
Moderately Prolonged QTc in Computer-Assessed ECG, Random Variation or Significant Risk Factor? A Literature Review
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
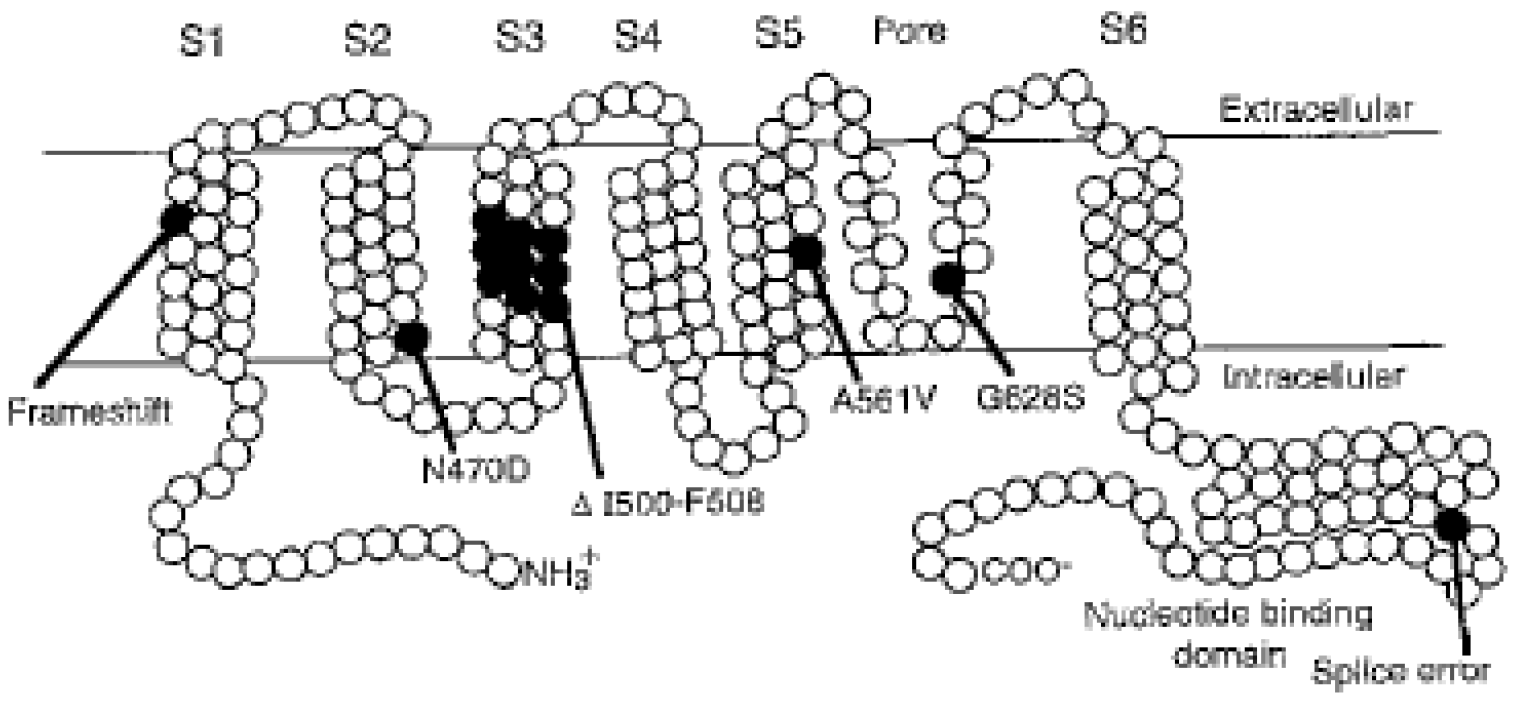
2. Molecular Genetic Studies
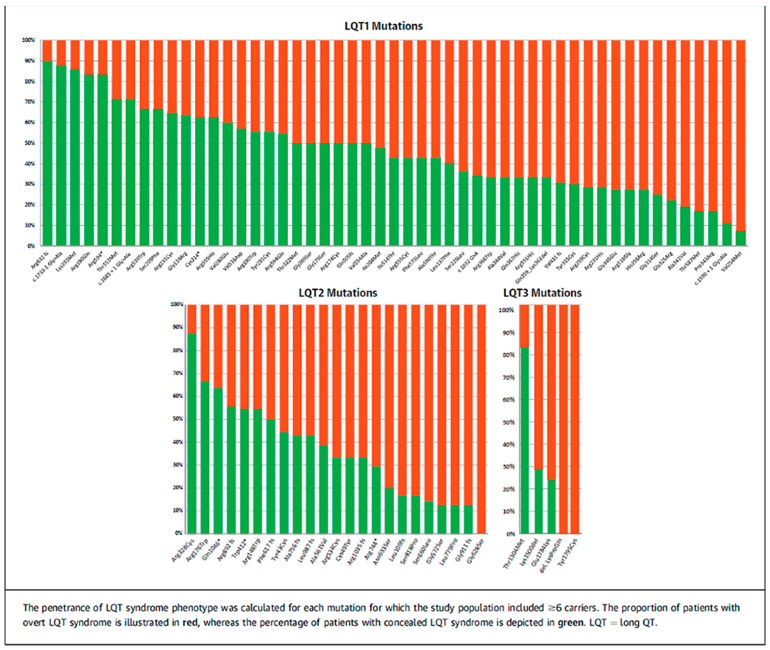
3. Mapping of Gene Penetrance Variation
4. Diagnostics and Treatment
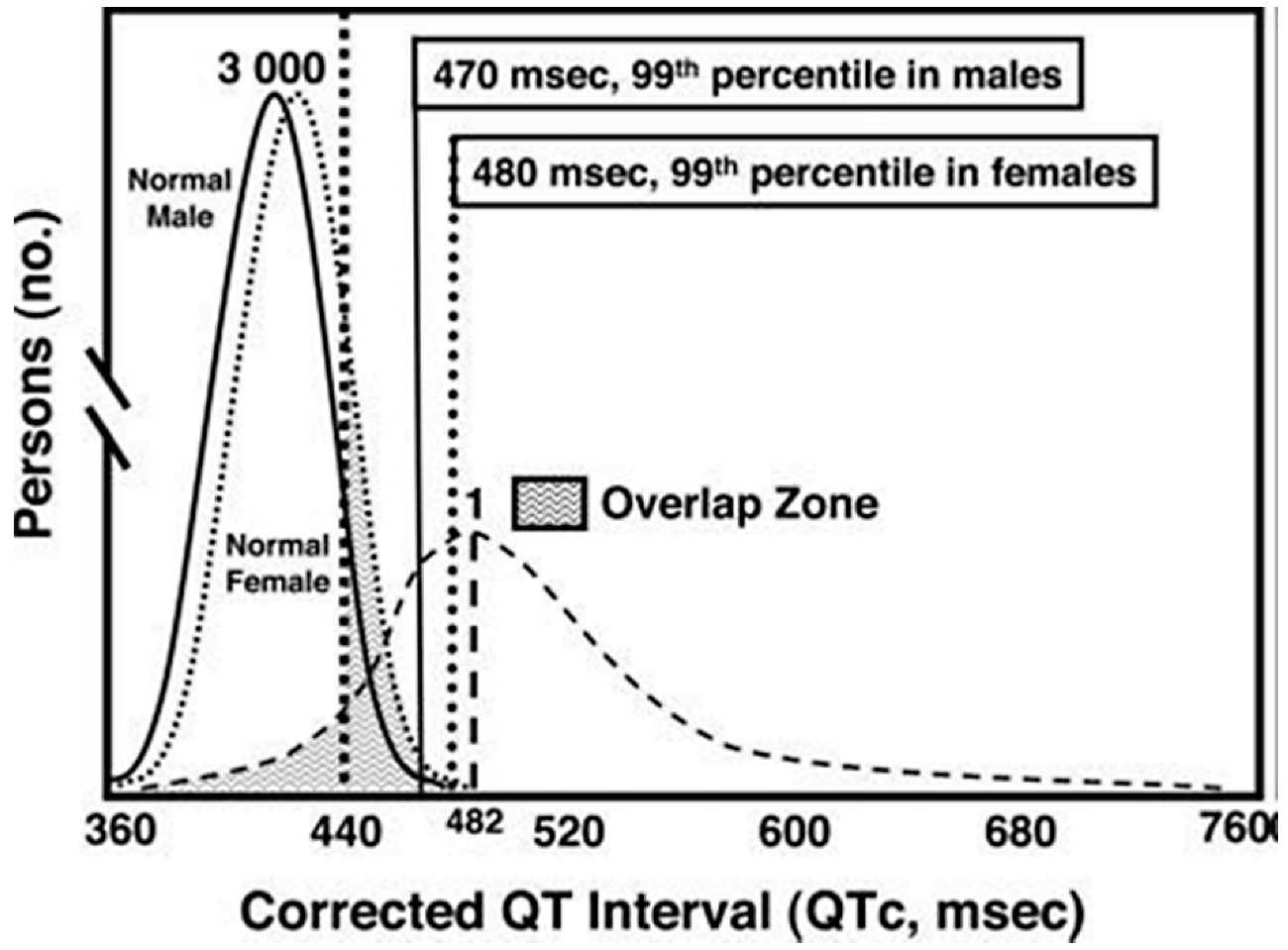
5. LQTS Incidence Is Highly Dependent on the Cohort Studied
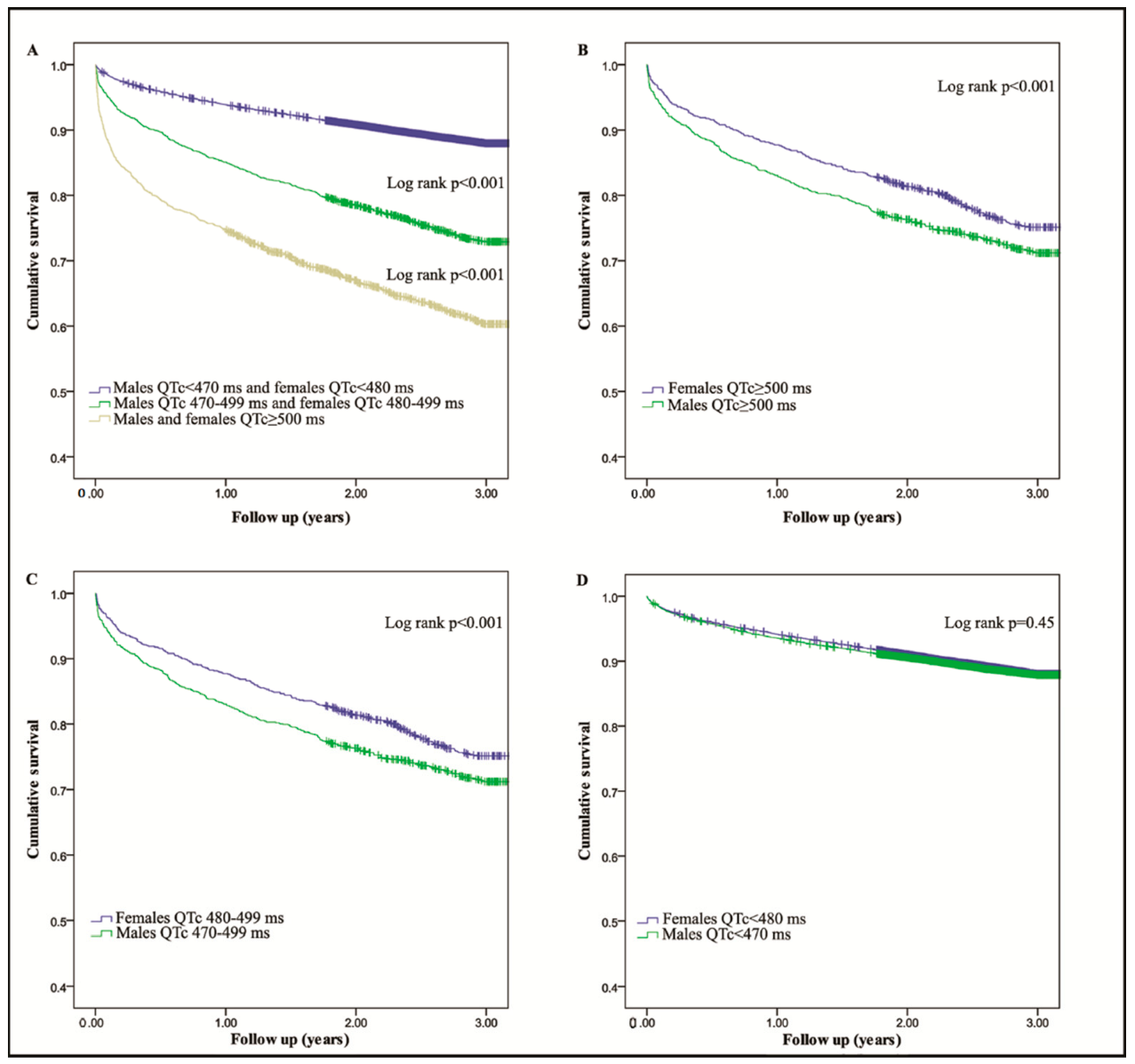
6. Materials and Results from a Community Hospital
7. Discussion
8. Summary
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ECG | Electrocardiogram |
| QT interval | Time in milliseconds between beginning of Q-wave and end of T-wave |
| QTc | QT interval adjusted for heat rate |
| cLQTS | Congenital long QT syndrome |
| aLQTS | Acquired long QT syndrome |
| LAE | Life-threatening arrhythmic event |
| LVH | Left ventricular hyperthrophy |
References
- Jervell, A.; Lange-Nielsen, F. Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval and sudden death. Am. Heart J. 1957, 54, 59–68. [Google Scholar] [CrossRef]
- Ljung, O. A simple formula for clinical interpretation of the QT interval. Acta Med. Scand. 1949, 134, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, P.J.; Periti, M.; Malliani, A. The long Q-T syndrome. Am. Heart J. 1975, 89, 378–390. [Google Scholar] [CrossRef]
- Schwartz, P.J. Idiopathic long QT syndrome: Progress and questions. Am. Heart J. 1985, 109, 399–411. [Google Scholar] [CrossRef]
- Moss, A.J.; Schwartz, P.J.; Crampton, R.S.; Tzivoni, D.; Locati, E.H.; MacCluer, J.; Hall, W.J.; Weitkamp, L.; Vincent, G.M.; Garson, A., Jr.; et al. The long QT syndrome. Prospective longitudinal study of 328 families. Circulation 1991, 84, 1136–1144. [Google Scholar] [CrossRef]
- Motte, G.; Abitbol, G.; Dessertenne, F.; Salma, R. Le syndrome QT long et syncopes par “torsades de pointe”. Arch. Mal. Coer. 1970, 63, 831–853. [Google Scholar]
- Curran, M.E.; Splawski, I.; Timothy, K.W.; Vincent, G.M.; Green, E.D.; Keating, M.T. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 1995, 80, 795–803. [Google Scholar] [CrossRef]
- Wang, Q.; Shen, J.; Splawski, I.; Atkinson, D.; Li, Z.; Robinson, J.L.; Moss, A.J.; Towbin, J.A.; Keating, M.T. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell 1995, 80, 805–811. [Google Scholar] [CrossRef]
- Wang, Q.; Curran, M.E.; Splawski, I.; Burn, T.C.; Millholland, J.M.; VanRaay, T.J.; Shen, J.; Timothy, K.W.; Vincent, G.M.; De Jager, T.; et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat. Genet. 1996, 12, 17–23. [Google Scholar] [CrossRef]
- Ingles, J.; Semsarian, C. Time to Rethink the Genetic Architecture of Long QT Syndrome. Circulation 2020, 141, 440–443. [Google Scholar] [CrossRef]
- Roden, D.M. Taking the “idio” out of “idiosyncratic”: Predicting torsades de pointes. Pacing Clin. Electrophysiol. PACE 1998, 21, 1029–1034. [Google Scholar] [CrossRef]
- Ray, W.A.; Murray, K.T.; Hall, K.; Arbogast, P.G.; Stein, C.M. Azithromycin and the risk of cardiovascular death. N. Engl. J. Med. 2012, 366, 1881–1890. [Google Scholar] [CrossRef]
- Goldenberg, I.; Horr, S.; Moss, A.J.; Lopes, C.M.; Barsheshet, A.; McNitt, S.; Zareba, W.; Andrews, M.L.; Robinson, J.L.; Locati, E.H.; et al. Risk for life-threatening cardiac events in patients with genotype-confirmed long-QT syndrome and normal-range corrected QT intervals. J. Am. Coll. Cardiol. 2011, 57, 51–59. [Google Scholar] [CrossRef]
- Taggart, N.W.; Haglund, C.M.; Tester, D.J.; Ackerman, M.J. Diagnostic miscues in congenital long-QT syndrome. Circulation 2007, 115, 2613–2620. [Google Scholar] [CrossRef]
- Mazzanti, A.; Maragna, R.; Vacanti, G.; Monteforte, N.; Bloise, R.; Marino, M.; Braghieri, L.; Gambelli, P.; Memmi, M.; Pagan, E.; et al. Interplay Between Genetic Substrate, QTc Duration, and Arrhythmia Risk in Patients with Long QT Syndrome. J. Am. Coll. Cardiol. 2018, 71, 1663–1671. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Moreno, C.; Kotta, M.C.; Pedrazzini, M.; Crotti, L.; Dagradi, F.; Castelletti, S.; Haugaa, K.H.; Denjoy, I.; Shkolnikova, M.A.; et al. Mutation location and IKs regulation in the arrhythmic risk of long QT syndrome type 1: The importance of the KCNQ1 S6 region. Eur. Heart J. 2021, 42, 4743–4755. [Google Scholar] [CrossRef]
- Pasquier, M.; Pantet, O.; Hugli, O.; Pruvot, E.; Buclin, T.; Waeber, G.; Aujesky, D. Prevalence and determinants of QT interval prolongation in medical inpatients. Intern. Med. J. 2012, 42, 933–940. [Google Scholar] [CrossRef]
- Seftchick, M.W.; Adler, P.H.; Hsieh, M.; Wolfson, A.B.; Chan, S.T.; Webster, B.W.; Frattaroli, G.D. The prevalence and factors associated with QTc prolongation among emergency department patients. Ann. Emerg. Med. 2009, 54, 763–768. [Google Scholar] [CrossRef]
- Tisdale, J.E.; Wroblewski, H.A.; Overholser, B.R.; Kingery, J.R.; Trujillo, T.N.; Kovacs, R.J. Prevalence of QT interval prolongation in patients admitted to cardiac care units and frequency of subsequent administration of QT interval-prolonging drugs: A prospective, observational study in a large urban academic medical center in the US. Drug Saf. 2012, 35, 459–470. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Stramba-Badiale, M.; Crotti, L.; Pedrazzini, M.; Besana, A.; Bosi, G.; Gabbarini, F.; Goulene, K.; Insolia, R.; Mannarino, S.; et al. Prevalence of the congenital long-QT syndrome. Circulation 2009, 120, 1761–1767. [Google Scholar] [CrossRef]
- Berge, K.E.; Haugaa, K.H.; Fruh, A.; Anfinsen, O.G.; Gjesdal, K.; Siem, G.; Oyen, N.; Greve, G.; Carlsson, A.; Rognum, T.O.; et al. Molecular genetic analysis of long QT syndrome in Norway indicating a high prevalence of heterozygous mutation carriers. Scand. J. Clin. Lab. Investig. 2008, 68, 362–368. [Google Scholar] [CrossRef]
- Locati, E.H.; Zareba, W.; Moss, A.J.; Schwartz, P.J.; Vincent, G.M.; Lehmann, M.H.; Towbin, J.A.; Priori, S.G.; Napolitano, C.; Robinson, J.L.; et al. Age- and sex-related differences in clinical manifestations in patients with congenital long-QT syndrome: Findings from the International LQTS Registry. Circulation 1998, 97, 2237–2244. [Google Scholar] [CrossRef]
- Mason, J.W.; Ramseth, D.J.; Chanter, D.O.; Moon, T.E.; Goodman, D.B.; Mendzelevski, B. Electrocardiographic reference ranges derived from 79,743 ambulatory subjects. J. Electrocardiol. 2007, 40, 228–234. [Google Scholar] [CrossRef]
- Priori, S.G.; Blomstrom-Lundqvist, C.; Mazzanti, A.; Blom, N.; Borggrefe, M.; Camm, J.; Elliott, P.M.; Fitzsimons, D.; Hatala, R.; Hindricks, G.; et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur. Heart J. 2015, 36, 2793–2867. [Google Scholar] [CrossRef]
- Priori, S.G.; Wilde, A.A.; Horie, M.; Cho, Y.; Behr, E.R.; Berul, C.; Blom, N.; Brugada, J.; Chiang, C.E.; Huikuri, H.; et al. HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: Document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm. Off. J. Heart Rhythm. Soc. 2013, 10, 1932–1963. [Google Scholar] [CrossRef]
- Goldenberg, I.; Moss, A.J.; Zareba, W. QT interval: How to measure it and what is “normal”. J. Cardiovasc. Electrophysiol. 2006, 17, 333–336. [Google Scholar] [CrossRef]
- Vandenberk, B.; Vandael, E.; Robyns, T.; Vandenberghe, J.; Garweg, C.; Foulon, V.; Ector, J.; Willems, R. Which QT Correction Formulae to Use for QT Monitoring? J. Am. Heart Assoc. 2016, 5, e003264. [Google Scholar] [CrossRef]
- Viskin, S.; Rosovski, U.; Sands, A.J.; Chen, E.; Kistler, P.M.; Kalman, J.M.; Rodriguez Chavez, L.; Iturralde Torres, P.; Cruz, F.F.; Centurion, O.A.; et al. Inaccurate electrocardiographic interpretation of long QT: The majority of physicians cannot recognize a long QT when they see one. Heart Rhythm. Off. J. Heart Rhythm. Soc. 2005, 2, 569–574. [Google Scholar] [CrossRef]
- Hnatkova, K.; Gang, Y.; Batchvarov, V.N.; Malik, M. Precision of QT interval measurement by advanced electrocardiographic equipment. Pacing Clin. Electrophysiol. PACE 2006, 29, 1277–1284. [Google Scholar] [CrossRef]
- Wilde, A.A.M.; Semsarian, C.; Marquez, M.F.; Sepehri Shamloo, A.; Ackerman, M.J.; Ashley, E.A.; Sternick, E.B.; Barajas-Martinez, H.; Behr, E.R.; Bezzina, C.R.; et al. European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) Expert Consensus Statement on the State of Genetic Testing for Cardiac Diseases. Heart Rhythm. Off. J. Heart Rhythm. Soc. 2022, 22, 1147–1148. [Google Scholar] [CrossRef]
- Gibbs, C.; Thalamus, J.; Heldal, K.; Holla, O.L.; Haugaa, K.H.; Hysing, J. Predictors of mortality in high-risk patients with QT prolongation in a community hospital. Europace 2018, 20, f99–f107. [Google Scholar] [CrossRef]
- Gibbs, C.; Thalamus, J.; Tveten, K.; Busk, O.L.; Hysing, J.; Haugaa, K.H.; Holla, O.L. Genetic and Phenotypic Characterization of Community Hospital Patients with QT Prolongation. J. Am. Heart Assoc. 2018, 7, e009706. [Google Scholar] [CrossRef]
- Tisdale, J.E.; Jaynes, H.A.; Kingery, J.R.; Mourad, N.A.; Trujillo, T.N.; Overholser, B.R.; Kovacs, R.J. Development and validation of a risk score to predict QT interval prolongation in hospitalized patients. Circ. Cardiovasc. Qual. Outcomes 2013, 6, 479–487. [Google Scholar] [CrossRef]
- Sorita, A.; Bos, J.M.; Morlan, B.W.; Tarrell, R.F.; Ackerman, M.J.; Caraballo, P.J. Impact of clinical decision support preventing the use of QT-prolonging medications for patients at risk for torsade de pointes. J. Am. Med. Inform. Assoc. JAMIA 2015, 22, e21–e27. [Google Scholar] [CrossRef]
- Zhang, Z.M.; Rautaharju, P.M.; Soliman, E.Z.; Manson, J.E.; Cain, M.E.; Martin, L.W.; Bavry, A.A.; Mehta, L.; Vitolins, M.; Prineas, R.J. Mortality risk associated with bundle branch blocks and related repolarization abnormalities (from the Women’s Health Initiative [WHI]). Am. J. Cardiol. 2012, 110, 1489–1495. [Google Scholar] [CrossRef]
- Imanishi, R.; Seto, S.; Ichimaru, S.; Nakashima, E.; Yano, K.; Akahoshi, M. Prognostic significance of incident complete left bundle branch block observed over a 40-year period. Am. J. Cardiol. 2006, 98, 644–648. [Google Scholar] [CrossRef]
- Haugaa, K.H.; Bos, J.M.; Borkenhagen, E.J.; Tarrell, R.F.; Morlan, B.W.; Caraballo, P.J.; Ackerman, M.J. Impact of left ventricular hypertrophy on QT prolongation and associated mortality. Heart Rhythm. Off. J. Heart Rhythm. Soc. 2014, 11, 1957–1965. [Google Scholar] [CrossRef]
- Joyce, D.D.; Bos, J.M.; Haugaa, K.H.; Tarrell, R.F.; Morlan, B.W.; Caraballo, P.J.; Ackerman, M.J. Frequency and cause of transient QT prolongation after surgery. Am. J. Cardiol. 2015, 116, 1605–1609. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Wolf, S. QT Interval Prolongation as Predictor of Sudden Death in Patients with Myocardial Infarction. Circulation 1978, 57, 1074–1077. [Google Scholar] [CrossRef] [PubMed]
- Karwatowska-Prokopczuk, E.; Wang, W.; Cheng, M.L.; Zeng, D.; Schwartz, P.J.; Belardinelli, L. The risk of sudden cardiac death in patients with non-ST elevation acute coronary syndrome and prolonged QTc interval: Effect of ranolazine. Europace 2013, 15, 429–436. [Google Scholar] [CrossRef]
- Straus, S.M.; Kors, J.A.; De Bruin, M.L.; Van der Hooft, C.S.; Hofman, A.; Heeringa, J.; Deckers, J.W.; Kingma, J.H.; Sturkenboom, M.C.; Stricker, B.H.; et al. Prolonged QTc interval and risk of sudden cardiac death in a population of older adults. J. Am. Coll. Cardiol. 2006, 47, 362–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chugh, S.S.; Reinier, K.; Singh, T.; Uy-Evanado, A.; Socoteanu, C.; Peters, D.; Mariani, R.; Gunson, K.; Jui, J. Determinants of prolonged QT interval and their contribution to sudden death risk in coronary artery disease: The Oregon Sudden Unexpected Death Study. Circulation 2009, 119, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Haugaa, K.H.; Bos, J.M.; Tarrell, R.F.; Morlan, B.W.; Caraballo, P.J.; Ackerman, M.J. Institution-wide QT alert system identifies patients with a high risk of mortality. Mayo. Clin. Proc. 2013, 88, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, C.; Thalamus, J.; Kristoffersen, D.T.; Svendsen, M.V.; Holla, O.L.; Heldal, K.; Haugaa, K.H.; Hysing, J. QT prolongation predicts short-term mortality independent of comorbidity. Europace 2019, 21, 1254–1260. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hysing, J.; Gibbs, C.; Holla, Ø.L.; Thalamus, J.; Haugaa, K.H. Moderately Prolonged QTc in Computer-Assessed ECG, Random Variation or Significant Risk Factor? A Literature Review. Cardiogenetics 2022, 12, 261-269. https://doi.org/10.3390/cardiogenetics12030025
Hysing J, Gibbs C, Holla ØL, Thalamus J, Haugaa KH. Moderately Prolonged QTc in Computer-Assessed ECG, Random Variation or Significant Risk Factor? A Literature Review. Cardiogenetics. 2022; 12(3):261-269. https://doi.org/10.3390/cardiogenetics12030025
Chicago/Turabian StyleHysing, Jan, Charlotte Gibbs, Øystein Lunde Holla, Jacob Thalamus, and Kristina H. Haugaa. 2022. "Moderately Prolonged QTc in Computer-Assessed ECG, Random Variation or Significant Risk Factor? A Literature Review" Cardiogenetics 12, no. 3: 261-269. https://doi.org/10.3390/cardiogenetics12030025