
Clinical Exome Sequencing Revealed a De Novo FLNC Mutation in a Child with Restrictive Cardiomyopathy

 , ,
, , {kind=link}
{kind=link}
Abstract
:1. Introduction
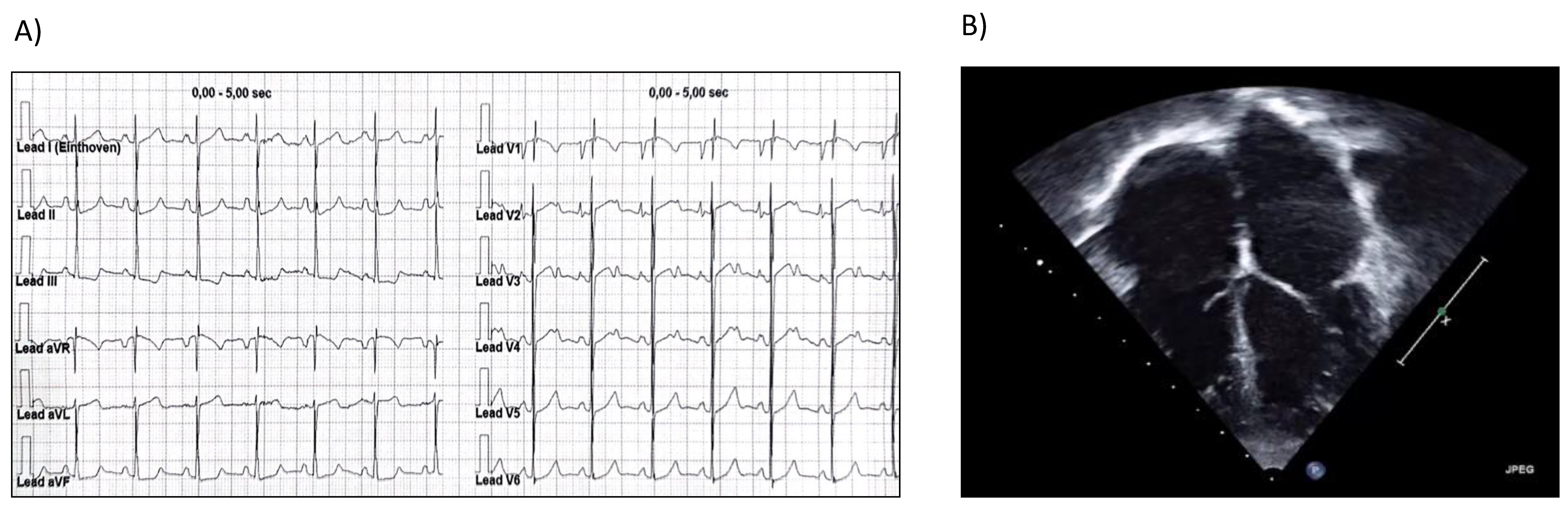
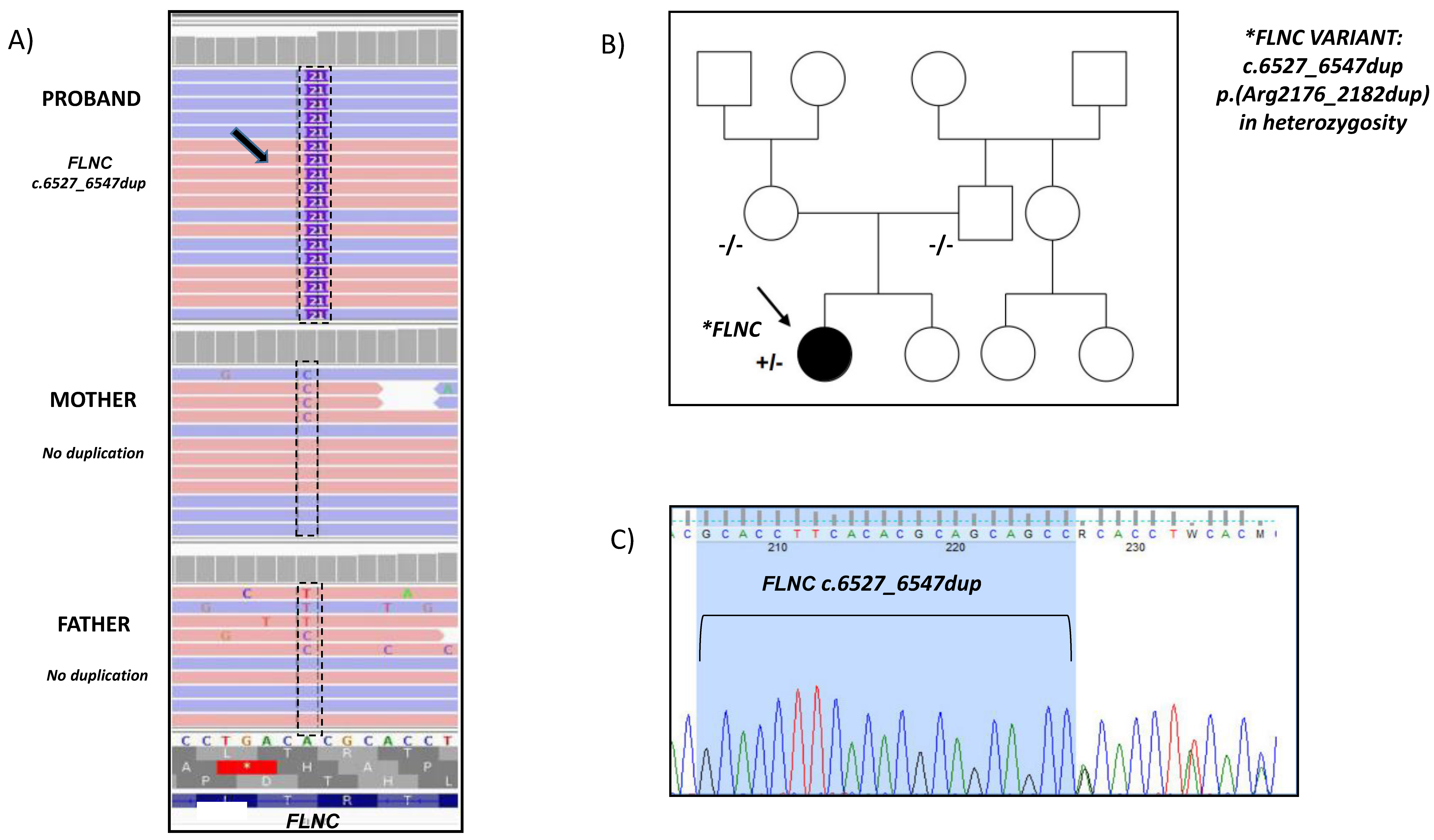
2. Case Report
3. Discussion
4. Conclusions
Key Clinical Message
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Richardson, P.; McKenna, W.; Bristow, M.; Maisch, B.; Mautner, B.; O′Connell, J.; Olsen, E.; Thiene, G.; Goodwin, J.; Gyarfas, I.; et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation 1996, 93, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Lipshultz, S.E.; Law, Y.M.; Asante-Korang, A.; Austin, E.D.; Dipchand, A.I.; Everitt, M.D.; Hsu, D.T.; Lin, K.Y.; Price, J.F.; Wilkinson, J.D.; et al. Cardiomyopathy in Children: Classification and Diagnosis: A Scientific Statement From the American Heart Association. Circulation 2019, 140, e9–e68. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.M.; Hsu, D.T.; Kantor, P.; Towbin, J.A.; Ware, S.M.; Colan, S.D.; Chung, W.K.; Jefferies, J.L.; Rossano, J.W.; Castleberry, C.D.; et al. Pediatric Cardiomyopathies. Circ. Res. 2017, 121, 855–873. [Google Scholar] [CrossRef] [PubMed]
- Towbin, J.A. Inherited cardiomyopathies. Circ. J. 2014, 78, 2347–2356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, H.N.; Cetta, F.; Driscoll, D.J.; Olson, T.M.; Ackerman, M.J.; Johnson, J.N. Idiopathic Restrictive Cardiomyopathy in Children and Young Adults. Am. J. Cardiol. 2018, 12, 1266–1270. [Google Scholar] [CrossRef]
- Mori, H.; Kogaki, S.; Ishida, H.; Yoshikawa, T.; Shindo, T.; Inuzuka, R.; Furutani, Y.; Ishido, M.; Nakanishi, T. Outcomes of Restrictive Cardiomyopathy in Japanese Children—A Retrospective Cohort Study. Circ. J. 2021, 121, 1266–1270. [Google Scholar] [CrossRef]
- Denfield, S.W.; Webber, S.A. Restrictive Cardiomyopathy in childhood. Heart Fail. Clin. 2010, 6, 445–452. [Google Scholar] [CrossRef]
- Hayashi, T.; Tanimoto, K.; Hirayama-Yamada, K.; Tsuda, E.; Ayusawa, M.; Nunoda, S.; Hosaki, A.; Kimuraet, A. Genetic background of Japanese patients with pediatric hypertrophic and restrictive cardiomyopathy. J. Hum. Genet. 2008, 63, 989–996. [Google Scholar] [CrossRef]
- Caleshu, F.C.; Sakhuja, R.; Nussbaum, R.L.; Schiller, N.B.; Ursell, P.C.; Eng, C.; De Marco, T.; McGlothlin, D.; Burchard, E.G.; Rameurthering, J.E. The link between the sarcomere and primary cardiomyopathies: Restrictive cardiomyopathy associated with multiple mutations in genes previously associated with hypertrophic or dilated cardiomyopathy. Am. J. Med. Genet. Part A 2011, 155A, 2229–2235. [Google Scholar] [CrossRef] [Green Version]
- Ploski, E.R.; Rydzanicz, M.; Ksiazczyk, T.M.; Franaszczyk, M.; Pollak, A.; Kosinska, J.; Michalak, E.; Stawinski, P.; Ziolkowska, L.; Bilinska, Z.T.; et al. Evidence for troponin C (TNNC1) as a gene for autosomal recessive restrictive cardiomyopathy with fatal outcome in infancy. Am. J. Med. Genet. Part A 2016, 170, 3241–3248. [Google Scholar] [CrossRef]
- Rath, A.; Weintraub, R. Overview of pediatric restrictive cardiomyopathy—2021. Prog. Pediatric. Cardiol. 2021, 62, 101415. [Google Scholar]
- Dalakas, M.C.; Park, K.Y.; Semino-Mora, C.; SLee, H.S.; Sivakumar, K.; Goldfarb, L.G. Desmin myopathy, a skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. N. Engl. J. Med. 2000, 342, 770–780. [Google Scholar] [CrossRef] [PubMed]
- Ader, F.; De Groote, P.; Réant, P.; Rooryck-Thambo, C.; Dupin-Deguine, D.; Rambaud, C.; Khraiche, D.; Perret, C.; Pruny, J.F.; Mathieu-Dramard, M.; et al. FLNC pathogenic variants in patients with cardiomyopathies: Prevalence and genotype-phenotype correlations. Clin. Genet. Wiley 2019, 96, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Gerull, B. Genetic Insights into Primary Restrictive Cardiomyopathy. Rev. J. Clin. Med. 2022, 11, 2094. [Google Scholar] [CrossRef]
- Ditaranto, R.; Caponetti, A.G.; Ferrara, V.; Parisi, V.; Minnucci, M.; Chiti, C.; Baldassarre, R.; Di Nicola, F.; Bonetti, S.; Hasan, T.; et al. Pediatric Restrictive Cardiomyopathies. Rev. Front. Pediatr. 2022, 25, 9:745365. [Google Scholar] [CrossRef]
- Ortiz-Genga, M.F.; Cuenca, S.; Dal Ferro, M.; Zorio, E.; Salgado-Aranda, R.; Climent, V.; Padrón-Barthe, L.; Duro-Aguado, I.; Jiménez-Jáimez, J.; Hidalgo-Olivares, V.M.; et al. Truncating FLNC Mutations Are Associated With High-Risk Dilated and Arrhythmogenic Cardiomyopathies. J. Am. Coll. Cardiol. 2016, 68, 2440–2451. [Google Scholar] [CrossRef]
- Muchtar, E.; Blauwet, L.A.; Gertz, M.A. Restrictive Cardiomyopathy Genetics, Pathogenesis, Clinical Manifestations, Diagnosis, and Therapy. Circ. Res. 2017, 121, 819–837. [Google Scholar] [CrossRef]
- Schubert, J.; Tariq, M.; Geddes, G.; Kindel, S.; Miller, E.M.; Ware, S.M. Novel pathogenic variants in filamin C identified in pediatric restrictive cardiomyopathy. Hum. Mutat. 2018, 39, 2083–2096. [Google Scholar] [CrossRef]
- Roldán-Sevilla, A.; Palomino-Doza, J.; de Juan, J.; Sánchez, V.; Domínguez-González, C.; Salguero-Bodes, R.; Arribas-Ynsaurriaga, F. Missense Mutations in the FLNC Gene Causing Familial Restrictive Cardiomyopathy. Circ. Genom. Precis. Med. 2019, 12, e002388. [Google Scholar] [CrossRef]
- Pantou, M.P.; Gourzi, P.; Gkouziouta, A.; Armenis, I.; Kaklamanis, L.; Zygouri, C.; Constantoulakis, P.; Adamopoulos, S.; Degiannis, D. A case report of recessive restrictive cardiomyopathy caused by a novel mutation in cardiac troponin I (TNNI3). BMC Med. Genet. 2019, 20, 61. [Google Scholar] [CrossRef]
- Xiao, F.; Wei, Q.; Wu, B.; Liu, X.; Mading, A.; Yang, L.; Li, Y.; Liu, F.; Pan, X.; Wang, H. Clinical exome sequencing revealed that FLNC variants contribute to the early diagnosis of cardiomyopathies in infant patients. Transl. Pediatr. 2020, 9, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Shi, A.; Lian, H.; Hu, S.; Nie, Y. Filamin C in cardiomyopathy: From physiological roles to DNA variants. Heart Fail. Rev. 2021, 17. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiselev, A.; Vaz, R.; Knyazeva, A.; Khudiakov, A.; Tarnovskaya, S.; Liu, J.; Sergushichev, A.; Kazakov, S.; Frishman, D.; Smolina, N.; et al. De novo mutations in FLNC leading to early-onset restrictive cardiomyopathy and congenital myopathy. Hum. Mutat. 2018, 39, 1161–1172. [Google Scholar] [CrossRef]
- Eden, M.; Frey, N. Cardiac Filaminopathies: Illuminating the Divergent Role of Filamin C Mutations in Human Cardiomyopathy. J. Clin. Med. 2021, 10, 577. [Google Scholar] [CrossRef]
- Brodehl, A.; Ferrier, R.A.; Hamilton, S.J.; Greenway, S.C.; Brundler, M.A.; Yu, W.; Gibson, W.T.; McKinnon, M.L.; McGillivray, B.; Alvarez, N.; et al. Schwartzentruber.FORGE Canada Consortium, Gerull, B. Mutations in FLNC are Associated with Familial Restrictive Cardiomyopathy. Hum. Mutat. 2016, 37, 269–279. [Google Scholar] [CrossRef]
- Hershberger, R.E.; Givertz, M.M.; Ho, C.Y.; Judge, D.P.; Kantor, P.F.; McBride, K.L.; Morales, A.; Taylor, M.R.G.; Vatta, M.; Ware, S.M. Genetic Evaluation of Cardiomyopathy—A Heart Failure Society of America Practice Guideline. Rev. J. Card Fail. 2018, 24, 281–302. [Google Scholar] [CrossRef] [Green Version]
- Girolami, F.; Iascone, M.; Pezzoli, L.; Passantino, S.; Limongelli, G.; Monda, E.; Rubino, M.; Adorisio, R.; Lombardi, M.; Ragni, L.; et al. Indicazioni all’esecuzione del test genetico nella diagnosi delle cardiomiopatie ad esordio pediatrico: Percorso clinico della Società Italiana di Cardiologia Pediatrica. G Ital. Cardiol. 2022, 23, 1–11. [Google Scholar]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Girolami, F.; Passantino, S.; Ballerini, A.; Gozzini, A.; Porcedda, G.; Olivotto, I.; Favilli, S. Clinical Exome Sequencing Revealed a De Novo FLNC Mutation in a Child with Restrictive Cardiomyopathy. Cardiogenetics 2022, 12, 206-211. https://doi.org/10.3390/cardiogenetics12020019
Girolami F, Passantino S, Ballerini A, Gozzini A, Porcedda G, Olivotto I, Favilli S. Clinical Exome Sequencing Revealed a De Novo FLNC Mutation in a Child with Restrictive Cardiomyopathy. Cardiogenetics. 2022; 12(2):206-211. https://doi.org/10.3390/cardiogenetics12020019
Chicago/Turabian StyleGirolami, Francesca, Silvia Passantino, Adelaide Ballerini, Alessia Gozzini, Giulio Porcedda, Iacopo Olivotto, and Silvia Favilli. 2022. "Clinical Exome Sequencing Revealed a De Novo FLNC Mutation in a Child with Restrictive Cardiomyopathy" Cardiogenetics 12, no. 2: 206-211. https://doi.org/10.3390/cardiogenetics12020019