
Mutation in the STXBP1 Gene Associated with Early Onset West Syndrome: A Case Report and Literature Review

 , ,
, ,
Abstract
:1. Introduction
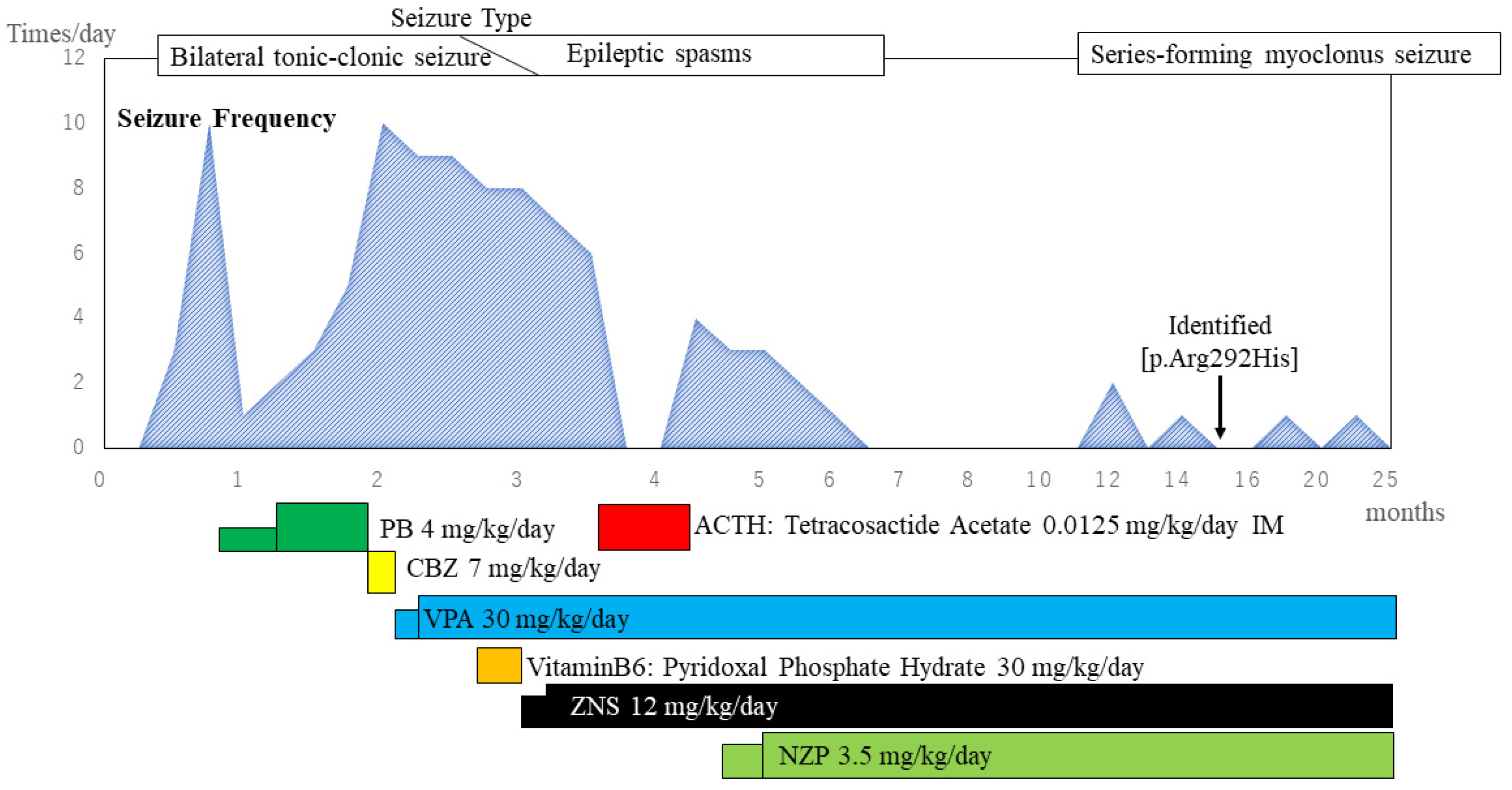
2. Case Presentation
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pressler, R.M.; Cilio, M.R.; Mizrahi, E.M.; Moshé, S.L.; Nunes, M.L.; Plouin, P.; Vanhatalo, S.; Yozawitz, E.; de Vries, L.S.; Puthenveettil Vinayan, K.; et al. The ILAE Classification of Seizures and the Epilepsies: Modification for Seizures in the Neonate. Position Paper by the ILAE Task Force on Neonatal Seizures. Epilepsia 2021, 62, 615–628. [Google Scholar] [CrossRef]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshé, S.L.; et al. ILAE Classification of the Epilepsies: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef]
- Lanoue, V.; Chai, Y.J.; Brouillet, J.Z.; Weckhuysen, S.; Palmer, E.E.; Collins, B.M.; Meunier, F.A. STXBP1 Encephalopathy: Connecting Neurodevelopmental Disorders with α-Synucleinopathies? Neurology 2019, 93, 114–123. [Google Scholar] [CrossRef]
- Hussain, S. Developing a PPI Inhibitor-Based Therapy for STXBP1 Haploinsufficiency-Associated Epileptic Disorders. Front. Mol. Neurosci. 2014, 7, 4–7. [Google Scholar] [CrossRef]
- Morrison-Levy, N.; Borlot, F.; Jain, P.; Whitney, R. Early-Onset Developmental and Epileptic Encephalopathies of Infancy: An Overview of the Genetic Basis and Clinical Features. Pediatric Neurol. 2021, 116, 85–94. [Google Scholar] [CrossRef]
- Pavone, P.; Polizzi, A.; Marino, S.D.; Corsello, G.; Falsaperla, R.; Marino, S.; Ruggieri, M. West Syndrome: A Comprehensive Review. Neurol. Sci. 2020, 41, 3547–3562. [Google Scholar] [CrossRef]
- Wheless, J.W.; Gibson, P.A.; Rosbeck, K.L.; Hardin, M.; O’Dell, C.; Whittemore, V.; Pellock, J.M. Infantile Spasms (West Syndrome): Update and Resources for Pediatricians and Providers to Share with Parents. BMC Pediatrics 2012, 12, 108. [Google Scholar] [CrossRef]
- Stamberger, H.; Nikanorova, M.; Accorsi, P.; Angriman, M.; Benkel-herrenbrueck, I.; Capovilla, G.; Erasmus, C.E.; Fannemel, M.; Giordano, L.; Helbig, K.L.; et al. A Neurodevelopmental Disorder Including Epilepsy. Neurology 2016, 86, 954–962. [Google Scholar] [CrossRef]
- Saitsu, H.; Kato, M.; Okada, I.; Orii, K.E.; Higuchi, T.; Hoshino, H.; Kubota, M.; Arai, H.; Tagawa, T.; Kimura, S.; et al. STXBP1 Mutations in Early Infantile Epileptic Encephalopathy with Suppression-Burst Pattern. Epilepsia 2010, 51, 2397–2405. [Google Scholar] [CrossRef]
- di Meglio, C.; Lesca, G.; Villeneuve, N.; Lacoste, C.; Abidi, A.; Cacciagli, P.; Altuzarra, C.; Roubertie, A.; Afenjar, A.; Renaldo-Robin, F.; et al. Epileptic Patients with de Novo STXBP1 Mutations: Key Clinical Features Based on 24 Cases. Epilepsia 2015, 56, 1931–1940. [Google Scholar] [CrossRef]
- Milh, M.; Villeneuve, N.; Chouchane, M.; Kaminska, A.; Laroche, C.; Barthez, M.A.; Gitiaux, C.; Bartoli, C.; Borges-Correia, A.; Cacciagli, P.; et al. Epileptic and Nonepileptic Features in Patients with Early Onset Epileptic Encephalopathy and STXBP1 Mutations. Epilepsia 2011, 52, 1828–1834. [Google Scholar] [CrossRef]
- O’Brien, S.; Ng-Cordell, E.; Astle, D.E.; Scerif, G.; Baker, K. STXBP1-Associated Neurodevelopmental Disorder: A Comparative Study of Behavioural Characteristics. J. Neurodev. Disord. 2019, 11, 17. [Google Scholar] [CrossRef]
- Deprez, L.; Weckhuysen, S.; Holmgren, P.; Suls, A.; van Dyck, T.; Goossens, D.; Del-Favero, J.; Jansen, A.; Verhaert, K.; Lagae, L.; et al. Clinical Spectrum of Early-Onset Epileptic Encephalopathies Associated with STXBP1 Mutations. Neurology 2010, 75, 1159–1165. [Google Scholar] [CrossRef]
- Weckhuysen, S.; Holmgren, P.; Hendrickx, R.; Jansen, A.C.; Hasaerts, D.; Dielman, C.; de Bellescize, J.; Boutry-Kryza, N.; Lesca, G.; von Spiczak, S.; et al. Reduction of Seizure Frequency after Epilepsy Surgery in a Patient with STXBP1 Encephalopathy and Clinical Description of Six Novel Mutation Carriers. Epilepsia 2013, 54, 74–80. [Google Scholar] [CrossRef]
- Saitsu, H.; Hoshino, H.; Kato, M.; Nishiyama, K.; Okada, I.; Yoneda, Y.; Tsurusaki, Y.; Doi, H.; Miyake, N.; Kubota, M.; et al. Paternal Mosaicism of an STXBP1 Mutation in OS. Clin. Genet. 2011, 80, 484–488. [Google Scholar] [CrossRef]
- Otsuka, M.; Oguni, H.; Liang, J.S.; Ikeda, H.; Imai, K.; Hirasawa, K.; Imai, K.; Tachikawa, E.; Shimojima, K.; Osawa, M.; et al. STXBP1 Mutations Cause Not Only Ohtahara Syndrome but Also West Syndrome-Result of Japanese Cohort Study. Epilepsia 2010, 51, 2449–2452. [Google Scholar] [CrossRef]
- Liu, S.; Wang, L.; Cai, X.T.; Zhou, H.; Yu, D.; Wang, Z. Therapeutic Benefits of ACTH and Levetiracetam in STXBP1 Encephalopathy with a de Novo Mutation. Medicine 2018, 97, e0663. [Google Scholar] [CrossRef]
- Xian, J.; Parthasarathy, S.; Ruggiero, S.M.; Balagura, G.; Fitch, E.; Helbig, K.; Gan, J.; Ganesan, S.; Kaufman, M.C.; Ellis, C.A.; et al. Assessing the Landscape of STXBP1 -Related Disorders in 534 Individuals. Brain 2022, 145, 1668–1683. [Google Scholar] [CrossRef]
- Li, T.; Cheng, M.; Wang, J.; Hong, S.; Li, M.; Liao, S.; Xie, L.; Jiang, L. De Novo Mutations of STXBP1 in Chinese Children with Early Onset Epileptic Encephalopathy. Genes Brain Behav. 2018, 17, e12492. [Google Scholar] [CrossRef]
- Zhang, Q.; Li, J.; Zhao, Y.; Bao, X.; Wei, L.; Wang, J. Gene Mutation Analysis of 175 Chinese Patients with Early-Onset Epileptic Encephalopathy. Clin. Genet. 2017, 91, 717–724. [Google Scholar] [CrossRef] [Green Version]
- Helbig, K.L.; Farwell Hagman, K.D.; Shinde, D.N.; Mroske, C.; Powis, Z.; Li, S.; Tang, S.; Helbig, I. Diagnostic Exome Sequencing Provides a Molecular Diagnosis for a Significant Proportion of Patients with Epilepsy. Genet. Med. 2016, 18, 898–905. [Google Scholar] [CrossRef]
- Trump, N.; McTague, A.; Brittain, H.; Papandreou, A.; Meyer, E.; Ngoh, A.; Palmer, R.; Morrogh, D.; Boustred, C.; Hurst, J.A.; et al. Improving Diagnosis and Broadening the Phenotypes in Early-Onset Seizure and Severe Developmental Delay Disorders through Gene Panel Analysis. J. Med. Genet. 2016, 53, 310–317. [Google Scholar] [CrossRef]
- Michaud, J.L.; Lachance, M.; Hamdan, F.F.; Carmant, L.; Lortie, A.; Diadori, P.; Major, P.; Meijer, I.A.; Lemyre, E.; Cossette, P.; et al. The Genetic Landscape of Infantile Spasms. Hum. Mol. Genet. 2014, 23, 4846–4858. [Google Scholar] [CrossRef]
- Saitsu, H.; Kato, M.; Mizuguchi, T.; Hamada, K.; Osaka, H.; Tohyama, J.; Uruno, K.; Kumada, S.; Nishiyama, K.; Nishimura, A.; et al. De Novo Mutations in the Gene Encoding STXBP1 (MUNC18-1) Cause Early Infantile Epileptic Encephalopathy. Nat. Genet. 2008, 40, 782–788. [Google Scholar] [CrossRef]
- Guiberson, N.G.L.; Pineda, A.; Abramov, D.; Kharel, P.; Carnazza, K.E.; Wragg, R.T.; Dittman, J.S.; Burré, J. Mechanism-Based Rescue of Munc18-1 Dysfunction in Varied Encephalopathies by Chemical Chaperones. Nat. Commun. 2018, 9, 3986. [Google Scholar] [CrossRef]
- Chai, Y.J.; Sierecki, E.; Tomatis, V.M.; Gormal, R.S.; Giles, N.; Morrow, I.C.; Xia, D.; Götz, J.; Parton, R.G.; Collins, B.M.; et al. Munc18-1 Is a Molecular Chaperone for α-Synuclein, Controlling Its Self-Replicating Aggregation. J. Cell Biol. 2016, 214, 705–718. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Stamberger H [6] | Li.T [10] | Zhang Q [11] | Helbig KL [12] | Trump N [13] | Michaud JL [14] | Our patient | |
|---|---|---|---|---|---|---|---|
| (Pt. 42) | (Pt. 8) | (Pt. 123) | (Pt. 77) | (Pt. 64) | (Pt. 9) | ||
| Sex/age at study | F | M/2 mo 3 d | F/0.8 mo | NR | M | M | F/2 Y |
| Age at onset | 8 Y | 15 d | 3 d | Childhood | NR | 7 d | 15 d |
| Seizure semiology:initial | Focal behavior arrest | Eye blinking, lip smacking | NR | Focal Seizures | NR | Focal Seizures Epileptic spasms | FBTCS, FMS, Epileptic spasms, Myoclonic |
| Development prior to Seizure onset | Global developmental delay, severe ID | ||||||
| EEG at onset | Sharp waves and poly spikes waves R central temporal regions (8 Y) | Burst-suppression during sleep, multifocal epileptic activity during awake status (2 mo) | NR | NR | Burst-suppression | Multifocal spikes | Multifocal spikes |
| Response to initial treatment | Oxcarbazepine: seizure free | No effect to PB | Refractory to AEDs | NR | NR | Refractory to AEDs (tried 7 drugs) | No effect to PB |
| MRI findings (age) | Normal (5.3 Y) | Normal (2 mo) | NR | NR | NR | Atrophy, Basal ganglia hyperintensities | Normal (3 mo) |
| Evolution of AEDs and other regimens | Not change | TPM, LEV, NZP, and Dex, MP, PRD | NR | NR | NR | NR | CBZ, Vitamin B6, VPA, ZNS, NZP |
| Clinical diagnosis | Non-convulsive status epilepticus | Ohtahara syndrome with evolution to West syndrome | Ohtahara syndrome | Early infantile epileptic encephalopathy | Early infantile epileptic encephalopathy | West syndrome | West syndrome |
| Current treatment | Oxcarbazepine | LEV, NZP | NR | NR | NR | NR | VPA, ZNS, NZP |
| Current status | Seizure free | Seizure free | Seizure not free | NR | NR | NR | Seizure not free |
| Clinical examination at the end of follow up | 9 Y. Non ambulatory, No speech, Truncal and limb ataxia, Hyperreflexia hypotonia, Pubertas praecox | 1 y 3 mo. Poor head control, unable to sit independently, poor social contact | Global developmental delay | Global developmental delay | Severe Global developmental delay | Global developmental delay, moderate ID from infancy | 2 y 1 mo. Global developmental delay |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takeda, K.; Miyamoto, Y.; Yamamoto, H.; Iwasaki, T.; Sumitomo, N.; Takeshita, E.; Ishii, A.; Hirose, S.; Shimizu, N. Mutation in the STXBP1 Gene Associated with Early Onset West Syndrome: A Case Report and Literature Review. Pediatr. Rep. 2022, 14, 386-395. https://doi.org/10.3390/pediatric14040046
Takeda K, Miyamoto Y, Yamamoto H, Iwasaki T, Sumitomo N, Takeshita E, Ishii A, Hirose S, Shimizu N. Mutation in the STXBP1 Gene Associated with Early Onset West Syndrome: A Case Report and Literature Review. Pediatric Reports. 2022; 14(4):386-395. https://doi.org/10.3390/pediatric14040046
Chicago/Turabian StyleTakeda, Kanako, Yusaku Miyamoto, Hisako Yamamoto, Toshiyuki Iwasaki, Noriko Sumitomo, Eri Takeshita, Atsushi Ishii, Shinichi Hirose, and Naoki Shimizu. 2022. "Mutation in the STXBP1 Gene Associated with Early Onset West Syndrome: A Case Report and Literature Review" Pediatric Reports 14, no. 4: 386-395. https://doi.org/10.3390/pediatric14040046