
Autophagy and Apoptosis in Inflammatory Bowel Disease


{kind=link}
{kind=link}
Abstract
:1. Introduction
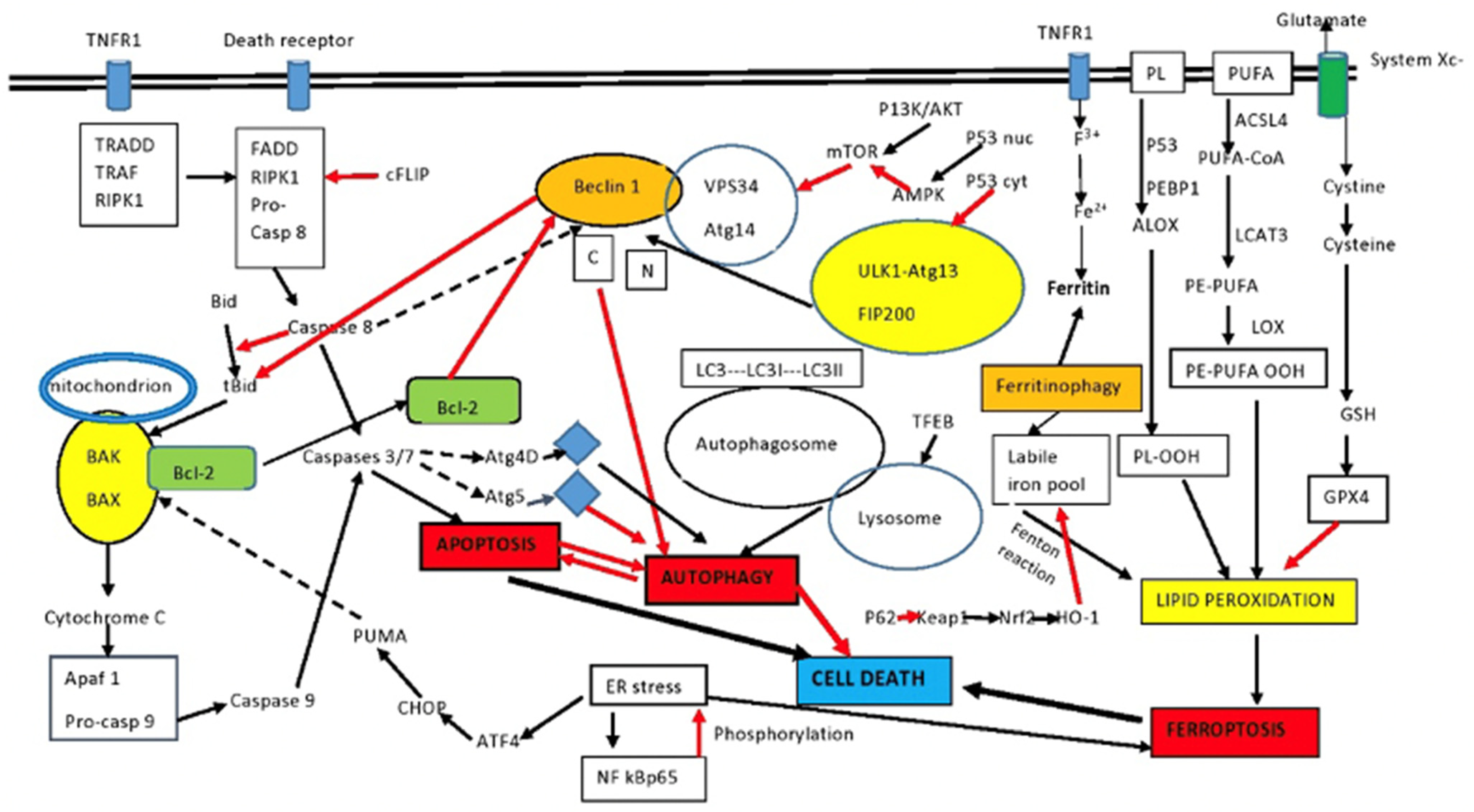
2. An Overview of Autophagy and Apoptosis
2.1. Autophagy
2.2. Mitophagy
2.3. Apoptosis
2.4. Ferroptosis
2.5. Interaction of Autophagy and Apoptosis
3. Genetics and Autophagy in IBD
3.1. ATG16L1
3.2. IRGM
3.3. LRRK2
3.4. ATG7 and ATG5
3.5. Other Autophagy-Associated Genes
3.6. NOD2
4. Autophagy and Apoptosis in IBD
4.1. Autophagy
4.2. Intestinal Functions Affected by Autophagy Dysregulation in IBD
4.2.1. Paneth Cell Autophagy
4.2.2. Inflammatory Cytokine Regulation
4.2.3. Antigen Presentation by Dendritic Cells
4.2.4. Goblet Cell Mucus Secretion
4.2.5. ER Stress Response
4.3. Autophagy Signal Pathways Implicated in IBD
4.3.1. mTOR
4.3.2. AMPK/mTOR
4.3.3. mTOR/NLRP3
4.3.4. AKT/mTOR
4.3.5. NF-kB Pathways
4.3.6. Nrf2/HO-1 Pathways
4.3.7. Additional Pathways
4.4. Role of Autophagy in CD
4.5. Role of Autophagy in UC
4.6. Lysosomes in IBD
4.7. Autophagy and Fibrosis in Experimental Colitis
4.8. Mitophagy in IBD
4.9. Apoptosis
4.9.1. The Role of TNFα
4.9.2. Other Factors Affect Apoptosis in IBD
4.10. Ferroptosis in IBD
Other Ferroptosis Regulators in Intestinal Diseases
4.11. Interaction of Autophagy–Apoptosis in IBD
Intersection of ERS, Autophagy and Apoptosis
5. Autophagy and Treatment of IBD
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Adams, S.M.; Close, E.D.; Shreenath, A.P. Ulcerative Colitis: Rapid Evidence Review. Am. Fam. Physician 2022, 105, 406–411. [Google Scholar]
- Ramos, G.P.; Papadakis, K.A. Mechanisms of Disease: Inflammatory Bowel Diseases. Mayo Clin. Proc. 2019, 94, 155–165. [Google Scholar] [CrossRef]
- Peterson, L.W.; Artis, D. Intestinal epithelial cells: Regulators of barrier function and immune homeostasis. Nat. Rev. Immunol. 2014, 14, 141–153. [Google Scholar] [CrossRef]
- Ray, K. IBD: The changing epidemiology of IBD. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 690. [Google Scholar] [CrossRef]
- Ng, S.C.; Leung, W.K.; Shi, H.Y.; Li, M.K.; Leung, C.M.; Ng, C.K.; Lo, F.H.; Hui, Y.T.; Tsang, S.W.; Chan, Y.K.; et al. Epidemiology of Inflammatory Bowel Disease from 1981 to 2014: Results from a Territory-Wide Population-Based Registry in Hong Kong. Inflamm. Bowel Dis. 2016, 22, 1954–1960. [Google Scholar] [CrossRef]
- Jarmakiewicz-Czaja, S.; Zielińska, M.; Sokal, A.; Filip, R. Genetic and Epigenetic Etiology of Inflammatory Bowel Disease: An Update. Genes 2022, 13, 2388. [Google Scholar] [CrossRef]
- Schroeder, B.O. Fight them or feed them: How the intestinal mucus layer manages the gut microbiota. Gastroenterol. Rep. 2019, 7, 3–12. [Google Scholar] [CrossRef]
- Pelaseyed, T.; Hansson, G.C. Membrane mucins of the intestine at a glance. J. Cell Sci. 2020, 133, jcs240929. [Google Scholar] [CrossRef]
- Yin, Y.B.; de Jonge, H.R.; Wu, X.; Yin, Y.L. Enteroids for Nutritional Studies. Mol. Nutr. Food Res. 2019, 63, e1801143. [Google Scholar] [CrossRef]
- Ho, J.; Chan, H.; Liang, Y.; Liu, X.; Zhang, L.; Li, Q.; Zhang, Y.; Zeng, J.; Ugwu, F.N.; Ho, I.H.T.; et al. Cathelicidin preserves intestinal barrier function in polymicrobial sepsis. Crit. Care. 2020, 24, 47. [Google Scholar] [CrossRef]
- Luissint, A.C.; Williams, H.C.; Kim, W.; Flemming, S.; Azcutia, V.; Hilgarth, R.S.; Leary, M.N.O.; Denning, T.L.; Nusrat, A.; Parkos, C.A. Macrophage-dependent neutrophil recruitment is impaired under conditions of increased intestinal permeability in JAM-A-deficient mice. Mucosal. Immunol. 2019, 12, 668–678. [Google Scholar] [CrossRef]
- Gardet, A.; Xavier, R.J. Common alleles that influence autophagy and the risk for inflammatory bowel disease. Curr. Opin. Immunol. 2012, 24, 522–529. [Google Scholar] [CrossRef]
- Pott, J.; Kabat, A.M.; Maloy, K.J. Intestinal Epithelial Cell Autophagy Is Required to Protect against TNF-Induced Apoptosis during Chronic Colitis in Mice. Cell Host Microbe. 2018, 23, 191–202. [Google Scholar] [CrossRef]
- Xiong, Y.J.; Deng, Z.B.; Liu, J.N.; Qiu, J.J.; Guo, L.; Feng, P.P.; Sui, J.R.; Chen, D.P.; Guo, H.S. Enhancement of epithelial cell autophagy induced by sinensetin alleviates epithelial barrier dysfunction in colitis. Pharmacol. Res. 2019, 148, 104461. [Google Scholar] [CrossRef]
- Petrović, A.; Bogojević, D.; Korać, A.; Golić, I.; Jovanović-Stojanov, S.; Martinović, V.; Ivanović-Matić, S.; Stevanović, J.; Poznanović, G.; Grigorov, I. Oxidative stress-dependent contribution of HMGB1 to the interplay between apoptosis and autophagy in diabetic rat liver. J. Physiol. Biochem. 2017, 73, 511–521. [Google Scholar] [CrossRef]
- Kabat, A.M.; Pott, J.; Maloy, K.J. The Mucosal Immune System and Its Regulation by Autophagy. Front. Immunol. 2016, 7, 240. [Google Scholar] [CrossRef]
- Zhang, H.; Zheng, L.; McGovern, D.P.; Hamill, A.M.; Ichikawa, R.; Kanazawa, Y.; Luu, J.; Kumagai, K.; Cilluffo, M.; Fukata, M.; et al. Myeloid ATG16L1 Facilitates Host-Bacteria Interactions in Maintaining Intestinal Homeostasis. J. Immunol. 2017, 198, 2133–2146. [Google Scholar] [CrossRef]
- Lassen, K.G.; Kuballa, P.; Conway, K.L.; Patel, K.K.; Becker, C.E.; Peloquin, J.M.; Villablanca, E.J.; Norman, J.M.; Liu, T.C.; Heath, R.J.; et al. Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc. Natl. Acad. Sci. USA 2014, 111, 7741–7746. [Google Scholar] [CrossRef]
- Cadwell, K.; Liu, J.Y.; Brown, S.L.; Miyoshi, H.; Loh, J.; Lennerz, J.K.; Kishi, C.; Kc, W.; Carrero, J.A.; Hunt, S.; et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008, 456, 259–263. [Google Scholar] [CrossRef]
- Ouellette, A.J. Paneth cells and innate mucosal immunity. Curr. Opin. Gastroenterol. 2010, 26, 547–553. [Google Scholar] [CrossRef]
- Paulus, G.L.; Xavier, R.J. Autophagy and checkpoints for intracellular pathogen defense. Curr. Opin. Gastroenterol. 2015, 31, 14–23. [Google Scholar] [CrossRef]
- Salzman, N.H.; Bevins, C.L. Dysbiosis—A consequence of Paneth cell dysfunction. Semin. Immunol. 2013, 25, 334–341. [Google Scholar] [CrossRef]
- Mizushima, N. A brief history of autophagy from cell biology to physiology and disease. Nat. Cell Biol. 2018, 20, 521–527. [Google Scholar] [CrossRef]
- Harnett, M.M.; Pineda, M.A.; Latré de Laté, P.; Eason, R.J.; Besteiro, S.; Harnett, W.; Langsley, G. From Christian de Duve to Yoshinori Ohsumi: More to autophagy than just dining at home. Biomed. J. 2017, 40, 9–22. [Google Scholar] [CrossRef]
- Corona Velazquez, A.F.; Jackson, W.T. So Many Roads: The Multifaceted Regulation of Autophagy Induction. Mol. Cell Biol. 2018, 38, e00303-18. [Google Scholar] [CrossRef]
- Liang, N.; He, Q.; Liu, X.; Sun, H. Multifaceted roles of ATM in autophagy: From nonselective autophagy to selective autophagy. Cell Biochem. Funct. 2019, 37, 177–184. [Google Scholar] [CrossRef]
- Zachari, M.; Ganley, I.G. The mammalian ULK1 complex and autophagy initiation. Essays Biochem. 2017, 61, 585–596. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef]
- Russell, R.C.; Tian, Y.; Yuan, H.; Park, H.W.; Chang, Y.Y.; Kim, J.; Kim, H.; Neufeld, T.P.; Dillin, A.; Guan, K.L. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 2013, 15, 741–750. [Google Scholar] [CrossRef]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef]
- Levine, B.; Sinha, S.; Kroemer, G. Bcl-2 family members: Dual regulators of apoptosis and autophagy. Autophagy 2008, 4, 600–606. [Google Scholar] [CrossRef]
- Galluzzi, L.; Green, D.R. Autophagy-Independent Functions of the Autophagy Machinery. Cell 2019, 177, 1682–1699. [Google Scholar] [CrossRef]
- Shahrabi, S.; Paridar, M.; Zeinvand-Lorestani, M.; Jalili, A.; Zibara, K.; Abdollahi, M.; Khosravi, A. Autophagy regulation and its role in normal and malignant hematopoiesis. J. Cell Physiol. 2019, 234, 21746–21757. [Google Scholar] [CrossRef]
- Thorburn, A. Autophagy and disease. J. Biol. Chem. 2018, 293, 5425–5430. [Google Scholar] [CrossRef]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef]
- Foerster, E.G.; Mukherjee, T.; Cabral-Fernandes, L.; Rocha, J.D.B.; Girardin, S.E.; Philpott, D.J. How autophagy controls the intestinal epithelial barrier. Autophagy 2022, 18, 86–103. [Google Scholar] [CrossRef]
- Kim, S.; Eun, H.S.; Jo, E.K. Roles of Autophagy-Related Genes in the Pathogenesis of Inflammatory Bowel Disease. Cells 2019, 8, 77. [Google Scholar] [CrossRef]
- Puertollano, R.; Ferguson, S.M.; Brugarolas, J.; Ballabio, A. The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J. 2018, 37, e98804. [Google Scholar] [CrossRef]
- Frank, M.; Duvezin-Caubet, S.; Koob, S.; Occhipinti, A.; Jagasia, R.; Petcherski, A.; Ruonala, M.O.; Priault, M.; Salin, B.; Reichert, A.S. Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim. Biophys. Acta 2012, 1823, 2297–2310. [Google Scholar] [CrossRef]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- Durcan, T.M.; Fon, E.A. The three ‘P’s of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes Dev. 2015, 29, 989–999. [Google Scholar] [CrossRef]
- Ryter, S.W.; Bhatia, D.; Choi, M.E. Autophagy: A Lysosome-Dependent Process with Implications in Cellular Redox Homeostasis and Human Disease. Antioxid. Redox Signal. 2019, 30, 138–159. [Google Scholar] [CrossRef]
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deak, M.; et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012, 2, 120080. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef]
- Koyano, F.; Okatsu, K.; Kosako, H.; Tamura, Y.; Go, E.; Kimura, M.; Kimura, Y.; Tsuchiya, H.; Yoshihara, H.; Hirokawa, T.; et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014, 510, 162–166. [Google Scholar] [CrossRef]
- Chen, Y.; Dorn, G.W. 2nd. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 2013, 340, 471–475. [Google Scholar] [CrossRef]
- Geisler, S.; Holmström, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef]
- Moore, A.S.; Holzbaur, E.L. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc. Natl. Acad. Sci. USA 2016, 113, E3349–E3358. [Google Scholar] [CrossRef]
- Richter, B.; Sliter, D.A.; Herhaus, L.; Stolz, A.; Wang, C.; Beli, P.; Zaffagnini, G.; Wild, P.; Martens, S.; Wagner, S.A.; et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc. Natl. Acad. Sci. USA 2016, 113, 4039–4044. [Google Scholar] [CrossRef]
- Xu, Y.; Shen, J.; Ran, Z. Emerging views of mitophagy in immunity and autoimmune diseases. Autophagy 2020, 16, 3–17. [Google Scholar] [CrossRef]
- Yao, R.Q.; Ren, C.; Xia, Z.F.; Yao, Y.M. Organelle-specific autophagy in inflammatory diseases: A potential therapeutic target underlying the quality control of multiple organelles. Autophagy 2021, 17, 385–401. [Google Scholar] [CrossRef]
- Van Opdenbosch, N.; Lamkanfi, M. Caspases in Cell Death, Inflammation, and Disease. Immunity 2019, 50, 1352–1364. [Google Scholar] [CrossRef]
- Patel, T.; Gores, G.J.; Kaufmann, S.H. The role of proteases during apoptosis. FASEB J. 1996, 10, 587–597. [Google Scholar] [CrossRef]
- Cosentino, K.; García-Sáez, A.J. Bax and Bak Pores: Are We Closing the Circle? Trends Cell Biol. 2017, 27, 266–275. [Google Scholar] [CrossRef]
- Woo, S.M.; Kwon, T.K. E3 ubiquitin ligases and deubiquitinases as modulators of TRAIL-mediated extrinsic apoptotic signaling pathway. BMB Rep. 2019, 52, 119–126. [Google Scholar] [CrossRef]
- Chipuk, J.E.; Bouchier-Hayes, L.; Green, D.R. Mitochondrial outer membrane permeabilization during apoptosis: The innocent bystander scenario. Cell Death Differ. 2006, 13, 1396–1402. [Google Scholar] [CrossRef]
- Hisamatsu, T.; Kanai, T.; Mikami, Y.; Yoneno, K.; Matsuoka, K.; Hibi, T. Immune aspects of the pathogenesis of inflammatory bowel disease. Pharmacol. Ther. 2013, 137, 283–297. [Google Scholar] [CrossRef]
- Garcia-Carbonell, R.; Yao, S.J.; Das, S.; Guma, M. Dysregulation of Intestinal Epithelial Cell RIPK Pathways Promotes Chronic Inflammation in the IBD Gut. Front. Immunol. 2019, 10, 1094. [Google Scholar] [CrossRef]
- Kuang, F.; Liu, J.; Tang, D.; Kang, R. Oxidative Damage and Antioxidant Defense in Ferroptosis. Front. Cell Dev. Biol. 2020, 8, 586578. [Google Scholar] [CrossRef]
- Macías-Rodríguez, R.U.; Inzaugarat, M.E.; Ruiz-Margáin, A.; Nelson, L.J.; Trautwein, C.; Cubero, F.J. Reclassifying Hepatic Cell Death during Liver Damage: Ferroptosis-A Novel Form of Non-Apoptotic Cell Death? Int. J. Mol. Sci. 2020, 21, 1651. [Google Scholar] [CrossRef]
- Ocansey, D.K.W.; Yuan, J.; Wei, Z.; Mao, F.; Zhang, Z. Role of ferroptosis in the pathogenesis and as a therapeutic target of inflammatory bowel disease (Review). Int. J. Mol. Med. 2023, 51, 53. [Google Scholar] [CrossRef]
- Mayr, L.; Grabherr, F.; Schwärzler, J.; Reitmeier, I.; Sommer, F.; Gehmacher, T.; Niederreiter, L.; He, G.W.; Ruder, B.; Kunz, K.T.R.; et al. Dietary lipids fuel GPX4-restricted enteritis resembling Crohn’s disease. Nat. Commun. 2020, 11, 1775. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Xu, S.; He, Y.; Lin, L.; Chen, P.; Chen, M.; Zhang, S. The emerging role of ferroptosis in intestinal disease. Cell Death Dis. 2021, 12, 289. [Google Scholar] [CrossRef]
- Bano, I.; Horky, P.; Abbas, S.Q.; Majid, M.; Bilal, A.H.M.; Ali, F.; Behl, T.; Hassan, S.S.U.; Bungau, S. Ferroptosis: A New Road towards Cancer Management. Molecules 2022, 27, 2129. [Google Scholar] [CrossRef]
- Gao, W.; Zhang, T.; Wu, H. Emerging Pathological Engagement of Ferroptosis in Gut Diseases. Oxid. Med. Cell Longev. 2021, 2021, 4246255. [Google Scholar] [CrossRef]
- Battaglia, A.M.; Chirillo, R.; Aversa, I.; Sacco, A.; Costanzo, F.; Biamonte, F. Ferroptosis and Cancer: Mitochondria Meet the “Iron Maiden” Cell Death. Cells 2020, 9, 1505. [Google Scholar] [CrossRef]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Organelle-specific regulation of ferroptosis. Cell Death Differ. 2021, 28, 2843–2856. [Google Scholar] [CrossRef]
- Lee, H.; Zandkarimi, F.; Zhang, Y.; Meena, J.K.; Kim, J.; Zhuang, L.; Tyagi, S.; Ma, L.; Westbrook, T.F.; Steinberg, G.R.; et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat. Cell Biol. 2020, 22, 225–234. [Google Scholar] [CrossRef]
- Huo, C.; Li, G.; Hu, Y.; Sun, H. The Impacts of Iron Overload and Ferroptosis on Intestinal Mucosal Homeostasis and Inflammation. Int. J. Mol. Sci. 2022, 23, 14195. [Google Scholar] [CrossRef]
- Lai, B.; Wu, C.H.; Wu, C.Y.; Luo, S.F.; Lai, J.H. Ferroptosis and Autoimmune Diseases. Front. Immunol. 2022, 13, 916664. [Google Scholar] [CrossRef]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef]
- Jia, L.; Dourmashkin, R.R.; Allen, P.D.; Gray, A.B.; Newland, A.C.; Kelsey, S.M. Inhibition of autophagy abrogates tumour necrosis factor alpha induced apoptosis in human T-lymphoblastic leukaemic cells. Br. J. Haematol. 1997, 98, 673–685. [Google Scholar] [CrossRef]
- Thorburn, J.; Moore, F.; Rao, A.; Barclay, W.W.; Thomas, L.R.; Grant, K.W.; Cramer, S.D.; Thorburn, A. Selective inactivation of a Fas-associated death domain protein (FADD)-dependent apoptosis and autophagy pathway in immortal epithelial cells. Mol. Biol. Cell. 2005, 16, 1189–1199. [Google Scholar] [CrossRef]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef]
- Decuypere, J.P.; Parys, J.B.; Bultynck, G. Regulation of the autophagic bcl-2/beclin 1 interaction. Cells 2012, 1, 284–312. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Kroemer, G. Mitochondria: Master regulators of danger signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 780–788. [Google Scholar] [CrossRef]
- Bassik, M.C.; Scorrano, L.; Oakes, S.A.; Pozzan, T.; Korsmeyer, S.J. Phosphorylation of BCL-2 regulates ER Ca2+ homeostasis and apoptosis. EMBO J. 2004, 23, 1207–1216. [Google Scholar] [CrossRef]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef]
- Wei, Y.; Sinha, S.; Levine, B. Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy 2008, 4, 949–951. [Google Scholar] [CrossRef]
- Ciechomska, I.A.; Goemans, G.C.; Skepper, J.N.; Tolkovsky, A.M. Bcl-2 complexed with Beclin-1 maintains full anti-apoptotic function. Oncogene 2009, 28, 2128–2141. [Google Scholar] [CrossRef]
- Lee, J.S.; Li, Q.; Lee, J.Y.; Lee, S.H.; Jeong, J.H.; Lee, H.R.; Chang, H.; Zhou, F.C.; Gao, S.J.; Liang, C.; et al. FLIP-mediated autophagy regulation in cell death control. Nat. Cell Biol. 2009, 11, 1355–1362. [Google Scholar] [CrossRef]
- Yousefi, S.; Perozzo, R.; Schmid, I.; Ziemiecki, A.; Schaffner, T.; Scapozza, L.; Brunner, T.; Simon, H.U. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat. Cell Biol. 2006, 8, 1124–1132. [Google Scholar] [CrossRef]
- Wirawan, E.; Vande Walle, L.; Kersse, K.; Cornelis, S.; Claerhout, S.; Vanoverberghe, I.; Roelandt, R.; De Rycke, R.; Verspurten, J.; Declercq, W.; et al. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis. 2010, 1, e18. [Google Scholar] [CrossRef]
- Booth, L.A.; Roberts, J.L.; Dent, P. The role of cell signaling in the crosstalk between autophagy and apoptosis in the regulation of tumor cell survival in response to sorafenib and neratinib. Semin. Cancer Biol. 2020, 66, 129–139. [Google Scholar] [CrossRef]
- Hooper, K.M.; Barlow, P.G.; Stevens, C.; Henderson, P. Inflammatory Bowel Disease Drugs: A Focus on Autophagy. J. Crohns Colitis. 2017, 11, 118–127. [Google Scholar] [CrossRef]
- Katsandegwaza, B.; Horsnell, W.; Smith, K. Inflammatory Bowel Disease: A Review of Pre-Clinical Murine Models of Human Disease. Int. J. Mol. Sci. 2022, 23, 9344. [Google Scholar] [CrossRef]
- Catana, C.S.; Magdas, C.; Tabaran, F.A.; Crăciun, E.C.; Deak, G.; Magdaş, V.A.; Cozma, V.; Gherman, C.M.; Berindan-Neagoe, I.; Dumitraşcu, D.L. Comparison of two models of inflammatory bowel disease in rats. Adv. Clin. Exp. Med. 2018, 27, 599–607. [Google Scholar] [CrossRef]
- Oh, S.Y.; Cho, K.A.; Kang, J.L.; Kim, K.H.; Woo, S.Y. Comparison of experimental mouse models of inflammatory bowel disease. Int. J. Mol. Med. 2014, 33, 333–340. [Google Scholar] [CrossRef]
- Silva, I.; Solas, J.; Pinto, R.; Mateus, V. Chronic Experimental Model of TNBS-Induced Colitis to Study Inflammatory Bowel Disease. Int. J. Mol. Sci. 2022, 23, 4739. [Google Scholar] [CrossRef]
- Mestas, J.; Hughes, C.C. Of mice and not men: Differences between mouse and human immunology. J. Immunol. 2004, 172, 2731–2738. [Google Scholar] [CrossRef]
- Hampe, J.; Franke, A.; Rosenstiel, P.; Till, A.; Teuber, M.; Huse, K.; Albrecht, M.; Mayr, G.; De La Vega, F.M.; Briggs, J.; et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat. Genet. 2007, 39, 207–211. [Google Scholar] [CrossRef]
- Prescott, N.J.; Fisher, S.A.; Franke, A.; Hampe, J.; Onnie, C.M.; Soars, D.; Bagnall, R.; Mirza, M.M.; Sanderson, J.; Forbes, A.; et al. A nonsynonymous SNP in ATG16L1 predisposes to ileal Crohn’s disease and is independent of CARD15 and IBD5. Gastroenterology 2007, 132, 1665–1671. [Google Scholar] [CrossRef]
- Rioux, J.D.; Xavier, R.J.; Taylor, K.D.; Silverberg, M.S.; Goyette, P.; Huett, A.; Green, T.; Kuballa, P.; Barmada, M.M.; Datta, L.W.; et al. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat. Genet. 2007, 39, 596–604. [Google Scholar] [CrossRef]
- Singh, S.B.; Davis, A.S.; Taylor, G.A.; Deretic, V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science 2006, 313, 1438–1441. [Google Scholar] [CrossRef]
- Taylor, G.A. IRG proteins: Key mediators of interferon-regulated host resistance to intracellular pathogens. Cell Microbiol. 2007, 9, 1099–1107. [Google Scholar] [CrossRef]
- Wellcome Trust Case Control Consortium. Genome-wide association study of 14,000 cases of seven common diseases and 3000 shared controls. Nature 2007, 447, 661–678. [Google Scholar] [CrossRef]
- Lassen, K.G.; McKenzie, C.I.; Mari, M.; Murano, T.; Begun, J.; Baxt, L.A.; Goel, G.; Villablanca, E.J.; Kuo, S.Y.; Huang, H.; et al. Genetic Coding Variant in GPR65 Alters Lysosomal pH and Links Lysosomal Dysfunction with Colitis Risk. Immunity 2016, 44, 1392–1405. [Google Scholar] [CrossRef]
- Morgan, A.R.; Lam, W.J.; Han, D.Y.; Fraser, A.G.; Ferguson, L.R. Association Analysis of ULK1 with Crohn’s Disease in a New Zealand Population. Gastroenterol. Res. Pract. 2012, 2012, 715309. [Google Scholar] [CrossRef]
- de Lange, K.M.; Barrett, J.C. Understanding inflammatory bowel disease via immunogenetics. J. Autoimmun. 2015, 64, 91–100. [Google Scholar] [CrossRef]
- Liu, J.Z.; van Sommeren, S.; Huang, H.; Ng, S.C.; Alberts, R.; Takahashi, A.; Ripke, S.; Lee, J.C.; Jostins, L.; Shah, T.; et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 2015, 47, 979–986. [Google Scholar] [CrossRef]
- Murano, T.; Najibi, M.; Paulus, G.L.C.; Adiliaghdam, F.; Valencia-Guerrero, A.; Selig, M.; Wang, X.; Jeffrey, K.; Xavier, R.J.; Lassen, K.G.; et al. Transcription factor TFEB cell-autonomously modulates susceptibility to intestinal epithelial cell injury in vivo. Sci. Rep. 2017, 7, 13938. [Google Scholar] [CrossRef]
- Smith, A.M.; Sewell, G.W.; Levine, A.P.; Chew, T.S.; Dunne, J.; O’Shea, N.R.; Smith, P.J.; Harrison, P.J.; Macdonald, C.M.; Bloom, S.L.; et al. Disruption of macrophage pro-inflammatory cytokine release in Crohn’s disease is associated with reduced optineurin expression in a subset of patients. Immunology 2015, 144, 45–55. [Google Scholar] [CrossRef]
- Takagawa, T.; Kitani, A.; Fuss, I.; Levine, B.; Brant, S.R.; Peter, I.; Tajima, M.; Nakamura, S.; Strober, W. An increase in LRRK2 suppresses autophagy and enhances Dectin-1-induced immunity in a mouse model of colitis. Sci. Transl. Med. 2018, 10, eaan8162. [Google Scholar] [CrossRef]
- Nighot, P.K.; Hu, C.A.; Ma, T.Y. Autophagy enhances intestinal epithelial tight junction barrier function by targeting claudin-2 protein degradation. J. Biol. Chem. 2015, 290, 7234–7246. [Google Scholar] [CrossRef]
- Saha, K.; Subramenium Ganapathy, A.; Wang, A.; Michael Morris, N.; Suchanec, E.; Ding, W.; Yochum, G.; Koltun, W.; Nighot, M.; Ma, T.; et al. Autophagy Reduces the Degradation and Promotes Membrane Localization of Occludin to Enhance the Intestinal Epithelial Tight Junction Barrier against Paracellular Macromolecule Flux. J. Crohns Colitis 2023, 17, 433–449. [Google Scholar] [CrossRef]
- Wong, M.; Ganapathy, A.S.; Suchanec, E.; Laidler, L.; Ma, T.; Nighot, P. Intestinal epithelial tight junction barrier regulation by autophagy-related protein ATG6/beclin 1. Am. J. Physiol. Cell Physiol. 2019, 316, 753–765. [Google Scholar] [CrossRef]
- Jung, H.; Leal-Ekman, J.S.; Lu, Q.; Stappenbeck, T.S. Atg14 protects the intestinal epithelium from TNF-triggered villus atrophy. Autophagy 2019, 15, 1990–2001. [Google Scholar] [CrossRef]
- Tran, S.; Juliani, J.; Fairlie, W.D.; Lee, E.F. The emerging roles of autophagy in intestinal epithelial cells and its links to inflammatory bowel disease. Biochem. Soc. Trans. 2023, 51, 811–826. [Google Scholar] [CrossRef]
- Girardelli, M.; Basaldella, F.; Paolera, S.D.; Vuch, J.; Tommasini, A.; Martelossi, S.; Crovella, S.; Bianco, A.M. Genetic profile of patients with early onset inflammatory bowel disease. Gene 2018, 645, 18–29. [Google Scholar] [CrossRef]
- Roberts, R.L.; Gearry, R.B.; Hollis-Moffatt, J.E.; Miller, A.L.; Reid, J.; Abkevich, V.; Timms, K.M.; Gutin, A.; Lanchbury, J.S.; Merriman, T.R.; et al. IL23R R381Q and ATG16L1 T300A are strongly associated with Crohn’s disease in a study of New Zealand Caucasians with inflammatory bowel disease. Am. J. Gastroenterol. 2007, 102, 2754–2761. [Google Scholar] [CrossRef]
- Yamazaki, K.; Onouchi, Y.; Takazoe, M.; Kubo, M.; Nakamura, Y.; Hata, A. Association analysis of genetic variants in IL23R, ATG16L1 and 5p13.1 loci with Crohn’s disease in Japanese patients. J. Hum. Genet. 2007, 52, 575–583. [Google Scholar] [CrossRef]
- Adolph, T.E.; Tomczak, M.F.; Niederreiter, L.; Ko, H.J.; Böck, J.; Martinez-Naves, E.; Glickman, J.N.; Tschurtschenthaler, M.; Hartwig, J.; Hosomi, S.; et al. Paneth cells as a site of origin for intestinal inflammation. Nature 2013, 503, 272–276. [Google Scholar] [CrossRef]
- Tschurtschenthaler, M.; Adolph, T.E.; Ashcroft, J.W.; Niederreiter, L.; Bharti, R.; Saveljeva, S.; Bhattacharyya, J.; Flak, M.B.; Shih, D.Q.; Fuhler, G.M.; et al. Defective ATG16L1-mediated removal of IRE1α drives Crohn’s disease-like ileitis. J. Exp. Med. 2017, 214, 401–422. [Google Scholar] [CrossRef]
- Liu, H.; Gao, P.; Jia, B.; Lu, N.; Zhu, B.; Zhang, F. IBD-Associated Atg16L1T300A Polymorphism Regulates Commensal Microbiota of the Intestine. Front. Immunol. 2022, 12, 772189. [Google Scholar] [CrossRef]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.; Yang, B.G.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M.; et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 2008, 456, 264–268. [Google Scholar] [CrossRef]
- Plantinga, T.S.; Crisan, T.O.; Oosting, M.; van de Veerdonk, F.L.; de Jong, D.J.; Philpott, D.J.; van der Meer, J.W.; Girardin, S.E.; Joosten, L.A.; Netea, M.G. Crohn’s disease-associated ATG16L1 polymorphism modulates pro-inflammatory cytokine responses selectively upon activation of NOD2. Gut 2011, 60, 1229–12235. [Google Scholar] [CrossRef]
- Samie, M.; Lim, J.; Verschueren, E.; Baughman, J.M.; Peng, I.; Wong, A.; Kwon, Y.; Senbabaoglu, Y.; Hackney, J.A.; Keir, M.; et al. Selective autophagy of the adaptor TRIF regulates innate inflammatory signaling. Nat. Immunol. 2018, 19, 246–254. [Google Scholar] [CrossRef]
- Nuij, V.J.A.A.; Peppelenbosch, M.P.; van der Woude, C.J.; Fuhler, G.M. Genetic polymorphism in ATG16L1 gene is associated with adalimumab use in inflammatory bowel disease. J. Transl. Med. 2017, 15, 248. [Google Scholar] [CrossRef]
- Murthy, A.; Li, Y.; Peng, I.; Reichelt, M.; Katakam, A.K.; Noubade, R.; Roose-Girma, M.; DeVoss, J.; Diehl, L.; Graham, R.R.; et al. A Crohn’s disease variant in Atg16l1 enhances its degradation by caspase 3. Nature 2014, 506, 456–462. [Google Scholar] [CrossRef]
- Wang, C.; Gong, G.; Sheh, A.; Muthupalani, S.; Bryant, E.M.; Puglisi, D.A.; Holcombe, H.; Conaway, E.A.; Parry, N.A.P.; Bakthavatchalu, V.; et al. Interleukin-22 drives nitric oxide-dependent DNA damage and dysplasia in a murine model of colitis-associated cancer. Mucosal Immunol. 2017, 10, 1504–1517. [Google Scholar] [CrossRef]
- Aden, K.; Tran, F.; Ito, G.; Sheibani-Tezerji, R.; Lipinski, S.; Kuiper, J.W.; Tschurtschenthaler, M.; Saveljeva, S.; Bhattacharyya, J.; Häsler, R.; et al. ATG16L1 orchestrates interleukin-22 signaling in the intestinal epithelium via cGAS-STING. J Exp. Med. 2018, 215, 2868–2886. [Google Scholar] [CrossRef]
- Slowicka, K.; Serramito-Gómez, I.; Boada-Romero, E.; Martens, A.; Sze, M.; Petta, I.; Vikkula, H.K.; De Rycke, R.; Parthoens, E.; Lippens, S.; et al. Physical and functional interaction between A20 and ATG16L1-WD40 domain in the control of intestinal homeostasis. Nat. Commun. 2019, 10, 1834. [Google Scholar] [CrossRef]
- Deretic, V. Autophagy in infection. Curr. Opin. Cell Biol. 2010, 22, 252–262. [Google Scholar] [CrossRef]
- Chauhan, S.; Mandell, M.A.; Deretic, V. IRGM governs the core autophagy machinery to conduct antimicrobial defense. Mol. Cell 2015, 58, 507–521. [Google Scholar] [CrossRef]
- Mehto, S.; Jena, K.K.; Nath, P.; Chauhan, S.; Kolapalli, S.P.; Das, S.K.; Sahoo, P.K.; Jain, A.; Taylor, G.A.; Chauhan, S. The Crohn’s Disease Risk Factor IRGM Limits NLRP3 Inflammasome Activation by Impeding Its Assembly and by Mediating Its Selective Autophagy. Mol. Cell 2019, 73, 429–445.e7. [Google Scholar] [CrossRef]
- Palomino-Morales, R.J.; Oliver, J.; Gómez-García, M.; López-Nevot, M.A.; Rodrigo, L.; Nieto, A.; Alizadeh, B.Z.; Martín, J. Association of ATG16L1 and IRGM genes polymorphisms with inflammatory bowel disease: A meta-analysis approach. Genes Immun. 2009, 10, 356–364. [Google Scholar] [CrossRef]
- Rufini, S.; Ciccacci, C.; Di Fusco, D.; Ruffa, A.; Pallone, F.; Novelli, G.; Biancone, L.; Borgiani, P. Autophagy and inflammatory bowel disease: Association between variants of the autophagy-related IRGM gene and susceptibility to Crohn’s disease. Dig. Liver Dis. 2015, 47, 744–750. [Google Scholar] [CrossRef]
- Moon, C.M.; Shin, D.J.; Kim, S.W.; Son, N.H.; Park, A.; Park, B.; Jung, E.S.; Kim, E.S.; Hong, S.P.; Kim, T.I.; et al. Associations between genetic variants in the IRGM gene and inflammatory bowel diseases in the Korean population. Inflamm. Bowel Dis. 2013, 19, 106–114. [Google Scholar] [CrossRef]
- Rogala, A.R.; Schoenborn, A.A.; Fee, B.E.; Cantillana, V.A.; Joyce, M.J.; Gharaibeh, R.Z.; Roy, S.; Fodor, A.A.; Sartor, R.B.; Taylor, G.A.; et al. Environmental factors regulate Paneth cell phenotype and host susceptibility to intestinal inflammation in Irgm1-deficient mice. Dis. Model. Mech. 2018, 11, dmm031070. [Google Scholar] [CrossRef]
- Liu, B.; Gulati, A.S.; Cantillana, V.; Henry, S.C.; Schmidt, E.A.; Daniell, X.; Grossniklaus, E.; Schoenborn, A.A.; Sartor, R.B.; Taylor, G.A. Irgm1-deficient mice exhibit Paneth cell abnormalities and increased susceptibility to acute intestinal inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G573–G584. [Google Scholar] [CrossRef]
- Lapaquette, P.; Glasser, A.L.; Huett, A.; Xavier, R.J.; Darfeuille-Michaud, A. Crohn’s disease-associated adherent-invasive E. coli are selectively favoured by impaired autophagy to replicate intracellularly. Cell Microbiol. 2010, 12, 99–113. [Google Scholar] [CrossRef]
- Rolhion, N.; Darfeuille-Michaud, A. Adherent-invasive Escherichia coli in inflammatory bowel disease. Inflamm. Bowel Dis. 2007, 13, 1277–1283. [Google Scholar] [CrossRef]
- Brest, P.; Lapaquette, P.; Souidi, M.; Lebrigand, K.; Cesaro, A.; Vouret-Craviari, V.; Mari, B.; Barbry, P.; Mosnier, J.F.; Hébuterne, X.; et al. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn’s disease. Nat. Genet. 2011, 43, 242–245. [Google Scholar] [CrossRef]
- Simon, T.G.; Van Der Sloot, K.W.J.; Chin, S.B.; Joshi, A.D.; Lochhead, P.; Ananthakrishnan, A.N.; Xavier, R.; Chung, R.T.; Khalili, H. IRGM Gene Variants Modify the Relationship Between Visceral Adipose Tissue and NAFLD in Patients With Crohn’s Disease. Inflamm. Bowel Dis. 2018, 24, 2247–2257. [Google Scholar] [CrossRef]
- Thachil, E.; Hugot, J.P.; Arbeille, B.; Paris, R.; Grodet, A.; Peuchmaur, M.; Codogno, P.; Barreau, F.; Ogier-Denis, E.; Berrebi, D.; et al. Abnormal activation of autophagy-induced crinophagy in Paneth cells from patients with Crohn’s disease. Gastroenterology 2012, 142, 1097–1099.e4. [Google Scholar] [CrossRef]
- Cheluvappa, R.; Luo, A.S.; Grimm, M.C. Autophagy suppression by appendicitis and appendectomy protects against colitis. Inflamm. Bowel Dis. 2014, 20, 847–855. [Google Scholar] [CrossRef]
- Gardet, A.; Benita, Y.; Li, C.; Sands, B.E.; Ballester, I.; Stevens, C.; Korzenik, J.R.; Rioux, J.D.; Daly, M.J.; Xavier, R.J.; et al. LRRK2 is involved in the IFN-gamma response and host response to pathogens. J. Immunol. 2010, 185, 5577–5585. [Google Scholar] [CrossRef]
- Zhang, F.R.; Huang, W.; Chen, S.M.; Sun, L.D.; Liu, H.; Li, Y.; Cui, Y.; Yan, X.X.; Yang, H.T.; Yang, R.D.; et al. Genomewide association study of leprosy. N. Engl. J. Med. 2009, 361, 2609–2618. [Google Scholar] [CrossRef]
- Toledo Pinto, T.G.; Batista-Silva, L.R.; Medeiros, R.C.A.; Lara, F.A.; Moraes, M.O. Type I Interferons, Autophagy and Host Metabolism in Leprosy. Front. Immunol. 2018, 9, 806. [Google Scholar] [CrossRef]
- Liu, Z.; Lee, J.; Krummey, S.; Lu, W.; Cai, H.; Lenardo, M.J. The kinase LRRK2 is a regulator of the transcription factor NFAT that modulates the severity of inflammatory bowel disease. Nat. Immunol. 2011, 12, 1063–1070. [Google Scholar] [CrossRef]
- Fujishima, Y.; Nishiumi, S.; Masuda, A.; Inoue, J.; Nguyen, N.M.; Irino, Y.; Komatsu, M.; Tanaka, K.; Kutsumi, H.; Azuma, T.; et al. Autophagy in the intestinal epithelium reduces endotoxin-induced inflammatory responses by inhibiting NF-κB activation. Arch. Biochem. Biophys. 2011, 506, 223–235. [Google Scholar] [CrossRef]
- Tsuboi, K.; Nishitani, M.; Takakura, A.; Imai, Y.; Komatsu, M.; Kawashima, H. Autophagy Protects against Colitis by the Maintenance of Normal Gut Microflora and Secretion of Mucus. J. Biol. Chem. 2015, 290, 20511–20526. [Google Scholar] [CrossRef]
- Wittkopf, N.; Günther, C.; Martini, E.; Waldner, M.; Amann, K.U.; Neurath, M.F.; Becker, C. Lack of intestinal epithelial atg7 affects paneth cell granule formation but does not compromise immune homeostasis in the gut. Clin. Dev. Immunol. 2012, 2012, 278059. [Google Scholar] [CrossRef]
- Burger, E.; Araujo, A.; López-Yglesias, A.; Rajala, M.W.; Geng, L.; Levine, B.; Hooper, L.V.; Burstein, E.; Yarovinsky, F. Loss of Paneth Cell Autophagy Causes Acute Susceptibility to Toxoplasma gondii-Mediated Inflammation. Cell Host Microbe 2018, 23, 177–190.e4. [Google Scholar] [CrossRef]
- Patel, K.K.; Miyoshi, H.; Beatty, W.L.; Head, R.D.; Malvin, N.P.; Cadwell, K.; Guan, J.L.; Saitoh, T.; Akira, S.; Seglen, P.O.; et al. Autophagy proteins control goblet cell function by potentiating reactive oxygen species production. EMBO J. 2013, 32, 3130–3144. [Google Scholar] [CrossRef]
- Yang, L.; Liu, C.; Zhao, W.; He, C.; Ding, J.; Dai, R.; Xu, K.; Xiao, L.; Luo, L.; Liu, S.; et al. Impaired Autophagy in Intestinal Epithelial Cells Alters Gut Microbiota and Host Immune Responses. Appl. Environ. Microbiol. 2018, 84, e00880-18. [Google Scholar] [CrossRef]
- Lee, H.Y.; Kim, J.; Quan, W.; Lee, J.C.; Kim, M.S.; Kim, S.H.; Bae, J.W.; Hur, K.Y.; Lee, M.S. Autophagy deficiency in myeloid cells increases susceptibility to obesity-induced diabetes and experimental colitis. Autophagy 2016, 12, 1390–1403. [Google Scholar] [CrossRef]
- Cabrera, S.; Fernández, A.F.; Mariño, G.; Aguirre, A.; Suárez, M.F.; Español, Y.; Vega, J.A.; Laurà, R.; Fueyo, A.; Fernández-García, M.S.; et al. ATG4B/autophagin-1 regulates intestinal homeostasis and protects mice from experimental colitis. Autophagy 2013, 9, 1188–1200. [Google Scholar] [CrossRef]
- Benjamin, J.L.; Sumpter, R., Jr.; Levine, B.; Hooper, L.V. Intestinal epithelial autophagy is essential for host defense against invasive bacteria. Cell Host Microbe 2013, 13, 723–734. [Google Scholar] [CrossRef]
- Asano, J.; Sato, T.; Ichinose, S.; Kajita, M.; Onai, N.; Shimizu, S.; Ohteki, T. Intrinsic Autophagy Is Required for the Maintenance of Intestinal Stem Cells and for Irradiation-Induced Intestinal Regeneration. Cell Rep. 2017, 20, 1050–1060. [Google Scholar] [CrossRef]
- Trentesaux, C.; Fraudeau, M.; Pitasi, C.L.; Lemarchand, J.; Jacques, S.; Duche, A.; Letourneur, F.; Naser, E.; Bailly, K.; Schmitt, A.; et al. Essential role for autophagy protein ATG7 in the maintenance of intestinal stem cell integrity. Proc. Natl. Acad. Sci. USA 2020, 117, 11136–11146. [Google Scholar] [CrossRef]
- Naama, M.; Telpaz, S.; Awad, A.; Ben-Simon, S.; Harshuk-Shabso, S.; Modilevsky, S.; Rubin, E.; Sawaed, J.; Zelik, L.; Zigdon, M.; et al. Autophagy controls mucus secretion from intestinal goblet cells by alleviating ER stress. Cell Host Microbe 2023, 31, 433–446.e4. [Google Scholar] [CrossRef]
- Ellinghaus, D.; Zhang, H.; Zeissig, S.; Lipinski, S.; Till, A.; Jiang, T.; Stade, B.; Bromberg, Y.; Ellinghaus, E.; Keller, A.; et al. Association between variants of PRDM1 and NDP52 and Crohn’s disease, based on exome sequencing and functional studies. Gastroenterology 2013, 145, 339–347. [Google Scholar] [CrossRef]
- Till, A.; Lipinski, S.; Ellinghaus, D.; Mayr, G.; Subramani, S.; Rosenstiel, P.; Franke, A. Autophagy receptor CALCOCO2/NDP52 takes center stage in Crohn disease. Autophagy 2013, 9, 1256–1257. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.; Adams, P.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- Cosin-Roger, J.; Simmen, S.; Melhem, H.; Atrott, K.; Frey-Wagner, I.; Hausmann, M.; de Vallière, C.; Spalinger, M.R.; Spielmann, P.; Wenger, R.H.; et al. Hypoxia ameliorates intestinal inflammation through NLRP3/mTOR downregulation and autophagy activation. Nat. Commun. 2017, 8, 98. [Google Scholar] [CrossRef]
- Paiva, N.M.; Pascoal, L.B.; Negreiros, L.M.V.; Portovedo, M.; Coope, A.; Ayrizono, M.L.S.; Coy, C.S.R.; Milanski, M.; Leal, R.F. Ileal pouch of ulcerative colitis and familial adenomatous polyposis patients exhibit modulation of autophagy markers. Sci. Rep. 2018, 8, 2619. [Google Scholar] [CrossRef]
- Mimouna, S.; Bazin, M.; Mograbi, B.; Darfeuille-Michaud, A.; Brest, P.; Hofman, P.; Vouret-Craviari, V. HIF1A regulates xenophagic degradation of adherent and invasive Escherichia coli (AIEC). Autophagy 2014, 10, 2333–2345. [Google Scholar] [CrossRef]
- Ortiz-Masiá, D.; Cosín-Roger, J.; Calatayud, S.; Hernández, C.; Alós, R.; Hinojosa, J.; Apostolova, N.; Alvarez, A.; Barrachina, M.D. Hypoxic macrophages impair autophagy in epithelial cells through Wnt1: Relevance in IBD. Mucosal Immunol. 2014, 7, 929–938. [Google Scholar] [CrossRef]
- Ryan, T.A.; Tumbarello, D.A. Optineurin: A Coordinator of Membrane-Associated Cargo Trafficking and Autophagy. Front. Immunol. 2018, 9, 1024. [Google Scholar] [CrossRef]
- Chew, T.S.; O’Shea, N.R.; Sewell, G.W.; Oehlers, S.H.; Mulvey, C.M.; Crosier, P.S.; Godovac-Zimmermann, J.; Bloom, S.L.; Smith, A.M.; Segal, A.W. Optineurin deficiency in mice contributes to impaired cytokine secretion and neutrophil recruitment in bacteria-driven colitis. Dis. Model. Mech. 2015, 8, 817–829. [Google Scholar] [CrossRef]
- Napolitano, G.; Ballabio, A. TFEB at a glance. J. Cell Sci. 2016, 129, 2475–2481. [Google Scholar] [CrossRef]
- Kaser, A.; Lee, A.H.; Franke, A.; Glickman, J.N.; Zeissig, S.; Tilg, H.; Nieuwenhuis, E.E.; Higgins, D.E.; Schreiber, S.; Glimcher, L.H.; et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 2008, 134, 743–756. [Google Scholar] [CrossRef]
- Hampe, J.; Cuthbert, A.; Croucher, P.J.; Mirza, M.M.; Mascheretti, S.; Fisher, S.; Frenzel, H.; King, K.; Hasselmeyer, A.; MacPherson, A.J.; et al. Association between insertion mutation in NOD2 gene and Crohn’s disease in German and British populations. Lancet 2001, 357, 1925–1928. [Google Scholar] [CrossRef]
- Hugot, J.P.; Chamaillard, M.; Zouali, H.; Lesage, S.; Cézard, J.P.; Belaiche, J.; Almer, S.; Tysk, C.; O’Morain, C.A.; Gassull, M.; et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 2001, 411, 599–603. [Google Scholar] [CrossRef]
- Ogura, Y.; Bonen, D.K.; Inohara, N.; Nicolae, D.L.; Chen, F.F.; Ramos, R.; Britton, H.; Moran, T.; Karaliuskas, R.; Duerr, R.H.; et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 2001, 411, 603–606. [Google Scholar] [CrossRef]
- Niess, J.H.; Klaus, J.; Stephani, J.; Pflüger, C.; Degenkolb, N.; Spaniol, U.; Mayer, B.; Lahr, G.; von Boyen, G.B. NOD2 polymorphism predicts response to treatment in Crohn’s disease--first steps to a personalized therapy. Dig. Dis. Sci. 2012, 57, 879–886. [Google Scholar] [CrossRef]
- Cooney, R.; Baker, J.; Brain, O.; Danis, B.; Pichulik, T.; Allan, P.; Ferguson, D.J.; Campbell, B.J.; Jewell, D.; Simmons, A. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat. Med. 2010, 16, 90–97. [Google Scholar] [CrossRef]
- Fritz, T.; Niederreiter, L.; Adolph, T.; Blumberg, R.S.; Kaser, A. Crohn’s disease: NOD2, autophagy and ER stress converge. Gut 2011, 60, 1580–1588. [Google Scholar] [CrossRef]
- Travassos, L.H.; Carneiro, L.A.; Ramjeet, M.; Hussey, S.; Kim, Y.G.; Magalhães, J.G.; Yuan, L.; Soares, F.; Chea, E.; Le Bourhis, L.; et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 2010, 11, 55–62. [Google Scholar] [CrossRef]
- Mak, W.Y.; Zhao, M.; Ng, S.C.; Burisch, J. The epidemiology of inflammatory bowel disease: East meets west. J. Gastroenterol. Hepatol. 2020, 35, 380–389. [Google Scholar] [CrossRef]
- Brant, S.R.; Okou, D.T.; Simpson, C.L.; Cutler, D.J.; Haritunians, T.; Bradfield, J.P.; Chopra, P.; Prince, J.; Begum, F.; Kumar, A.; et al. Genome-Wide Association Study Identifies African-Specific Susceptibility Loci in African Americans with Inflammatory Bowel Disease. Gastroenterology 2017, 152, 206–217.e2. [Google Scholar] [CrossRef]
- Larabi, A.; Barnich, N.; Nguyen, H.T.T. New insights into the interplay between autophagy, gut microbiota and inflammatory responses in IBD. Autophagy 2020, 16, 38–51. [Google Scholar] [CrossRef]
- Kaser, A.; Blumberg, R.S. Autophagy, microbial sensing, endoplasmic reticulum stress, and epithelial function in inflammatory bowel disease. Gastroenterology 2011, 140, 1738–1747. [Google Scholar] [CrossRef]
- Xavier, R.J.; Huett, A.; Rioux, J.D. Autophagy as an important process in gut homeostasis and Crohn’s disease pathogenesis. Gut 2008, 57, 717–7120. [Google Scholar] [CrossRef]
- El-Khider, F.; McDonald, C. Links of Autophagy Dysfunction to Inflammatory Bowel Disease Onset. Dig. Dis. 2016, 34, 27–34. [Google Scholar] [CrossRef]
- Elliott, T.R.; Hudspith, B.N.; Rayment, N.B.; Prescott, N.J.; Petrovska, L.; Hermon-Taylor, J.; Brostoff, J.; Boussioutas, A.; Mathew, C.G.; Sanderson, J.D. Defective macrophage handling of Escherichia coli in Crohn’s disease. J. Gastroenterol. Hepatol. 2015, 30, 1265–1274. [Google Scholar] [CrossRef]
- Kernbauer, E.; Cadwell, K. Autophagy, viruses, and intestinal immunity. Curr. Opin. Gastroenterol. 2014, 30, 539–546. [Google Scholar] [CrossRef]
- Lapaquette, P.; Bringer, M.A.; Darfeuille-Michaud, A. Defects in autophagy favour adherent-invasive Escherichia coli persistence within macrophages leading to increased pro-inflammatory response. Cell Microbiol. 2012, 14, 791–807. [Google Scholar] [CrossRef]
- Vaishnava, S.; Yamamoto, M.; Severson, K.M.; Ruhn, K.A.; Yu, X.; Koren, O.; Ley, R.; Wakeland, E.K.; Hooper, L.V. The antibacterial lectin RegIIIgamma promotes the spatial segregation of microbiota and host in the intestine. Science 2011, 334, 255–258. [Google Scholar] [CrossRef]
- Rocha, J.D.; Schlossmacher, M.G.; Philpott, D.J. LRRK2 and Nod2 promote lysozyme sorting in Paneth cells. Nat. Immunol. 2015, 16, 898–900. [Google Scholar] [CrossRef]
- Cadwell, K.; Patel, K.K.; Komatsu, M.; Virgin, H.W., 4th; Stappenbeck, T.S. A common role for Atg16L1, Atg5 and Atg7 in small intestinal Paneth cells and Crohn disease. Autophagy 2009, 5, 250–252. [Google Scholar] [CrossRef]
- Smythies, L.E.; Shen, R.; Bimczok, D.; Novak, L.; Clements, R.H.; Eckhoff, D.E.; Bouchard, P.; George, M.D.; Hu, W.K.; Dandekar, S.; et al. Inflammation anergy in human intestinal macrophages is due to Smad-induced IkappaBalpha expression and NF-kappaB inactivation. J. Biol. Chem. 2010, 285, 19593–19604. [Google Scholar] [CrossRef]
- Randall-Demllo, S.; Chieppa, M.; Eri, R. Intestinal epithelium and autophagy: Partners in gut homeostasis. Front. Immunol. 2013, 4, 301. [Google Scholar] [CrossRef]
- Scharl, M.; Wojtal, K.A.; Becker, H.M.; Fischbeck, A.; Frei, P.; Arikkat, J.; Pesch, T.; Kellermeier, S.; Boone, D.L.; Weber, A.; et al. Protein tyrosine phosphatase nonreceptor type 2 regulates autophagosome formation in human intestinal cells. Inflamm. Bowel Dis. 2012, 18, 1287–1302. [Google Scholar] [CrossRef]
- Lee, H.K.; Mattei, L.M.; Steinberg, B.E.; Alberts, P.; Lee, Y.H.; Chervonsky, A.; Mizushima, N.; Grinstein, S.; Iwasaki, A. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity 2010, 32, 227–239. [Google Scholar] [CrossRef]
- Liu, E.; Van Grol, J.; Subauste, C.S. Atg5 but not Atg7 in dendritic cells enhances IL-2 and IFN-γ production by Toxoplasma gondii-reactive CD4+ T cells. Microbes Infect. 2015, 17, 275–284. [Google Scholar] [CrossRef]
- Swidsinski, A.; Loening-Baucke, V.; Herber, A. Mucosal flora in Crohn’s disease and ulcerative colitis—An overview. J. Physiol. Pharmacol. 2009, 60, 61–71. [Google Scholar]
- Chen, G.Y.; Stappenbeck, T.S. Mucus, it is not just a static barrier. Sci. Signal. 2014, 7, 11. [Google Scholar] [CrossRef]
- Baxt, L.A.; Xavier, R.J. Role of Autophagy in the Maintenance of Intestinal Homeostasis. Gastroenterology 2015, 149, 553–562. [Google Scholar] [CrossRef]
- Deuring, J.J.; Fuhler, G.M.; Konstantinov, S.R.; Peppelenbosch, M.P.; Kuipers, E.J.; de Haar, C.; van der Woude, C.J. Genomic ATG16L1 risk allele-restricted Paneth cell ER stress in quiescent Crohn’s disease. Gut 2014, 63, 1081–1091. [Google Scholar] [CrossRef]
- Lopes, F.; Keita, Å.V.; Saxena, A.; Reyes, J.L.; Mancini, N.L.; Al Rajabi, A.; Wang, A.; Baggio, C.H.; Dicay, M.; van Dalen, R.; et al. ER-stress mobilization of death-associated protein kinase-1-dependent xenophagy counteracts mitochondria stress-induced epithelial barrier dysfunction. J. Biol. Chem. 2018, 293, 3073–3087. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, S.; Dai, C.; Tang, S.; Yang, X.; Li, D.; Zhao, K.; Xiao, X. Quinocetone triggered ER stress-induced autophagy via ATF6/DAPK1-modulated mAtg9a trafficking. Cell Biol. Toxicol. 2016, 32, 141–152. [Google Scholar] [CrossRef]
- Stengel, S.T.; Fazio, A.; Lipinski, S.; Jahn, M.T.; Aden, K.; Ito, G.; Wottawa, F.; Kuiper, J.W.P.; Coleman, O.I.; Tran, F.; et al. Activating Transcription Factor 6 Mediates Inflammatory Signals in Intestinal Epithelial Cells Upon Endoplasmic Reticulum Stress. Gastroenterology 2020, 159, 1357–1374. [Google Scholar] [CrossRef]
- Kouroku, Y.; Fujita, E.; Tanida, I.; Ueno, T.; Isoai, A.; Kumagai, H.; Ogawa, S.; Kaufman, R.J.; Kominami, E.; Momoi, T. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 2007, 14, 230–239. [Google Scholar] [CrossRef]
- Moon, H.S.; Kim, B.; Gwak, H.; Suh, D.H.; Song, Y.S. Autophagy and protein kinase RNA-like endoplasmic reticulum kinase (PERK)/eukaryotic initiation factor 2 alpha kinase (eIF2α) pathway protect ovarian cancer cells from metformin-induced apoptosis. Mol. Carcinog. 2016, 55, 346–356. [Google Scholar] [CrossRef]
- Rouschop, K.M.; van den Beucken, T.; Dubois, L.; Niessen, H.; Bussink, J.; Savelkouls, K.; Keulers, T.; Mujcic, H.; Landuyt, W.; Voncken, J.W.; et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J. Clin. Investig. 2010, 120, 127–141. [Google Scholar] [CrossRef]
- Kimball, S.R.; Jefferson, L.S. Induction of REDD1 gene expression in the liver in response to endoplasmic reticulum stress is mediated through a PERK, eIF2α phosphorylation, ATF4-dependent cascade. Biochem. Biophys. Res. Commun. 2012, 427, 485–489. [Google Scholar] [CrossRef]
- Angelidou, I.; Chrysanthopoulou, A.; Mitsios, A.; Arelaki, S.; Arampatzioglou, A.; Kambas, K.; Ritis, D.; Tsironidou, V.; Moschos, I.; Dalla, V.; et al. REDD1/Autophagy Pathway Is Associated with Neutrophil-Driven IL-1β Inflammatory Response in Active Ulcerative Colitis. J. Immunol. 2018, 200, 3950–3961. [Google Scholar] [CrossRef]
- Corazzari, M.; Rapino, F.; Ciccosanti, F.; Giglio, P.; Antonioli, M.; Conti, B.; Fimia, G.M.; Lovat, P.E.; Piacentini, M. Oncogenic BRAF induces chronic ER stress condition resulting in increased basal autophagy and apoptotic resistance of cutaneous melanoma. Cell Death Differ. 2015, 22, 946–958. [Google Scholar] [CrossRef]
- Rather, R.A.; Bhagat, M.; Singh, S.K. Oncogenic BRAF, endoplasmic reticulum stress, and autophagy: Crosstalk and therapeutic targets in cutaneous melanoma. Mutat. Res. Rev. Mutat. Res. 2020, 785, 108321. [Google Scholar] [CrossRef]
- Diamanti, M.A.; Gupta, J.; Bennecke, M.; De Oliveira, T.; Ramakrishnan, M.; Braczynski, A.K.; Richter, B.; Beli, P.; Hu, Y.; Saleh, M.; et al. IKKα controls ATG16L1 degradation to prevent ER stress during inflammation. J. Exp. Med. 2017, 214, 423–437. [Google Scholar] [CrossRef]
- Ibrahim, I.M.; Abdelmalek, D.H.; Elfiky, A.A. GRP78: A cell’s response to stress. Life Sci. 2019, 226, 156–163. [Google Scholar] [CrossRef]
- Ravanan, P.; Srikumar, I.F.; Talwar, P. Autophagy: The spotlight for cellular stress responses. Life Sci. 2017, 188, 53–67. [Google Scholar] [CrossRef]
- Tréton, X.; Pédruzzi, E.; Cazals-Hatem, D.; Grodet, A.; Panis, Y.; Groyer, A.; Moreau, R.; Bouhnik, Y.; Daniel, F.; Ogier-Denis, E. Altered endoplasmic reticulum stress affects translation in inactive colon tissue from patients with ulcerative colitis. Gastroenterology 2011, 141, 1024–1035. [Google Scholar] [CrossRef]
- Zhang, H.S.; Chen, Y.; Fan, L.; Xi, Q.L.; Wu, G.H.; Li, X.X.; Yuan, T.L.; He, S.Q.; Yu, Y.; Shao, M.L.; et al. The Endoplasmic Reticulum Stress Sensor IRE1α in Intestinal Epithelial Cells Is Essential for Protecting against Colitis. J. Biol. Chem. 2015, 290, 15327–15336. [Google Scholar] [CrossRef]
- Doherty, J.; Baehrecke, E.H. Life, death and autophagy. Nat. Cell Biol. 2018, 20, 1110–1117. [Google Scholar] [CrossRef]
- Keestra-Gounder, A.M.; Byndloss, M.X.; Seyffert, N.; Young, B.M.; Chávez-Arroyo, A.; Tsai, A.Y.; Cevallos, S.A.; Winter, M.G.; Pham, O.H.; Tiffany, C.R.; et al. NOD1 and NOD2 signalling links ER stress with inflammation. Nature 2016, 532, 394–397. [Google Scholar] [CrossRef]
- Kaneko, M.; Niinuma, Y.; Nomura, Y. Activation signal of nuclear factor-kappa B in response to endoplasmic reticulum stress is transduced via IRE1 and tumor necrosis factor receptor-associated factor 2. Biol. Pharm. Bull. 2003, 26, 931–935. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef]
- Ding, W.X.; Ni, H.M.; Gao, W.; Yoshimori, T.; Stolz, D.B.; Ron, D.; Yin, X.M. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am. J. Pathol. 2007, 171, 513–524. [Google Scholar] [CrossRef]
- Moretti, J.; Roy, S.; Bozec, D.; Martinez, J.; Chapman, J.R.; Ueberheide, B.; Lamming, D.W.; Chen, Z.J.; Horng, T.; Yeretssian, G.; et al. STING Senses Microbial Viability to Orchestrate Stress-Mediated Autophagy of the Endoplasmic Reticulum. Cell 2017, 171, 809–823. [Google Scholar] [CrossRef]
- Zhou, M.; Xu, W.; Wang, J.; Yan, J.; Shi, Y.; Zhang, C.; Ge, W.; Wu, J.; Du, P.; Chen, Y. Boosting mTOR-dependent autophagy via upstream TLR4-MyD88-MAPK signalling and downstream NF-κB pathway quenches intestinal inflammation and oxidative stress injury. EBioMedicine 2018, 35, 345–360. [Google Scholar] [CrossRef]
- Guan, Y.; Zhang, L.; Li, X.; Zhang, X.; Liu, S.; Gao, N.; Li, L.; Gao, G.; Wei, G.; Chen, Z.; et al. Repression of Mammalian Target of Rapamycin Complex 1 Inhibits Intestinal Regeneration in Acute Inflammatory Bowel Disease Models. J. Immunol. 2015, 195, 339–346. [Google Scholar] [CrossRef]
- Gutiérrez-Martínez, I.Z.; Rubio, J.F.; Piedra-Quintero, Z.L.; Lopez-Mendez, O.; Serrano, C.; Reyes-Maldonado, E.; Salinas-Lara, C.; Betanzos, A.; Shibayama, M.; Silva-Olivares, A.; et al. mTORC1 Prevents Epithelial Damage During Inflammation and Inhibits Colitis-Associated Colorectal Cancer Development. Transl. Oncol. 2019, 12, 24–35. [Google Scholar] [CrossRef]
- Liu, Y.; Liao, R.; Qiang, Z.; Yang, W.; Cao, J.; Zeng, H. Exogenous H2S Protects Colon Cells in Ulcerative Colitis by Inhibiting NLRP3 and Activating Autophagy. DNA Cell Biol. 2021, 40, 748–756. [Google Scholar] [CrossRef]
- Deng, J.; Zeng, L.; Lai, X.; Li, J.; Liu, L.; Lin, Q.; Chen, Y. Metformin protects against intestinal barrier dysfunction via AMPKα1-dependent inhibition of JNK signalling activation. J. Cell Mol. Med. 2018, 22, 546–557. [Google Scholar] [CrossRef]
- Gao, Q.; Bi, P.; Luo, D.; Guan, Y.; Zeng, W.; Xiang, H.; Mi, Q.; Yang, G.; Li, X.; Yang, B. Nicotine-induced autophagy via AMPK/mTOR pathway exerts protective effect in colitis mouse model. Chem. Biol. Interact. 2020, 317, 108943. [Google Scholar] [CrossRef]
- Villani, A.C.; Lemire, M.; Fortin, G.; Louis, E.; Silverberg, M.S.; Collette, C.; Baba, N.; Libioulle, C.; Belaiche, J.; Bitton, A.; et al. Common variants in the NLRP3 region contribute to Crohn’s disease susceptibility. Nat. Genet. 2009, 41, 71–76. [Google Scholar] [CrossRef]
- Shi, C.S.; Shenderov, K.; Huang, N.N.; Kabat, J.; Abu-Asab, M.; Fitzgerald, K.A.; Sher, A.; Kehrl, J.H. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 2012, 13, 255–263. [Google Scholar] [CrossRef]
- Jiang, W.; Han, Y.P.; Hu, M.; Bao, X.Q.; Yan, Y.; Chen, G. A study on regulatory mechanism of miR-223 in ulcerative colitis through PI3K/Akt-mTOR signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 4865–4872. [Google Scholar] [CrossRef]
- Wang, B.; Gong, Z.; Zhan, J.; Yang, L.; Zhou, Q.; Yuan, X. Xianglian Pill Suppresses Inflammation and Protects Intestinal Epithelial Barrier by Promoting Autophagy in DSS Induced Ulcerative Colitis Mice. Front. Pharmacol. 2021, 11, 594847. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, W.; Niu, J.; Yang, G.; Luo, J.; Lan, D.; Wu, J.; Li, M.; Sun, Y.; Wang, K.; et al. Heat-shock transcription factor 2 promotes sodium butyrate-induced autophagy by inhibiting mTOR in ulcerative colitis. Exp. Cell Res. 2020, 388, 111820. [Google Scholar] [CrossRef]
- Zhu, Y.; Shi, Y.; Ke, X.; Xuan, L.; Ma, Z. RNF8 induces autophagy and reduces inflammation by promoting AKT degradation via ubiquitination in ulcerative colitis mice. J. Biochem. 2020, 168, 445–453. [Google Scholar] [CrossRef]
- Nivon, M.; Fort, L.; Muller, P.; Richet, E.; Simon, S.; Guey, B.; Fournier, M.; Arrigo, A.P.; Hetz, C.; Atkin, J.D.; et al. NFκB is a central regulator of protein quality control in response to protein aggregation stresses via autophagy modulation. Mol. Biol. Cell. 2016, 27, 1712–1727. [Google Scholar] [CrossRef]
- Platta, H.W.; Abrahamsen, H.; Thoresen, S.B.; Stenmark, H. Nedd4-dependent lysine-11-linked polyubiquitination of the tumour suppressor Beclin 1. Biochem. J. 2012, 441, 399–406. [Google Scholar] [CrossRef]
- Gerster, R.; Eloranta, J.J.; Hausmann, M.; Ruiz, P.A.; Cosin-Roger, J.; Terhalle, A.; Ziegler, U.; Kullak-Ublick, G.A.; von Eckardstein, A.; Rogler, G. Anti-inflammatory Function of High-Density Lipoproteins via Autophagy of IκB Kinase. Cell Mol. Gastroenterol. Hepatol. 2014, 1, 171–187. [Google Scholar] [CrossRef]
- Shibata, Y.; Oyama, M.; Kozuka-Hata, H.; Han, X.; Tanaka, Y.; Gohda, J.; Inoue, J. p47 negatively regulates IKK activation by inducing the lysosomal degradation of polyubiquitinated NEMO. Nat. Commun. 2012, 3, 1061. [Google Scholar] [CrossRef]
- Honjo, H.; Watanabe, T.; Arai, Y.; Kamata, K.; Minaga, K.; Komeda, Y.; Yamashita, K.; Kudo, M. ATG16L1 negatively regulates RICK/RIP2-mediated innate immune responses. Int. Immunol. 2021, 33, 91–105. [Google Scholar] [CrossRef]
- Gao, P.; Liu, H.; Huang, H.; Sun, Y.; Jia, B.; Hou, B.; Zhou, X.; Strober, W.; Zhang, F. The Crohn Disease-associated ATG16L1T300A polymorphism regulates inflammatory responses by modulating TLR- and NLR-mediated signaling. Autophagy 2022, 18, 2561–2575. [Google Scholar] [CrossRef]
- Wang, D.; Shao, S.; Zhang, Y.; Zhao, D.; Wang, M. Insight Into Polysaccharides From Panax ginseng C. A. Meyer in Improving Intestinal Inflammation: Modulating Intestinal Microbiota and Autophagy. Front. Immunol. 2021, 12, 683911. [Google Scholar] [CrossRef]
- Jain, A.; Lamark, T.; Sjøttem, E.; Larsen, K.B.; Awuh, J.A.; Øvervatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef]
- Kaushal, G.P.; Chandrashekar, K.; Juncos, L.A. Molecular Interactions Between Reactive Oxygen Species and Autophagy in Kidney Disease. Int. J. Mol. Sci. 2019, 20, 3791. [Google Scholar] [CrossRef]
- Li, J.; Wang, H.; Zheng, Z.; Luo, L.; Wang, P.; Liu, K.; Namani, A.; Jiang, Z.; Wang, X.J.; Tang, X. Mkp-1 cross-talks with Nrf2/Ho-1 pathway protecting against intestinal inflammation. Free Radic. Biol. Med. 2018, 124, 541–549. [Google Scholar] [CrossRef]
- Pompili, S.; Sferra, R.; Gaudio, E.; Viscido, A.; Frieri, G.; Vetuschi, A.; Latella, G. Can Nrf2 Modulate the Development of Intestinal Fibrosis and Cancer in Inflammatory Bowel Disease? Int. J. Mol. Sci. 2019, 20, 4061. [Google Scholar] [CrossRef]
- Farzaei, M.H.; El-Senduny, F.F.; Momtaz, S.; Parvizi, F.; Iranpanah, A.; Tewari, D.; Naseri, R.; Abdolghaffari, A.H.; Rezaei, N. An update on dietary consideration in inflammatory bowel disease: Anthocyanins and more. Expert. Rev. Gastroenterol. Hepatol. 2018, 12, 1007–1024. [Google Scholar] [CrossRef]
- Fu, X.Y. STAT3 in immune responses and inflammatory bowel diseases. Cell Res. 2006, 16, 214–219. [Google Scholar] [CrossRef]
- Zhang, Y.G.; Zhu, X.; Lu, R.; Messer, J.S.; Xia, Y.; Chang, E.B.; Sun, J. Intestinal epithelial HMGB1 inhibits bacterial infection via STAT3 regulation of autophagy. Autophagy 2019, 15, 1935–1953. [Google Scholar] [CrossRef]
- Yuan, Y.; Ding, D.; Zhang, N.; Xia, Z.; Wang, J.; Yang, H.; Guo, F.; Li, B. TNF-α induces autophagy through ERK1/2 pathway to regulate apoptosis in neonatal necrotizing enterocolitis model cells IEC-6. Cell Cycle. 2018, 17, 1390–1402. [Google Scholar] [CrossRef]
- Kubota, M.; Kakimoto, K.; Nakagawa, T.; Koubayashi, E.; Nakazawa, K.; Tawa, H.; Hirata, Y.; Okada, T.; Kawakami, K.; Asai, A.; et al. Autophagy deficiency exacerbates colitis through excessive oxidative stress and MAPK signaling pathway activation. PLoS ONE 2019, 14, e0225066. [Google Scholar] [CrossRef]
- Chen, S.L.; Li, C.M.; Li, W.; Liu, Q.S.; Hu, S.Y.; Zhao, M.Y.; Hu, D.S.; Hao, Y.W.; Zeng, J.H.; Zhang, Y. How autophagy, a potential therapeutic target, regulates intestinal inflammation. Front. Immunol. 2023, 14, 1087677. [Google Scholar] [CrossRef]
- Zhang, C.; Yan, J.; Xiao, Y.; Shen, Y.; Wang, J.; Ge, W.; Chen, Y. Inhibition of Autophagic Degradation Process Contributes to Claudin-2 Expression Increase and Epithelial Tight Junction Dysfunction in TNF-α Treated Cell Monolayers. Int. J. Mol. Sci. 2017, 18, 157. [Google Scholar] [CrossRef]
- Guo, J.; Yang, Z.; Yang, X.; Li, T.; Liu, M.; Tang, H. miR-346 functions as a pro-survival factor under ER stress by activating mitophagy. Cancer Lett. 2018, 413, 69–81. [Google Scholar] [CrossRef]
- Guo, W.; Sun, Y.; Liu, W.; Wu, X.; Guo, L.; Cai, P.; Wu, X.; Wu, X.; Shen, Y.; Shu, Y.; et al. Small molecule-driven mitophagy-mediated NLRP3 inflammasome inhibition is responsible for the prevention of colitis-associated cancer. Autophagy 2014, 10, 972–985. [Google Scholar] [CrossRef]
- Matsuzawa-Ishimoto, Y.; Shono, Y.; Gomez, L.E.; Hubbard-Lucey, V.M.; Cammer, M.; Neil, J.; Dewan, M.Z.; Lieberman, S.R.; Lazrak, A.; Marinis, J.M.; et al. Autophagy protein ATG16L1 prevents necroptosis in the intestinal epithelium. J. Exp. Med. 2017, 214, 3687–3705. [Google Scholar] [CrossRef]
- Bel, S.; Pendse, M.; Wang, Y.; Li, Y.; Ruhn, K.A.; Hassell, B.; Leal, T.; Winter, S.E.; Xavier, R.J.; Hooper, L.V. Paneth cells secrete lysozyme via secretory autophagy during bacterial infection of the intestine. Science 2017, 357, 1047–1052. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, X.; Zuo, Z.; Zhang, Q.; Pan, Y.; Zeng, B.; Li, W.; Wei, H.; Liu, Z. Rip2 Is Required for Nod2-Mediated Lysozyme Sorting in Paneth Cells. J. Immunol. 2017, 198, 3729–3736. [Google Scholar] [CrossRef]
- Zhang, Q.; Pan, Y.; Yan, R.; Zeng, B.; Wang, H.; Zhang, X.; Li, W.; Wei, H.; Liu, Z. Commensal bacteria direct selective cargo sorting to promote symbiosis. Nat. Immunol. 2015, 16, 918–926. [Google Scholar] [CrossRef]
- Ke, P.; Shao, B.Z.; Xu, Z.Q.; Chen, X.W.; Liu, C. Intestinal Autophagy and Its Pharmacological Control in Inflammatory Bowel Disease. Front. Immunol. 2017, 7, 695. [Google Scholar] [CrossRef]
- Ma, Y.; Galluzzi, L.; Zitvogel, L.; Kroemer, G. Autophagy and cellular immune responses. Immunity 2013, 39, 211–227. [Google Scholar] [CrossRef]
- Whang, M.I.; Tavares, R.M.; Benjamin, D.I.; Kattah, M.G.; Advincula, R.; Nomura, D.K.; Debnath, J.; Malynn, B.A.; Ma, A. The Ubiquitin Binding Protein TAX1BP1 Mediates Autophagasome Induction and the Metabolic Transition of Activated T Cells. Immunity 2017, 46, 405–420. [Google Scholar] [CrossRef]
- Jia, W.; He, Y.W. Temporal regulation of intracellular organelle homeostasis in T lymphocytes by autophagy. J. Immunol. 2011, 186, 5313–5322. [Google Scholar] [CrossRef]
- Kabat, A.M.; Harrison, O.J.; Riffelmacher, T.; Moghaddam, A.E.; Pearson, C.F.; Laing, A.; Abeler-Dörner, L.; Forman, S.P.; Grencis, R.K.; Sattentau, Q.; et al. The autophagy gene Atg16l1 differentially regulates Treg and TH2 cells to control intestinal inflammation. Elife 2016, 5, 12444. [Google Scholar] [CrossRef]
- Kovacs, J.R.; Li, C.; Yang, Q.; Li, G.; Garcia, I.G.; Ju, S.; Roodman, D.G.; Windle, J.J.; Zhang, X.; Lu, B. Autophagy promotes T-cell survival through degradation of proteins of the cell death machinery. Cell Death Differ. 2012, 19, 144–152. [Google Scholar] [CrossRef]
- Puleston, D.J.; Zhang, H.; Powell, T.J.; Lipina, E.; Sims, S.; Panse, I.; Watson, A.S.; Cerundolo, V.; Townsend, A.R.; Klenerman, P.; et al. Autophagy is a critical regulator of memory CD8+ T cell formation. Elife 2014, 3, 03706. [Google Scholar] [CrossRef]
- Schlie, K.; Westerback, A.; DeVorkin, L.; Hughson, L.R.; Brandon, J.M.; MacPherson, S.; Gadawski, I.; Townsend, K.N.; Poon, V.I.; Elrick, M.A.; et al. Survival of effector CD8+ T cells during influenza infection is dependent on autophagy. J. Immunol. 2015, 194, 4277–4286. [Google Scholar] [CrossRef]
- Wildenberg, M.E.; Vos, A.C.; Wolfkamp, S.C.; Duijvestein, M.; Verhaar, A.P.; Te Velde, A.A.; van den Brink, G.R.; Hommes, D.W. Autophagy attenuates the adaptive immune response by destabilizing the immunologic synapse. Gastroenterology 2012, 142, 1493–1503. [Google Scholar] [CrossRef]
- Xu, X.; Araki, K.; Li, S.; Han, J.H.; Ye, L.; Tan, W.G.; Konieczny, B.T.; Bruinsma, M.W.; Martinez, J.; Pearce, E.L.; et al. Autophagy is essential for effector CD8+ T cell survival and memory formation. Nat. Immunol. 2014, 15, 1152–1161. [Google Scholar] [CrossRef]
- Chu, H.; Khosravi, A.; Kusumawardhani, I.P.; Kwon, A.H.; Vasconcelos, A.C.; Cunha, L.D.; Mayer, A.E.; Shen, Y.; Wu, W.L.; Kambal, A.; et al. Gene-microbiota interactions contribute to the pathogenesis of inflammatory bowel disease. Science 2016, 352, 1116–1120. [Google Scholar] [CrossRef]
- Shen, Y.; Giardino Torchia, M.L.; Lawson, G.W.; Karp, C.L.; Ashwell, J.D.; Mazmanian, S.K. Outer membrane vesicles of a human commensal mediate immune regulation and disease protection. Cell Host Microbe 2012, 12, 509–520. [Google Scholar] [CrossRef]
- Qiao, D.; Zhang, Z.; Zhang, Y.; Chen, Q.; Chen, Y.; Tang, Y.; Sun, X.; Tang, Z.; Dai, Y. Regulation of Endoplasmic Reticulum Stress-Autophagy: A Potential Therapeutic Target for Ulcerative Colitis. Front. Pharmacol. 2021, 12, 697360. [Google Scholar] [CrossRef]
- Hu, X.; Deng, J.; Yu, T.; Chen, S.; Ge, Y.; Zhou, Z.; Guo, Y.; Ying, H.; Zhai, Q.; Chen, Y.; et al. ATF4 Deficiency Promotes Intestinal Inflammation in Mice by Reducing Uptake of Glutamine and Expression of Antimicrobial Peptides. Gastroenterology 2019, 156, 1098–1111. [Google Scholar] [CrossRef]
- Vernia, F.; Valvano, M.; Longo, S.; Cesaro, N.; Viscido, A.; Latella, G. Vitamin D in Inflammatory Bowel Diseases. Mechanisms of Action and Therapeutic Implications. Nutrients 2022, 14, 269. [Google Scholar] [CrossRef]
- Wu, S.; Zhang, Y.G.; Lu, R.; Xia, Y.; Zhou, D.; Petrof, E.O.; Claud, E.C.; Chen, D.; Chang, E.B.; Carmeliet, G.; et al. Intestinal epithelial vitamin D receptor deletion leads to defective autophagy in colitis. Gut 2015, 64, 1082–1094. [Google Scholar] [CrossRef]
- Battistini, C.; Ballan, R.; Herkenhoff, M.E.; Saad, S.M.I.; Sun, J. Vitamin D Modulates Intestinal Microbiota in Inflammatory Bowel Diseases. Int. J. Mol. Sci. 2020, 22, 362. [Google Scholar] [CrossRef]
- Sun, J. VDR/vitamin D receptor regulates autophagic activity through ATG16L1. Autophagy 2016, 12, 1057–1058. [Google Scholar] [CrossRef]
- Lu, R.; Zhang, Y.G.; Xia, Y.; Sun, J. Imbalance of autophagy and apoptosis in intestinal epithelium lacking the vitamin D receptor. FASEB J. 2019, 33, 11845–11856. [Google Scholar] [CrossRef]
- Zhu, T.; Liu, T.J.; Shi, Y.Y.; Zhao, Q. Vitamin D/VDR signaling pathway ameliorates 2,4,6-trinitrobenzene sulfonic acid-induced colitis by inhibiting intestinal epithelial apoptosis. Int. J. Mol. Med. 2015, 35, 1213–1218. [Google Scholar] [CrossRef]
- Li, W.; Lin, Y.; Luo, Y.; Wang, Y.; Lu, Y.; Li, Y.; Guo, H. Vitamin D Receptor Protects against Radiation-Induced Intestinal Injury in Mice via Inhibition of Intestinal Crypt Stem/Progenitor Cell Apoptosis. Nutrients 2021, 13, 2910. [Google Scholar] [CrossRef]
- Bakke, D.; Sun, J. Ancient Nuclear Receptor VDR With New Functions: Microbiome and Inflammation. Inflamm. Bowel Dis. 2018, 24, 1149–1154. [Google Scholar] [CrossRef]
- Law, A.D.; Dutta, U.; Kochhar, R.; Vaishnavi, C.; Kumar, S.; Noor, T.; Bhadada, S.; Singh, K. Vitamin D deficiency in adult patients with ulcerative colitis: Prevalence and relationship with disease severity, extent, and duration. Indian. J. Gastroenterol. 2019, 38, 6–14. [Google Scholar] [CrossRef]
- Wu, M.Y.; Liu, L.; Wang, E.J.; Xiao, H.T.; Cai, C.Z.; Wang, J.; Su, H.; Wang, Y.; Tan, J.; Zhang, Z.; et al. PI3KC3 complex subunit NRBF2 is required for apoptotic cell clearance to restrict intestinal inflammation. Autophagy 2021, 17, 1096–1111. [Google Scholar] [CrossRef]
- Zhang, S.L.; Li, Z.Y.; Wang, D.S.; Xu, T.Y.; Fan, M.B.; Cheng, M.H.; Miao, C.Y. Aggravated ulcerative colitis caused by intestinal Metrnl deficiency is associated with reduced autophagy in epithelial cells. Acta Pharmacol. Sin. 2020, 41, 763–770. [Google Scholar] [CrossRef]
- Shao, B.Z.; Yao, Y.; Zhai, J.S.; Zhu, J.H.; Li, J.P.; Wu, K. The Role of Autophagy in Inflammatory Bowel Disease. Front. Physiol. 2021, 12, 621132. [Google Scholar] [CrossRef]
- Banerjee, S.; Ghosh, S.; Sinha, K.; Chowdhury, S.; Sil, P.C. Sulphur dioxide ameliorates colitis related pathophysiology and inflammation. Toxicology 2019, 412, 63–78. [Google Scholar] [CrossRef]
- Shen, T.; Li, S.; Cai, L.D.; Liu, J.L.; Wang, C.Y.; Gan, W.J.; Li, X.M.; Wang, J.R.; Sun, L.N.; Deng, M.; et al. Erbin exerts a protective effect against inflammatory bowel disease by suppressing autophagic cell death. Oncotarget 2018, 9, 12035–12049. [Google Scholar] [CrossRef]
- Hausmann, M.; Obermeier, F.; Schreiter, K.; Spottl, T.; Falk, W.; Schölmerich, J.; Herfarth, H.; Saftig, P.; Rogler, G. Cathepsin D is up-regulated in inflammatory bowel disease macrophages. Clin. Exp. Immunol. 2004, 136, 157–167. [Google Scholar] [CrossRef]
- Fischbeck, A.; Leucht, K.; Frey-Wagner, I.; Bentz, S.; Pesch, T.; Kellermeier, S.; Krebs, M.; Fried, M.; Rogler, G.; Hausmann, M.; et al. Sphingomyelin induces cathepsin D-mediated apoptosis in intestinal epithelial cells and increases inflammation in DSS colitis. Gut 2011, 60, 55–65. [Google Scholar] [CrossRef]
- Menzel, K.; Hausmann, M.; Obermeier, F.; Schreiter, K.; Dunger, N.; Bataille, F.; Falk, W.; Scholmerich, J.; Herfarth, H.; Rogler, G. Cathepsins B, L and D in inflammatory bowel disease macrophages and potential therapeutic effects of cathepsin inhibition in vivo. Clin. Exp. Immunol. 2006, 146, 169–180. [Google Scholar] [CrossRef]
- Macias-Ceja, D.C.; Cosín-Roger, J.; Ortiz-Masiá, D.; Salvador, P.; Hernández, C.; Esplugues, J.V.; Calatayud, S.; Barrachina, M.D. Stimulation of autophagy prevents intestinal mucosal inflammation and ameliorates murine colitis. Br. J. Pharmacol. 2017, 174, 2501–2511. [Google Scholar] [CrossRef]
- Cosin-Roger, J.; Canet, F.; Macias-Ceja, D.C.; Gisbert-Ferrándiz, L.; Ortiz-Masiá, D.; Esplugues, J.V.; Alós, R.; Navarro, F.; Barrachina, M.D.; Calatayud, S. Autophagy Stimulation as a Potential Strategy against Intestinal Fibrosis. Cells 2019, 8, 1078. [Google Scholar] [CrossRef]
- Salvador, P.; Macías-Ceja, D.C.; Gisbert-Ferrándiz, L.; Hernández, C.; Bernardo, D.; Alós, R.; Navarro-Vicente, F.; Esplugues, J.V.; Ortiz-Masiá, D.; Barrachina, M.D.; et al. CD16+ Macrophages Mediate Fibrosis in Inflammatory Bowel Disease. J. Crohns Colitis. 2018, 12, 589–599. [Google Scholar] [CrossRef]
- Mathur, R.; Alam, M.M.; Zhao, X.F.; Liao, Y.; Shen, J.; Morgan, S.; Huang, T.; Lee, H.; Lee, E.; Huang, Y.; et al. Induction of autophagy in Cx3cr1+ mononuclear cells limits IL-23/IL-22 axis-mediated intestinal fibrosis. Mucosal Immunol. 2019, 12, 612–623. [Google Scholar] [CrossRef]
- Butera, A.; Quaranta, M.T.; Crippa, L.; Spinello, I.; Saulle, E.; Di Carlo, N.; Campanile, D.; Boirivant, M.; Labbaye, C. CD147 Targeting by AC-73 Induces Autophagy and Reduces Intestinal Fibrosis Associated with TNBS Chronic Colitis. J. Crohns Colitis. 2022, 16, 1751–1761. [Google Scholar] [CrossRef]
- Haq, S.; Grondin, J.; Banskota, S.; Khan, W.I. Autophagy: Roles in intestinal mucosal homeostasis and inflammation. J. Biomed. Sci. 2019, 26, 19. [Google Scholar] [CrossRef]
- Wu, Y.; Tang, L.; Wang, B.; Sun, Q.; Zhao, P.; Li, W. The role of autophagy in maintaining intestinal mucosal barrier. J. Cell Physiol. 2019, 234, 19406–19419. [Google Scholar] [CrossRef]
- Dombi, E.; Mortiboys, H.; Poulton, J. Modulating Mitophagy in Mitochondrial Disease. Curr. Med. Chem. 2018, 25, 5597–5612. [Google Scholar] [CrossRef]
- Kathiria, A.S.; Butcher, L.D.; Feagins, L.A.; Souza, R.F.; Boland, C.R.; Theiss, A.L. Prohibitin 1 modulates mitochondrial stress-related autophagy in human colonic epithelial cells. PLoS ONE 2012, 7, e31231. [Google Scholar] [CrossRef]
- Vincent, G.; Novak, E.A.; Siow, V.S.; Cunningham, K.E.; Griffith, B.D.; Comerford, T.E.; Mentrup, H.L.; Stolz, D.B.; Loughran, P.; Ranganathan, S.; et al. Nix-Mediated Mitophagy Modulates Mitochondrial Damage During Intestinal Inflammation. Antioxid. Redox Signal. 2020, 33, 1–19. [Google Scholar] [CrossRef]
- Carlberg, C.; Campbell, M.J. Vitamin D receptor signaling mechanisms: Integrated actions of a well-defined transcription factor. Steroids 2013, 78, 127–136. [Google Scholar] [CrossRef]
- Chetcuti Zammit, S.; Ellul, P.; Girardin, G.; Valpiani, D.; Nielsen, K.R.; Olsen, J.; Goldis, A.; Lazar, D.; Shonová, O.; Nováková, M.; et al. Vitamin D deficiency in a European inflammatory bowel disease inception cohort: An Epi-IBD study. Eur. J. Gastroenterol. Hepatol. 2018, 30, 1297–1303. [Google Scholar] [CrossRef]
- Ma, Z.; Wu, J.; Wu, Y.; Sun, X.; Rao, Z.; Sun, N.; Fu, Y.; Zhang, Z.; Li, J.; Xiao, M.; et al. Parkin increases the risk of colitis by downregulation of VDR via autophagy-lysosome degradation. Int. J. Biol. Sci. 2023, 19, 1633–1644. [Google Scholar] [CrossRef]
- Berger, E.; Rath, E.; Yuan, D.; Waldschmitt, N.; Khaloian, S.; Allgäuer, M.; Staszewski, O.; Lobner, E.M.; Schöttl, T.; Giesbertz, P.; et al. Mitochondrial function controls intestinal epithelial stemness and proliferation. Nat. Commun. 2016, 7, 13171. [Google Scholar] [CrossRef]
- Khaloian, S.; Rath, E.; Hammoudi, N.; Gleisinger, E.; Blutke, A.; Giesbertz, P.; Berger, E.; Metwaly, A.; Waldschmitt, N.; Allez, M.; et al. Mitochondrial impairment drives intestinal stem cell transition into dysfunctional Paneth cells predicting Crohn’s disease recurrence. Gut 2020, 69, 1939–1951. [Google Scholar] [CrossRef]
- Rodríguez-Colman, M.J.; Schewe, M.; Meerlo, M.; Stigter, E.; Gerrits, J.; Pras-Raves, M.; Sacchetti, A.; Hornsveld, M.; Oost, K.C.; Snippert, H.J.; et al. Interplay between metabolic identities in the intestinal crypt supports stem cell function. Nature 2017, 543, 424–427. [Google Scholar] [CrossRef]
- Liu, C.; Wang, J.; Yang, Y.; Liu, X.; Zhu, Y.; Zou, J.; Peng, S.; Le, T.H.; Chen, Y.; Zhao, S.; et al. Ginsenoside Rd ameliorates colitis by inducing p62-driven mitophagy-mediated NLRP3 inflammasome inactivation in mice. Biochem. Pharmacol. 2018, 155, 366–379. [Google Scholar] [CrossRef]
- Mai, C.T.; Wu, M.M.; Wang, C.L.; Su, Z.R.; Cheng, Y.Y.; Zhang, X.J. Palmatine attenuated dextran sulfate sodium (DSS)-induced colitis via promoting mitophagy-mediated NLRP3 inflammasome inactivation. Mol. Immunol. 2019, 105, 76–85. [Google Scholar] [CrossRef]
- Patel, J. IL-10 reprogramming of metabolism in macrophages through mitophagy. Cardiovasc. Res. 2017, 113, 40–41. [Google Scholar] [CrossRef]
- Singh, S.B.; Ornatowski, W.; Vergne, I.; Naylor, J.; Delgado, M.; Roberts, E.; Ponpuak, M.; Master, S.; Pilli, M.; White, E.; et al. Human IRGM regulates autophagy and cell-autonomous immunity functions through mitochondria. Nat. Cell Biol. 2010, 12, 1154–1165. [Google Scholar] [CrossRef]
- Eckmann, L.; Nebelsiek, T.; Fingerle, A.A.; Dann, S.M.; Mages, J.; Lang, R.; Robine, S.; Kagnoff, M.F.; Schmid, R.M.; Karin, M.; et al. Opposing functions of IKKbeta during acute and chronic intestinal inflammation. Proc. Natl. Acad. Sci. USA. 2008, 105, 15058–15063. [Google Scholar] [CrossRef]
- Patankar, J.V.; Becker, C. Cell death in the gut epithelium and implications for chronic inflammation. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 543–556. [Google Scholar] [CrossRef]
- Iwamoto, M.; Koji, T.; Makiyama, K.; Kobayashi, N.; Nakane, P.K. Apoptosis of crypt epithelial cells in ulcerative colitis. J. Pathol. 1996, 180, 152–159. [Google Scholar] [CrossRef]
- Pedersen, J.; LaCasse, E.C.; Seidelin, J.B.; Coskun, M.; Nielsen, O.H. Inhibitors of apoptosis (IAPs) regulate intestinal immunity and inflammatory bowel disease (IBD) inflammation. Trends Mol. Med. 2014, 20, 652–665. [Google Scholar] [CrossRef]
- Parker, A.; Vaux, L.; Patterson, A.M.; Modasia, A.; Muraro, D.; Fletcher, A.G.; Byrne, H.M.; Maini, P.K.; Watson, A.J.M.; Pin, C. Elevated apoptosis impairs epithelial cell turnover and shortens villi in TNF-driven intestinal inflammation. Cell Death Dis. 2019, 10, 108. [Google Scholar] [CrossRef]
- Vetuschi, A.; Latella, G.; Sferra, R.; Caprilli, R.; Gaudio, E. Increased proliferation and apoptosis of colonic epithelial cells in dextran sulfate sodium-induced colitis in rats. Dig. Dis. Sci. 2002, 47, 1447–1457. [Google Scholar] [CrossRef]
- Sträter, J.; Möller, P. Expression and function of death receptors and their natural ligands in the intestine. Ann. N. Y Acad. Sci. 2000, 915, 162–170. [Google Scholar] [CrossRef]
- Möller, P.; Walczak, H.; Reidl, S.; Sträter, J.; Krammer, P.H. Paneth cells express high levels of CD95 ligand transcripts: A unique property among gastrointestinal epithelia. Am. J. Pathol. 1996, 149, 9–13. [Google Scholar]
- Sträter, J.; Wellisch, I.; Riedl, S.; Walczak, H.; Koretz, K.; Tandara, A.; Krammer, P.H.; Möller, P. CD95 (APO-1/Fas)-mediated apoptosis in colon epithelial cells: A possible role in ulcerative colitis. Gastroenterology 1997, 113, 160–167. [Google Scholar] [CrossRef]
- Dirisina, R.; Katzman, R.B.; Goretsky, T.; Managlia, E.; Mittal, N.; Williams, D.B.; Qiu, W.; Yu, J.; Chandel, N.S.; Zhang, L.; et al. p53 and PUMA independently regulate apoptosis of intestinal epithelial cells in patients and mice with colitis. Gastroenterology 2011, 141, 1036–1045. [Google Scholar] [CrossRef]
- Sidi, S.; Sanda, T.; Kennedy, R.D.; Hagen, A.T.; Jette, C.A.; Hoffmans, R.; Pascual, J.; Imamura, S.; Kishi, S.; Amatruda, J.F.; et al. Chk1 suppresses a caspase-2 apoptotic response to DNA damage that bypasses p53, Bcl-2, and caspase-3. Cell 2008, 133, 864–877. [Google Scholar] [CrossRef]
- Greenow, K.R.; Clarke, A.R.; Jones, R.H. Chk1 deficiency in the mouse small intestine results in p53-independent crypt death and subsequent intestinal compensation. Oncogene 2009, 28, 1443–1453. [Google Scholar] [CrossRef]
- Watari, A.; Hasegawa, M.; Yagi, K.; Kondoh, M. Checkpoint Kinase 1 Activation Enhances Intestinal Epithelial Barrier Function via Regulation of Claudin-5 Expression. PLoS ONE 2016, 11, e0145631. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, X.; Li, Y.; Li, Y.; Wang, Y.; Zhu, L.; Chen, P.; Tian, Z.; Qiu, Y.; Feng, R.; et al. DNA Damage-Regulated Autophagy Modulator 1 (DRAM1) Mediates Autophagy and Apoptosis of Intestinal Epithelial Cells in Inflammatory Bowel Disease. Dig. Dis. Sci. 2021, 66, 3375–3390. [Google Scholar] [CrossRef]
- Zeissig, S.; Bojarski, C.; Buergel, N.; Mankertz, J.; Zeitz, M.; Fromm, M.; Schulzke, J.D. Downregulation of epithelial apoptosis and barrier repair in active Crohn’s disease by tumour necrosis factor alpha antibody treatment. Gut 2004, 53, 1295–1302. [Google Scholar] [CrossRef]
- Lin, A.; Karin, M. NF-kappaB in cancer: A marked target. Semin. Cancer Biol. 2003, 13, 107–114. [Google Scholar] [CrossRef]
- Vereecke, L.; Beyaert, R.; van Loo, G. The ubiquitin-editing enzyme A20 (TNFAIP3) is a central regulator of immunopathology. Trends Immunol. 2009, 30, 383–391. [Google Scholar] [CrossRef]
- Kaser, A.; Zeissig, S.; Blumberg, R.S. Inflammatory bowel disease. Annu. Rev. Immunol. 2010, 28, 573–621. [Google Scholar] [CrossRef]
- Qiu, W.; Wu, B.; Wang, X.; Buchanan, M.E.; Regueiro, M.D.; Hartman, D.J.; Schoen, R.E.; Yu, J.; Zhang, L. PUMA-mediated intestinal epithelial apoptosis contributes to ulcerative colitis in humans and mice. J. Clin. Investig. 2011, 121, 1722–1732. [Google Scholar] [CrossRef]
- Lin, W.; Ma, C.; Su, F.; Jiang, Y.; Lai, R.; Zhang, T.; Sun, K.; Fan, L.; Cai, Z.; Li, Z.; et al. Raf kinase inhibitor protein mediates intestinal epithelial cell apoptosis and promotes IBDs in humans and mice. Gut 2017, 66, 597–610. [Google Scholar] [CrossRef]
- Vlantis, K.; Wullaert, A.; Polykratis, A.; Kondylis, V.; Dannappel, M.; Schwarzer, R.; Welz, P.; Corona, T.; Walczak, H.; Weih, F.; et al. NEMO Prevents RIP Kinase 1-Mediated Epithelial Cell Death and Chronic Intestinal Inflammation by NF-κB-Dependent and -Independent Functions. Immunity 2016, 44, 553–567. [Google Scholar] [CrossRef]
- Wong, J.; Garcia-Carbonell, R.; Zelic, M.; Ho, S.B.; Boland, B.S.; Yao, S.J.; Desai, S.A.; Das, S.; Planell, N.; Harris, P.A.; et al. RIPK1 Mediates TNF-Induced Intestinal Crypt Apoptosis During Chronic NF-κB Activation. Cell Mol. Gastroenterol. Hepatol. 2020, 9, 295–312. [Google Scholar] [CrossRef]
- Yao, X.; Cadwell, K. Tumor Necrosis Factor-α-Induced Apoptosis in the Intestinal Epithelium due to Chronic Nuclear Factor Kappa B Signaling Is Mediated by Receptor Interacting Serine/Threonine Kinase 1. Cell. Mol. Gastroenterol. Hepatol. 2020, 9, 337–338. [Google Scholar] [CrossRef]
- Garcia-Carbonell, R.; Wong, J.; Kim, J.Y.; Close, L.A.; Boland, B.S.; Wong, T.L.; Harris, P.A.; Ho, S.B.; Das, S.; Ernst, P.B.; et al. Elevated A20 promotes TNF-induced and RIPK1-dependent intestinal epithelial cell death. Proc. Natl. Acad. Sci. USA 2018, 115, E9192–E9200. [Google Scholar] [CrossRef]
- Kattah, M.G.; Shao, L.; Rosli, Y.Y.; Shimizu, H.; Whang, M.I.; Advincula, R.; Achacoso, P.; Shah, S.; Duong, B.H.; Onizawa, M.; et al. A20 and ABIN-1 synergistically preserve intestinal epithelial cell survival. J. Exp. Med. 2018, 215, 1839–1852. [Google Scholar] [CrossRef]
- Zaidi, D.; Huynh, H.Q.; Carroll, M.W.; Baksh, S.; Wine, E. Tumor necrosis factor α-induced protein 3 (A20) is dysregulated in pediatric Crohn disease. Clin. Exp. Gastroenterol. 2018, 11, 217–231. [Google Scholar] [CrossRef]
- Geng, J.; Ito, Y.; Shi, L.; Amin, P.; Chu, J.; Ouchida, A.T.; Mookhtiar, A.K.; Zhao, H.; Xu, D.; Shan, B.; et al. Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nat. Commun. 2017, 8, 359. [Google Scholar] [CrossRef]
- Guo, X.; Yin, H.; Chen, Y.; Li, L.; Li, J.; Liu, Q. TAK1 regulates caspase 8 activation and necroptotic signaling via multiple cell death checkpoints. Cell Death Dis. 2016, 7, e2381. [Google Scholar] [CrossRef]
- Totzke, J.; Gurbani, D.; Raphemot, R.; Hughes, P.F.; Bodoor, K.; Carlson, D.A.; Loiselle, D.R.; Bera, A.K.; Eibschutz, L.S.; Perkins, M.M.; et al. Takinib, a Selective TAK1 Inhibitor, Broadens the Therapeutic Efficacy of TNF-α Inhibition for Cancer and Autoimmune Disease. Cell Chem. Biol. 2017, 24, 1029–1039. [Google Scholar] [CrossRef]
- Goretsky, T.; Dirisina, R.; Sinh, P.; Mittal, N.; Managlia, E.; Williams, D.B.; Posca, D.; Ryu, H.; Katzman, R.B.; Barrett, T.A. p53 mediates TNF-induced epithelial cell apoptosis in IBD. Am. J. Pathol. 2012, 181, 1306–1315. [Google Scholar] [CrossRef]
- Li, M.; Zhang, S.; Qiu, Y.; He, Y.; Chen, B.; Mao, R.; Cui, Y.; Zeng, Z.; Chen, M. Upregulation of miR-665 promotes apoptosis and colitis in inflammatory bowel disease by repressing the endoplasmic reticulum stress components XBP1 and ORMDL3. Cell Death Dis. 2017, 8, e2699. [Google Scholar] [CrossRef]
- Dias, C.B.; Milanski, M.; Portovedo, M.; Horita, V.; Ayrizono Mde, L.; Planell, N.; Coy, C.S.; Velloso, L.A.; Meirelles, L.R.; Leal, R.F. Defective apoptosis in intestinal and mesenteric adipose tissue of Crohn’s disease patients. PLoS ONE 2014, 9, e98547. [Google Scholar] [CrossRef]
- Itoh, J.; de La Motte, C.; Strong, S.A.; Levine, A.D.; Fiocchi, C. Decreased Bax expression by mucosal T cells favours resistance to apoptosis in Crohn’s disease. Gut 2001, 49, 35–41. [Google Scholar] [CrossRef]
- Santaolalla, R.; Mañé, J.; Pedrosa, E.; Lorén, V.; Fernández-Bañares, F.; Mallolas, J.; Carrasco, A.; Salas, A.; Rosinach, M.; Forné, M.; et al. Apoptosis resistance of mucosal lymphocytes and IL-10 deficiency in patients with steroid-refractory Crohn’s disease. Inflamm. Bowel Dis. 2011, 17, 1490–1500. [Google Scholar] [CrossRef]
- Qian, J.; Zhao, W.; Miao, X.; Li, L.; Zhang, D. Sam68 modulates apoptosis of intestinal epithelial cells via mediating NF-κB activation in ulcerative colitis. Mol. Immunol. 2016, 75, 48–59. [Google Scholar] [CrossRef]
- Tang, B.; Zhu, J.; Fang, S.; Wang, Y.; Vinothkumar, R.; Li, M.; Weng, Q.; Zheng, L.; Yang, Y.; Qiu, R.; et al. Pharmacological inhibition of MELK restricts ferroptosis and the inflammatory response in colitis and colitis-propelled carcinogenesis. Free Radic. Biol. Med. 2021, 172, 312–329. [Google Scholar] [CrossRef]
- Zhang, X.; Ma, Y.; Ji, J.; Zhao, X.; Yuan, J.; Wang, H.; Lv, G. High-fat diet alleviates colitis by inhibiting ferroptosis via solute carrier family seven member 11. J. Nutr. Biochem. 2022, 109, 109106. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, P.; Chen, W.; Chen, G. Ferroptosis mediated DSS-induced ulcerative colitis associated with Nrf2/HO-1 signaling pathway. Immunol. Lett. 2020, 225, 9–15. [Google Scholar] [CrossRef]
- Huang, F.; Zhang, S.; Li, X.; Huang, Y.; He, S.; Luo, L. STAT3-mediated ferroptosis is involved in ulcerative colitis. Free Radic. Biol. Med. 2022, 188, 375–385. [Google Scholar] [CrossRef]
- Cui, D.J.; Chen, C.; Yuan, W.Q.; Yang, Y.H.; Han, L. Integrative analysis of ferroptosis-related genes in ulcerative colitis. J. Int. Med. Res. 2021, 49, 3000605211042975. [Google Scholar] [CrossRef]
- Luo, L.; Zhang, S.; Guo, N.; Li, H.; He, S. ACSF2-mediated ferroptosis is involved in ulcerative colitis. Life Sci. 2023, 313, 121272. [Google Scholar] [CrossRef]
- Xu, M.; Tao, J.; Yang, Y.; Tan, S.; Liu, H.; Jiang, J.; Zheng, F.; Wu, B. Ferroptosis involves in intestinal epithelial cell death in ulcerative colitis. Cell Death Dis. 2020, 11, 86. [Google Scholar] [CrossRef]
- Qian, R.; Tang, M.; Ouyang, Z.; Cheng, H.; Xing, S. Identification of ferroptosis-related genes in ulcerative colitis: A diagnostic model with machine learning. Ann. Transl. Med. 2023, 11, 177. [Google Scholar] [CrossRef]
- Xu, C.; Liu, Z.; Xiao, J. Ferroptosis: A Double-Edged Sword in Gastrointestinal Disease. Int. J. Mol. Sci. 2021, 22, 12403. [Google Scholar] [CrossRef]
- Dong, S.; Lu, Y.; Peng, G.; Li, J.; Li, W.; Li, M.; Wang, H.; Liu, L.; Zhao, Q. Furin inhibits epithelial cell injury and alleviates experimental colitis by activating the Nrf2-Gpx4 signaling pathway. Dig. Liver Dis. 2021, 53, 1276–1285. [Google Scholar] [CrossRef]
- Wang, S.; Liu, W.; Wang, J.; Bai, X. Curculigoside inhibits ferroptosis in ulcerative colitis through the induction of GPX4. Life Sci. 2020, 259, 118356. [Google Scholar] [CrossRef]
- Rahman, M.S.; Alam, M.B.; Kim, Y.K.; Madina, M.H.; Fliss, I.; Lee, S.H.; Yoo, J.C. Activation of Nrf2/HO-1 by Peptide YD1 Attenuates Inflammatory Symptoms through Suppression of TLR4/MYyD88/NF-κB Signaling Cascade. Int. J. Mol. Sci. 2021, 22, 5161. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, J.; Li, J.; Zhu, J.; Wang, R.; Xi, Q.; Wu, H.; Shi, T.; Chen, W. Astragalus polysaccharide prevents ferroptosis in a murine model of experimental colitis and human Caco-2 cells via inhibiting NRF2/HO-1 pathway. Eur. J. Pharmacol. 2021, 911, 174518. [Google Scholar] [CrossRef]
- Mei, Y.; Wang, Z.; Zhang, Y.; Wan, T.; Xue, J.; He, W.; Luo, Y.; Xu, Y.; Bai, X.; Wang, Q.; et al. FA-97, a New Synthetic Caffeic Acid Phenethyl Ester Derivative, Ameliorates DSS-Induced Colitis Against Oxidative Stress by Activating Nrf2/HO-1 Pathway. Front. Immunol. 2020, 10, 2969. [Google Scholar] [CrossRef]
- Khor, T.O.; Huang, M.T.; Kwon, K.H.; Chan, J.Y.; Reddy, B.S.; Kong, A.N. Nrf2-deficient mice have an increased susceptibility to dextran sulfate sodium-induced colitis. Cancer Res. 2006, 66, 11580–11584. [Google Scholar] [CrossRef]
- Khor, T.O.; Huang, M.T.; Prawan, A.; Liu, Y.; Hao, X.; Yu, S.; Cheung, W.K.; Chan, J.Y.; Reddy, B.S.; Yang, C.S.; et al. Increased susceptibility of Nrf2 knockout mice to colitis-associated colorectal cancer. Cancer Prev. Res. 2008, 1, 187–191. [Google Scholar] [CrossRef]
- Su, D.; Wang, X.; Ma, Y.; Hao, J.; Wang, J.; Lu, Y.; Liu, Y.; Wang, X.; Zhang, L. Nrf2-induced miR-23a-27a-24-2 cluster modulates damage repair of intestinal mucosa by targeting the Bach1/HO-1 axis in inflammatory bowel diseases. Free Radic. Biol. Med. 2021, 163, 1–9. [Google Scholar] [CrossRef]
- Stenke, E.; Aviello, G.; Singh, A.; Martin, S.; Winter, D.; Sweeney, B.; McDermott, M.; Bourke, B.; Hussey, S.; Knaus, U.G. NADPH oxidase 4 is protective and not fibrogenic in intestinal inflammation. Redox Biol. 2020, 37, 101752. [Google Scholar] [CrossRef]
- Wittig, B.M.; Sabat, R.; Holzlöhner, P.; Witte-Händel, E.; Heilmann, K.; Witte, K.; Triebus, J.; Tzankov, A.; Laman, J.D.; Bokemeyer, B.; et al. Absence of specific alternatively spliced exon of CD44 in macrophages prevents colitis. Mucosal Immunol. 2018, 11, 846–860. [Google Scholar] [CrossRef]
- de Silva, P.S.; Olsen, A.; Christensen, J.; Schmidt, E.B.; Overvaad, K.; Tjonneland, A.; Hart, A.R. An association between dietary arachidonic acid, measured in adipose tissue, and ulcerative colitis. Gastroenterology 2010, 139, 1912–1917. [Google Scholar] [CrossRef]
- Vargas-Robles, H.; Castro-Ochoa, K.F.; Citalán-Madrid, A.F.; Schnoor, M. Beneficial effects of nutritional supplements on intestinal epithelial barrier functions in experimental colitis models in vivo. World J. Gastroenterol. 2019, 25, 4181–4198. [Google Scholar] [CrossRef]
- Alim, I.; Caulfield, J.T.; Chen, Y.; Swarup, V.; Geschwind, D.H.; Ivanova, E.; Seravalli, J.; Ai, Y.; Sansing, L.H.; Ste Marie, E.J.; et al. Selenium Drives a Transcriptional Adaptive Program to Block Ferroptosis and Treat Stroke. Cell 2019, 177, 1262–1279. [Google Scholar] [CrossRef]
- Kroschwald, S.; Chiu, C.Y.; Heydeck, D.; Rohwer, N.; Gehring, T.; Seifert, U.; Lux, A.; Rothe, M.; Weylandt, K.H.; Kuhn, H. Female mice carrying a defective Alox15 gene are protected from experimental colitis via sustained maintenance of the intestinal epithelial barrier function. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 866–880. [Google Scholar] [CrossRef]
- Jupp, J.; Hillier, K.; Elliott, D.H.; Fine, D.R.; Bateman, A.C.; Johnson, P.A.; Cazaly, A.M.; Penrose, J.F.; Sampson, A.P. Colonic expression of leukotriene-pathway enzymes in inflammatory bowel diseases. Inflamm. Bowel Dis. 2007, 13, 537–546. [Google Scholar] [CrossRef]
- Wenzel, S.E.; Tyurina, Y.Y.; Zhao, J.; St Croix, C.M.; Dar, H.H.; Mao, G.; Tyurin, V.A.; Anthonymuthu, T.S.; Kapralov, A.A.; Amoscato, A.A.; et al. PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell 2017, 171, 628–641. [Google Scholar] [CrossRef]
- Anthonymuthu, T.S.; Tyurina, Y.Y.; Sun, W.Y.; Mikulska-Ruminska, K.; Shrivastava, I.H.; Tyurin, V.A.; Cinemre, F.B.; Dar, H.H.; VanDemark, A.P.; Holman, T.R.; et al. Resolving the paradox of ferroptotic cell death: Ferrostatin-1 binds to 15LOX/PEBP1 complex, suppresses generation of peroxidized ETE-PE, and protects against ferroptosis. Redox Biol. 2021, 38, 101744. [Google Scholar] [CrossRef]
- Zhang, X.; Ma, Y.; Lv, G.; Wang, H. Ferroptosis as a therapeutic target for inflammation-related intestinal diseases. Front. Pharmacol. 2023, 14, 1095366. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, L.; Zhang, X.; Cui, W.; Liu, Y.; Sun, Q.R.; He, Q.; Zhao, S.; Zhang, G.A.; Wang, Y.; et al. Epigenetic regulation of ferroptosis by H2B monoubiquitination and p53. EMBO Rep. 2019, 20, 47563. [Google Scholar] [CrossRef]
- Panda, S.K.; Peng, V.; Sudan, R.; Ulezko Antonova, A.; Di Luccia, B.; Ohara, T.E.; Fachi, J.L.; Grajales-Reyes, G.E.; Jaeger, N.; Trsan, T.; et al. Repression of the aryl-hydrocarbon receptor prevents oxidative stress and ferroptosis of intestinal intraepithelial lymphocytes. Immunity 2023, 56, 797–812. [Google Scholar] [CrossRef]
- Zhang, D.; Li, Y.; Du, C.; Sang, L.; Liu, L.; Li, Y.; Wang, F.; Fan, W.; Tang, P.; Zhang, S.; et al. Evidence of pyroptosis and ferroptosis extensively involved in autoimmune diseases at the single-cell transcriptome level. J. Transl. Med. 2022, 20, 363. [Google Scholar] [CrossRef]
- Zhu, X.; Messer, J.S.; Wang, Y.; Lin, F.; Cham, C.M.; Chang, J.; Billiar, T.R.; Lotze, M.T.; Boone, D.L.; Chang, E.B. Cytosolic HMGB1 controls the cellular autophagy/apoptosis checkpoint during inflammation. J. Clin. Investig. 2015, 125, 1098–1110. [Google Scholar] [CrossRef]
- Hooper, K.M.; Barlow, P.G.; Henderson, P.; Stevens, C. Interactions Between Autophagy and the Unfolded Protein Response: Implications for Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2019, 25, 661–671. [Google Scholar] [CrossRef]
- Arab, H.H.; Al-Shorbagy, M.Y.; Saad, M.A. Activation of autophagy and suppression of apoptosis by dapagliflozin attenuates experimental inflammatory bowel disease in rats: Targeting AMPK/mTOR, HMGB1/RAGE and Nrf2/HO-1 pathways. Chem. Biol. Interact. 2021, 335, 109368. [Google Scholar] [CrossRef]
- Blander, J.M. Death in the intestinal epithelium-basic biology and implications for inflammatory bowel disease. FEBS J. 2016, 283, 2720–2730. [Google Scholar] [CrossRef]
- Levin, A.D.; Koelink, P.J.; Bloemendaal, F.M.; Vos, A.C.; D’Haens, G.R.; van den Brink, G.R.; Wildenberg, M.E. Autophagy Contributes to the Induction of Anti-TNF Induced Macrophages. J. Crohns Colitis. 2016, 10, 323–329. [Google Scholar] [CrossRef]
- Miao, Y.; Lv, Q.; Qiao, S.; Yang, L.; Tao, Y.; Yan, W.; Wang, P.; Cao, N.; Dai, Y.; Wei, Z. Alpinetin improves intestinal barrier homeostasis via regulating AhR/suv39h1/TSC2/mTORC1/autophagy pathway. Toxicol. Appl. Pharmacol. 2019, 384, 114772. [Google Scholar] [CrossRef]
- Hu, C.A.; Hou, Y.; Yi, D.; Qiu, Y.; Wu, G.; Kong, X.; Yin, Y. Autophagy and tight junction proteins in the intestine and intestinal diseases. Anim. Nutr. 2015, 1, 123–127. [Google Scholar] [CrossRef]
- Crook, N.E.; Clem, R.J.; Miller, L.K. An apoptosis-inhibiting baculovirus gene with a zinc finger-like motif. J. Virol. 1993, 67, 2168–2174. [Google Scholar] [CrossRef]
- Eckelman, B.P.; Salvesen, G.S.; Scott, F.L. Human inhibitor of apoptosis proteins: Why XIAP is the black sheep of the family. EMBO Rep. 2006, 7, 988–994. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature. 2008, 451, 1069–1075. [Google Scholar] [CrossRef]
- Huang, X.; Wu, Z.; Mei, Y.; Wu, M. XIAP inhibits autophagy via XIAP-Mdm2-p53 signalling. EMBO J. 2013, 32, 2204–2216. [Google Scholar] [CrossRef]
- Zeissig, Y.; Petersen, B.S.; Milutinovic, S.; Bosse, E.; Mayr, G.; Peuker, K.; Hartwig, J.; Keller, A.; Kohl, M.; Laass, M.W.; et al. XIAP variants in male Crohn’s disease. Gut 2015, 64, 66–76. [Google Scholar] [CrossRef]
- Zhao, Y.; Guo, Q.; Zhao, K.; Zhou, Y.; Li, W.; Pan, C.; Qiang, L.; Li, Z.; Lu, N. Small molecule GL-V9 protects against colitis-associated colorectal cancer by limiting NLRP3 inflammasome through autophagy. Oncoimmunology 2017, 7, e1375640. [Google Scholar] [CrossRef]
- Zheng, X.; Hu, M.; Zang, X.; Fan, Q.; Liu, Y.; Che, Y.; Guan, X.; Hou, Y.; Wang, G.; Hao, H. Kynurenic acid/GPR35 axis restricts NLRP3 inflammasome activation and exacerbates colitis in mice with social stress. Brain Behav. Immun. 2019, 79, 244–255. [Google Scholar] [CrossRef]
- Ding, W.; Ding, Z.; Wang, Y.; Zhu, Y.; Gao, Q.; Cao, W.; Du, R. Evodiamine Attenuates Experimental Colitis Injury Via Activating Autophagy and Inhibiting NLRP3 Inflammasome Assembly. Front. Pharmacol. 2020, 11, 573870. [Google Scholar] [CrossRef]
- Samoilă, I.; Dinescu, S.; Costache, M. Interplay between Cellular and Molecular Mechanisms Underlying Inflammatory Bowel Diseases Development-A Focus on Ulcerative Colitis. Cells 2020, 9, 1647. [Google Scholar] [CrossRef]
- Liu, R.; Li, X.; Ma, H.; Yang, Q.; Shang, Q.; Song, L.; Zheng, Z.; Zhang, S.; Pan, Y.; Huang, P.; et al. Spermidine endows macrophages anti-inflammatory properties by inducing mitochondrial superoxide-dependent AMPK activation, Hif-1α upregulation and autophagy. Free Radic. Biol. Med. 2020, 161, 339–350. [Google Scholar] [CrossRef]
- Tartakover Matalon, S.; Ringel, Y.; Konikoff, F.; Drucker, L.; Pery, S.; Naftali, T. Cannabinoid receptor 2 agonist promotes parameters implicated in mucosal healing in patients with inflammatory bowel disease. United Eur. Gastroenterol. J. 2020, 8, 271–283. [Google Scholar] [CrossRef]
- Ke, P.; Shao, B.Z.; Xu, Z.Q.; Wei, W.; Han, B.Z.; Chen, X.W.; Su, D.F.; Liu, C. Activation of Cannabinoid Receptor 2 Ameliorates DSS-Induced Colitis through Inhibiting NLRP3 Inflammasome in Macrophages. PLoS ONE 2016, 11, e0155076. [Google Scholar] [CrossRef]
- Wang, Z.; Shi, L.; Hua, S.; Qi, C.; Fang, M. IL-33 ameliorates experimental colitis involving regulation of autophagy of macrophages in mice. Cell Biosci. 2019, 9, 10. [Google Scholar] [CrossRef]
- Zheng, Y.; Yu, Y.; Chen, X.F.; Yang, S.L.; Tang, X.L.; Xiang, Z.G. Intestinal Macrophage Autophagy and its Pharmacological Application in Inflammatory Bowel Disease. Front. Pharmacol. 2021, 12, 803686. [Google Scholar] [CrossRef]
- Tschurtschenthaler, M.; Adolph, T.E. The Selective Autophagy Receptor Optineurin in Crohn’s Disease. Front. Immunol. 2018, 9, 766. [Google Scholar] [CrossRef]
- Qin, Z.; Wan, J.J.; Sun, Y.; Wu, T.; Wang, P.Y.; Du, P.; Su, D.F.; Yang, Y.; Liu, X. Nicotine protects against DSS colitis through regulating microRNA-124 and STAT3. J. Mol. Med. 2017, 95, 221–233. [Google Scholar] [CrossRef]
- Shao, B.Z.; Ke, P.; Xu, Z.Q.; Wei, W.; Cheng, M.H.; Han, B.Z.; Chen, X.W.; Su, D.F.; Liu, C. Autophagy Plays an Important Role in Anti-inflammatory Mechanisms Stimulated by Alpha7 Nicotinic Acetylcholine Receptor. Front. Immunol. 2017, 8, 553. [Google Scholar] [CrossRef]
- Shao, B.Z.; Wang, S.L.; Pan, P.; Yao, J.; Wu, K.; Li, Z.S.; Bai, Y.; Linghu, E.Q. Targeting NLRP3 Inflammasome in Inflammatory Bowel Disease: Putting out the Fire of Inflammation. Inflammation 2019, 42, 1147–1159. [Google Scholar] [CrossRef]
- Lin, X.T.; Zheng, X.B.; Fan, D.J.; Yao, Q.Q.; Hu, J.C.; Lian, L.; Wu, X.J.; Lan, P.; He, X.S. MicroRNA-143 Targets ATG2B to Inhibit Autophagy and Increase Inflammatory Responses in Crohn’s Disease. Inflamm. Bowel Dis. 2018, 24, 781–791. [Google Scholar] [CrossRef]
- Luo, X.; Gong, H.B.; Gao, H.Y.; Wu, Y.P.; Sun, W.Y.; Li, Z.Q.; Wang, G.; Liu, B.; Liang, L.; Kurihara, H.; et al. Oxygenated phosphatidylethanolamine navigates phagocytosis of ferroptotic cells by interacting with TLR2. Cell Death Differ. 2021, 28, 1971–1989. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, J.; Ma, J.; Ma, J.; Liu, J.; Wang, F.; Tang, X. Inhibiting Ferroptosis: A Novel Approach for Ulcerative Colitis Therapeutics. Oxid. Med. Cell Longev. 2022, 2022, 9678625. [Google Scholar] [CrossRef]
- Li, S.; He, Y.; Chen, K.; Sun, J.; Zhang, L.; He, Y.; Yu, H.; Li, Q. RSL3 Drives Ferroptosis through NF-κB Pathway Activation and GPX4 Depletion in Glioblastoma. Oxid. Med. Cell Longev. 2021, 2021, 2915019. [Google Scholar] [CrossRef]
- Tan, W.; Dai, F.; Yang, D.; Deng, Z.; Gu, R.; Zhao, X.; Cheng, Y. MiR-93-5p promotes granulosa cell apoptosis and ferroptosis by the NF-kB signaling pathway in polycystic ovary syndrome. Front. Immunol. 2022, 13, 967151. [Google Scholar] [CrossRef]
- Qiang, Z.; Dong, H.; Xia, Y.; Chai, D.; Hu, R.; Jiang, H. Nrf2 and STAT3 Alleviates Ferroptosis-Mediated IIR-ALI by Regulating SLC7A11. Oxid. Med. Cell. Longev. 2020, 2020, 5146982. [Google Scholar] [CrossRef]
- Zhang, Z.; Tang, J.; Song, J.; Xie, M.; Liu, Y.; Dong, Z.; Liu, X.; Li, X.; Zhang, M.; Chen, Y.; et al. Elabela alleviates ferroptosis, myocardial remodeling, fibrosis and heart dysfunction in hypertensive mice by modulating the IL-6/STAT3/GPX4 signaling. Free Radic. Biol. Med. 2022, 181, 130–142. [Google Scholar] [CrossRef]
- Li, J.; Tian, X.; Liu, J.; Mo, Y.; Guo, X.; Qiu, Y.; Liu, Y.; Ma, X.; Wang, Y.; Xiong, Y. Therapeutic material basis and underling mechanisms of Shaoyao Decoction-exerted alleviation effects of colitis based on GPX4-regulated ferroptosis in epithelial cells. Chin. Med. 2022, 17, 96. [Google Scholar] [CrossRef]
- Liu, Y.; Mi, Y.; Wang, Y.; Meng, Q.; Xu, L.; Liu, Y.; Zhou, D.; Wang, Y.; Liang, D.; Li, W.; et al. Loureirin C inhibits ferroptosis after cerebral ischemia reperfusion through regulation of the Nrf2 pathway in mice. Phytomedicine 2023, 113, 154729. [Google Scholar] [CrossRef]
- Hong, Y.; Wu, B.; Xu, Z.; Du, J.; Gao, Y.; Wen, K.; Sun, X.L. Exploration of the effect of Xuejie San on expressions of Notch1, Nrf2, GPX4 and PTGS2 in colon tissues of Crohn’s disease rats based on a bioinformatic analysis. J. Liaoning Univ. TCM 2022, 24, 38–42. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, Z.; Du, J.; Yang, X.; Wang, X.; Wen, K.; Sun, X. Xue-Jie-San restricts ferroptosis in Crohn’s disease via inhibiting FGL1/NF-κB/STAT3 positive feedback loop. Front. Pharmacol. 2023, 14, 1148770. [Google Scholar] [CrossRef]
- Louis, E.; Belaïche, J. Hydroxychloroquine (Plaquenil) for recurrence prevention of Crohn’s disease after curative surgery. Gastroenterol. Clin. Biol. 1995, 19, 233–234. [Google Scholar]
- Mutalib, M.; Borrelli, O.; Blackstock, S.; Kiparissi, F.; Elawad, M.; Shah, N.; Lindley, K. The use of sirolimus (rapamycin) in the management of refractory inflammatory bowel disease in children. J. Crohns Colitis. 2014, 8, 1730–1734. [Google Scholar] [CrossRef]
- Zhong, M.; Cui, B.; Xiang, J.; Wu, X.; Wen, Q.; Li, Q.; Zhang, F. Rapamycin is Effective for Upper but not for Lower Gastrointestinal Crohn’s Disease-Related Stricture: A Pilot Study. Front. Pharmacol. 2021, 11, 617535. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, H.; Yang, H.; Zhou, Y.; Tang, L. Autophagy induction by rapamycin ameliorates experimental colitis and improves intestinal epithelial barrier function in IL-10 knockout mice. Int. Immunopharmacol. 2020, 81, 105977. [Google Scholar] [CrossRef]
- Shoji-Kawata, S.; Sumpter, R.; Leveno, M.; Campbell, G.R.; Zou, Z.; Kinch, L.; Wilkins, A.D.; Sun, Q.; Pallauf, K.; MacDuff, D.; et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature 2013, 494, 201–206. [Google Scholar] [CrossRef]
- Fairlie, W.D.; Tran, S.; Lee, E.F. Crosstalk between apoptosis and autophagy signaling pathways. Int. Rev. Cell Mol. Biol. 2020, 352, 115–158. [Google Scholar] [CrossRef]
- Dong, X.; Liang, Q.; Pan, Y.Z.; Wang, X.; Kuo, Y.C.; Chiang, W.C.; Zhang, X.; Williams, N.S.; Rizo, J.; Levine, B.; et al. Novel Bcl-2 Inhibitors Selectively Disrupt the Autophagy-Specific Bcl-2-Beclin 1 Protein-Protein Interaction. ACS Med. Chem. Lett. 2022, 13, 1510–1516. [Google Scholar] [CrossRef]
- Calis, S.; Dogan, B.; Durdagi, S.; Celebi, A.; Yapicier, O.; Kilic, T.; Turanli, E.T.; Avsar, T. A novel BH3 mimetic Bcl-2 inhibitor promotes autophagic cell death and reduces in vivo Glioblastoma tumor growth. Cell Death Discov. 2022, 8, 433. [Google Scholar] [CrossRef]
- Hooper, K.M.; Casanova, V.; Kemp, S.; Staines, K.A.; Satsangi, J.; Barlow, P.G.; Henderson, P.; Stevens, C. The Inflammatory Bowel Disease Drug Azathioprine Induces Autophagy via mTORC1 and the Unfolded Protein Response Sensor PERK. Inflamm. Bowel Dis. 2019, 25, 1481–1496. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kouroumalis, E.; Tsomidis, I.; Voumvouraki, A. Autophagy and Apoptosis in Inflammatory Bowel Disease. Gastroenterol. Insights 2023, 14, 598-636. https://doi.org/10.3390/gastroent14040042
Kouroumalis E, Tsomidis I, Voumvouraki A. Autophagy and Apoptosis in Inflammatory Bowel Disease. Gastroenterology Insights. 2023; 14(4):598-636. https://doi.org/10.3390/gastroent14040042
Chicago/Turabian StyleKouroumalis, Elias, Ioannis Tsomidis, and Argyro Voumvouraki. 2023. "Autophagy and Apoptosis in Inflammatory Bowel Disease" Gastroenterology Insights 14, no. 4: 598-636. https://doi.org/10.3390/gastroent14040042