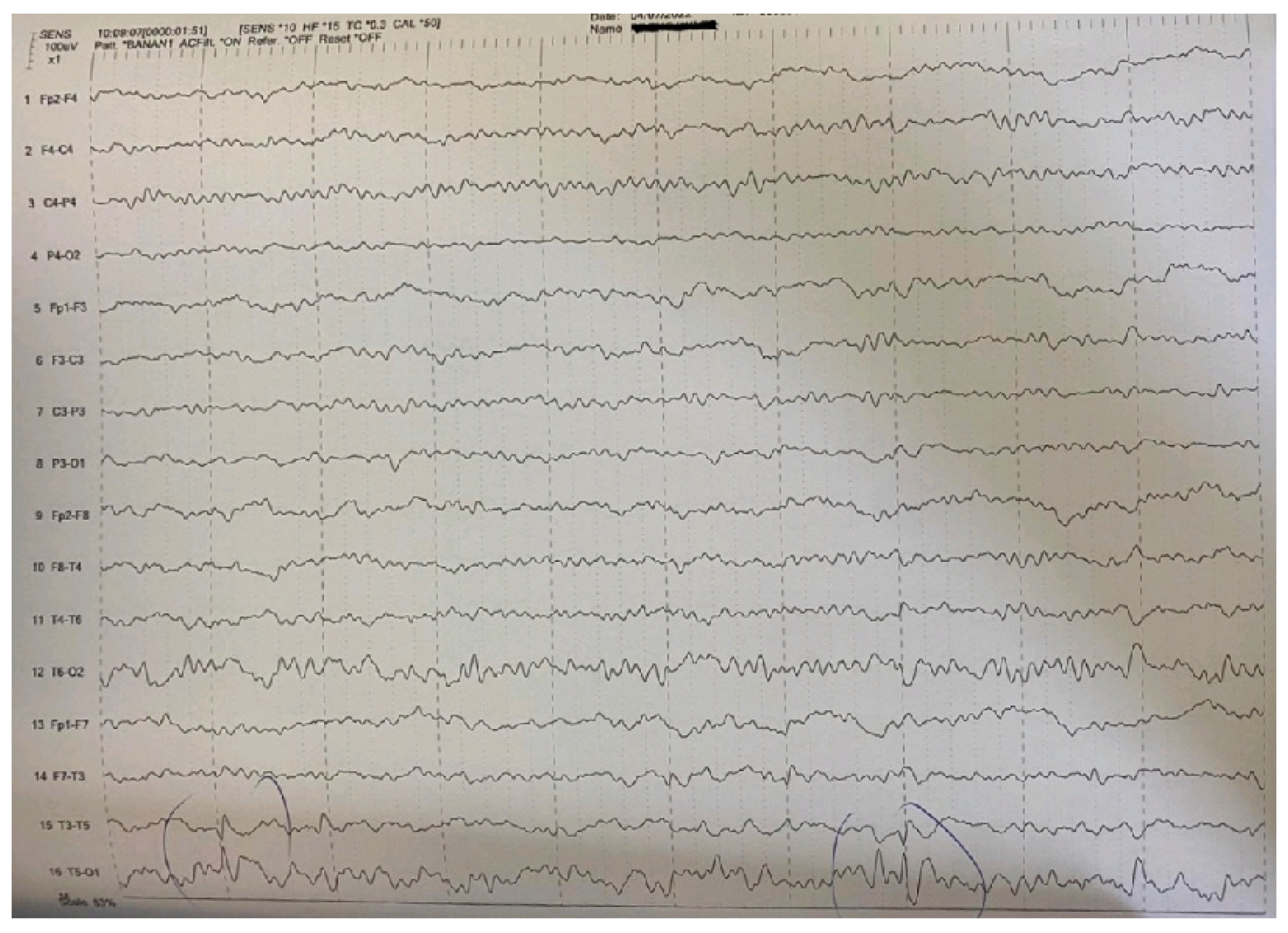
| Main interictal EEG | Multifocal spikes | High-amplitude sharp waves and spike-wave complexes in temporal-occipital areas of the left hemisphere in the EEG. Enhancement of focal abnormalities in temporal-occipital areas of the left hemisphere in the EEG during sleep. |
| Clinical Epilepsy Syndromes | Less Common Presentations |
|---|---|
| Febrile seizures (simple or complex) | Epilepsy with focal seizures |
| Febrile seizures plus (FS+) | Myoclonic–astatic epilepsy (MAE, Doose syndrome) |
| Generalized epilepsy | Lennox–Gastaut syndrome |
| Generalized epilepsy with febrile seizures plus (GEFS+) | Infantile spasms |
| Dravet syndrome | Vaccine-related encephalopathy and seizures |
| Severe myoclonic epilepsy, borderline (SMEB) | |
| Intractable childhood epilepsy with generalized tonic–clonic seizures (ICE-GTC) | |
| Infantile partial seizures with variable foci |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Katsaras, G.; Samartzi, P.; Tsitsani, P. A Case Report of a 5-Year-Old Girl with Self-Limited Epilepsy with Autonomic Seizures. Pediatr. Rep. 2023, 15, 494-501. https://doi.org/10.3390/pediatric15030045
Katsaras G, Samartzi P, Tsitsani P. A Case Report of a 5-Year-Old Girl with Self-Limited Epilepsy with Autonomic Seizures. Pediatric Reports. 2023; 15(3):494-501. https://doi.org/10.3390/pediatric15030045
Chicago/Turabian StyleKatsaras, Georgios, Petrina Samartzi, and Pelagia Tsitsani. 2023. "A Case Report of a 5-Year-Old Girl with Self-Limited Epilepsy with Autonomic Seizures" Pediatric Reports 15, no. 3: 494-501. https://doi.org/10.3390/pediatric15030045