Revisiting the Latency of Uridine Diphosphate-Glucuronosyltransferases (UGTs)—How Does the Endoplasmic Reticulum Membrane Influence Their Function?


Abstract
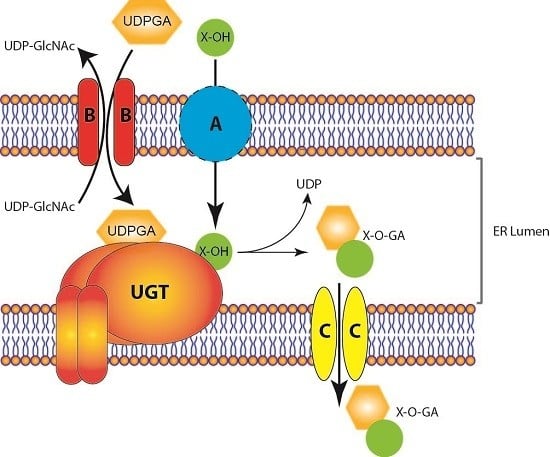
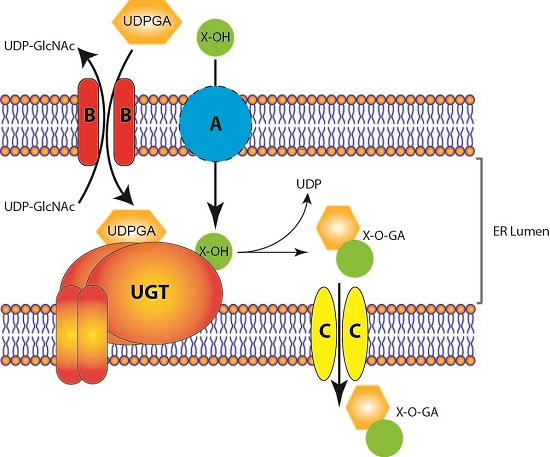
:
{kind=link}
{kind=link}
1. Introduction
2. UGTs and Latency
3. Compartmentation Hypothesis
4. Conformation Hypothesis
5. Adenine Nucleotide Inhibition Effect
6. Conclusions and Future Direction
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Radominska-Pandya, A.; Czernik, P.J.; Little, J.M.; Battaglia, E.; Mackenzie, P.I. Structural and functional studies of UDP-glucuronosyltransferases. Drug Metab. Rev. 1999, 31, 817–899. [Google Scholar] [CrossRef] [PubMed]
- Dutton, G.J. Glucuronidation of drugs and other compounds; CRC Press: Boca Raton, FL, USA, 1980; ISBN 9780849352959. [Google Scholar]
- Fry, D.J.; Wishart, G.J. Apparent induction by phenobarbital of uridine diphosphate glucuronyltransferase activity in nuclear envelopes of embryonic-chick liver. Biochem. Soc. Trans. 1976, 4, 265–266. [Google Scholar] [CrossRef] [PubMed]
- Wishart, G.J.; Fry, D.J. Uridine diphosphate glucuronyltransferase activity in nuclei and nuclear envelopes of rat liver and its apparent induction by phenobarbital. Biochem. Soc. Trans. 1977, 5, 705–706. [Google Scholar] [CrossRef]
- Fremont, J.J.; Wang, R.W.; King, C.D. Coimmunoprecipitation of UDP-glucuronosyltransferase isoforms and cytochrome P450 3A4. Mol. Pharmacol. 2005, 67, 260–262. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, R.; Nakajima, M.; Oda, S.; Yamanaka, H.; Ikushiro, S.; Sakaki, T.; Yokoi, T. Interactions between human UDP-glucuronosyltransferase (UGT) 2B7 and UGT1A enzymes. J. Pharm. Sci. 2010, 99, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, R.; Nakajima, M.; Yamanaka, H.; Katoh, M.; Yokoi, T. Interactions between human UGT1A1, UGT1A4, and UGT1A6 affect their enzymatic activities. Drug Metab. Dispos. 2007, 35, 1781–1787. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.S.; Sappal, B.S.; Kalpana, G.V.; Lee, S.W.; Chowdhury, J.R.; Chowdhury, N.R. Homodimerization of human bilirubin-uridine-diphosphoglucuronate glucuronosyltransferase-1 (UGT1A1) and its functional implications. J. Biol. Chem. 2001, 276, 42108–42115. [Google Scholar] [CrossRef] [PubMed]
- Ikushiro, S.; Emi, Y.; Iyanagi, T. Protein-protein interactions between UDP-glucuronosyltransferase isozymes in rat hepatic microsomes. Biochemistry 1997, 36, 7154–7161. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.C.; Mackenzie, P.I.; Miners, J.O. Homodimerization of UDP-glucuronosyltransferase 2B7 (UGT2B7) and identification of a putative dimerization domain by protein homology modeling. Biochem. Pharmacol. 2011, 82, 2016–2023. [Google Scholar] [CrossRef] [PubMed]
- Operana, T.N.; Tukey, R.H. Oligomerization of the UDP-glucuronosyltransferase 1A proteins: Homo- and heterodimerization analysis by fluorescence resonance energy transfer and co-immunoprecipitation. J. Biol. Chem. 2007, 282, 4821–4829. [Google Scholar] [CrossRef] [PubMed]
- Taura, K.I.; Yamada, H.; Hagino, Y.; Ishii, Y.; Mori, M.A.; Oguri, K. Interaction between cytochrome P450 and other drug-metabolizing enzymes: Evidence for an association of CYP1A1 with microsomal epoxide hydrolase and UDP-glucuronosyltransferase. Biochem. Biophys. Res. Commun. 2000, 273, 1048–1052. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.T. Detoxication Mechanisms, 2nd ed.; Chapman & Hall: London, UK, 1959. [Google Scholar]
- Ishii, Y.; Iwanaga, M.; Nishimura, Y.; Takeda, S.; Ikushiro, S.; Nagata, K.; Yamazoe, Y.; Mackenzie, P.I.; Yamada, H. Protein-protein interactions between rat hepatic cytochromes P450 (P450s) and UDP-glucuronosyltransferases (UGTs): Evidence for the functionally active UGT in P450-UGT complex. Drug Metab. Pharmacokinet. 2007, 22, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Ishii, Y.; Iwanaga, M.; Mackenzie, P.I.; Nagata, K.; Yamazoe, Y.; Oguri, K.; Yamada, H. Modulation of UDP-glucuronosyltransferase function by cytochrome P450: Evidence for the alteration of UGT2B7-catalyzed glucuronidation of morphine by CYP3A4. Mol. Pharmacol. 2005, 67, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Abbott, F.V.; Palmour, R.M. Morphine-6-glucuronide: Analgesic effects and receptor binding profile in rats. Life Sci. 1988, 43, 1685–1695. [Google Scholar] [CrossRef]
- Vore, M.; Slikker, W. Steroid D-ring glucuronides: A new class of cholestatic agents. Trends Pharmac. Sci. 1985, 6, 256–259. [Google Scholar] [CrossRef]
- Riches, Z.; Collier, A.C. Posttranscriptional regulation of uridine diphosphate glucuronosyltransferases. Expert. Opin. Drug Metab. Toxicol. 2015, 11, 949–965. [Google Scholar] [CrossRef] [PubMed]
- Caspersen, C.S.; Reznik, B.; Weldy, P.L.; Abildskov, K.M.; Stark, R.I.; Garland, M. Molecular cloning of the baboon UDP-glucuronosyltransferase 1A gene family: Evolution of the primate UGT1 locus and relevance for models of human drug metabolism. Pharmacogenet. Genomics 2007, 17, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Halac, E.; Bonevard, E. Solubilization and activation of liver UDP glucuronyltransferase by EDTA. Biochim. Biophys. Acta 1963, 67, 498–500. [Google Scholar] [CrossRef]
- Heirwegh, K.P.; Meuwissen, J.A. Activation in vitro and solubilization of glucuronyltransferase (assayed with bilirubin as acceptor) with digitonin. Biochem. J. 1968, 110, 31P–32P. [Google Scholar] [CrossRef] [PubMed]
- Isselbacher, K.J.; Chrabas, M.F.; Quinn, R.C. The solubilization and partial purification of a glucuronyl transferase from rabbit liver microsomes. J. Biol. Chem. 1962, 237, 3033–3036. [Google Scholar] [PubMed]
- Lueders, K.K.; Kuff, E.L. Spontaneous and detergent activation of a glucuronyltransferase in vitro. Arch. Biochem. Biophys. 1967, 120, 198–203. [Google Scholar] [CrossRef]
- Pogell, B.M.; Leloir, L.F. Nucleotide activation of liver microsomal glucuronidation. J. Biol. Chem. 1961, 236, 293–298. [Google Scholar] [PubMed]
- Stevenson, I.; Greenwood, D.; McEwen, J. Hepatic UDP-glucuronyltransferase in Wistar and Gunn rats - in vitro activation by diethylnitrosamine. Biochem. Biophys. Res. Commun. 1968, 32, 866–872. [Google Scholar] [CrossRef]
- Tomlinson, G.A.; Yaffe, S.J. The formation of bilirubin and p-nitrophenyl glucuronides by rabbit liver. Biochem. J. 1966, 99, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Van Roy, F.P.; Heirwegh, K.P. Determination of bilirubin glucuronide and assay of glucuronyltransferase with bilirubin as acceptor. Biochem. J. 1968, 107, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Winsnes, A. Studies on the activation in vitro of glucuronyltransferase. Biochim. Biophys. Acta 1969, 191, 279–291. [Google Scholar] [CrossRef]
- Burchell, A.; Waddell, I.D. The molecular basis of the hepatic microsomal glucose-6-phosphatase system. Biochim. Biophys. Acta 1991, 1092, 129–137. [Google Scholar] [CrossRef]
- Lin, J.H.; Wong, B.K. Complexities of glucuronidation affecting in vitro in vivo extrapolation. Curr. Drug Metab. 2002, 3, 623–646. [Google Scholar] [CrossRef] [PubMed]
- Berry, C.; Stellon, A.; Hallinan, T. Guinea pig liver microsomal UDP-glucuronyltransferase: Compartmented or phospholipid-constrained? Biochim. Biophys. Acta 1975, 403, 335–344. [Google Scholar] [CrossRef]
- Dannenberg, A.; Wong, T.; Zakim, D. Effect of brief treatment at alkaline pH on the properties of UDP-glucuronosyltransferase. Arch. Biochem. Biophys. 1990, 277, 312–317. [Google Scholar] [CrossRef]
- Dannenberg, A.J.; Kavecansky, J.; Scarlata, S.; Zakim, D. Organization of microsomal UDP-glucuronosyltransferase. Activation by treatment at high pressure. Biochemistry 1990, 29, 5961–5967. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.B.; Campanale, K.; Ackermann, B.L.; VandenBranden, M.; Wrighton, S.A. In vitro glucuronidation using human liver microsomes and the pore-forming peptide alamethicin. Drug Metab. Dispos. 2000, 28, 560–566. [Google Scholar] [PubMed]
- Graham, A.B.; Wood, G.C. The phospholipid-dependence of UDP-glucuronyltransferase. Biochem. Biophys. Res. Commun. 1969, 37, 567–575. [Google Scholar] [CrossRef]
- Mulder, G.J. The effect of phenobarbital on the submicrosomal distribution of uridine diphosphate glucuronyltransferase from rat liver. Biochem. J. 1970, 117, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Vessey, D.A.; Zakim, D. Regulation of microsomal enzymes by phospholipids. II. Acitvation of hepatic uridine diphosphate-glucuronyltransferase. J. Biol. Chem. 1971, 246, 4649–4656. [Google Scholar] [PubMed]
- Winsnes, A. Kinetic properties of different forms of hepatic UDP glucuronyltransferase. Biochim. Biophys. Acta 1972, 284, 394–405. [Google Scholar] [CrossRef]
- Zakim, D.; Dannenberg, A.J. How does the microsomal membrane regulate UDP-glucuronosyltransferases? Biochem. Pharmacol. 1992, 43, 1385–1393. [Google Scholar] [CrossRef]
- Coughtrie, M.W.H.; Blair, J.N.R.; Hume, R.; Burchell, A. Improved procedure for the preparation of hepatic microsomes to be used in the in vitro diagnosis of inherited disorders of the glucose-6-phosphatase system. Clin. Chem. 1991, 37, 739–742. [Google Scholar] [PubMed]
- Coughtrie, M.W.H. A Molecular Analysis of Biological Variations in UDP-Glucuronosyltransferase Activities. Ph.D. Thesis, University of Dundee, Dundee, UK, 1986. [Google Scholar]
- Coughtrie, M.W.H.; Burchell, B.; Bend, J.R. Purification and properties of rat kidney UDP-glucuronosyltransferase. Biochem. Pharmacol. 1987, 36, 245–251. [Google Scholar] [CrossRef]
- Berry, C.; Hallinan, T. Summary of a novel, three-component regulatory model for uridine diphosphate glucuronyltransferase. Biochem. Soc. Trans. 1976, 4, 650–652. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.R.; McCarthy, L.R.; Corser, R.B.; Barr, G.C.; Burchell, B. Cloning of cDNAs coding for rat hepatic microsomal UDP-glucuronyltransferases. Gene 1984, 34, 147–153. [Google Scholar] [CrossRef]
- Mackenzie, P.I.; Gonzalez, F.J.; Owens, I.S. Cloning and characterization of DNA complementary to rat liver UDP-glucuronosyltransferase mRNA. J. Biol. Chem. 1984, 259, 12153–12160. [Google Scholar]
- Mackenzie, P.I.; Owens, I.S. Cleavage of nascent UDP glucuronosyltransferase from rat liver by dog pancreatic microsomes. Biochem. Biophys. Res. Commun. 1984, 122, 1441–1449. [Google Scholar] [CrossRef]
- Rowland, A.; Miners, J.O.; Mackenzie, P.I. The UDP-glucuronosyltransferases: Their role in drug metabolism and detoxification. Int. J. Biochem. Cell Biol. 2013, 45, 1121–1132. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.R.; Nilsson, T.; Peterson, P.A. Identification of a consensus motif for retention of transmembrane proteins in the endoplasmic reticulum. EMBO J. 1990, 9, 3153–3162. [Google Scholar] [PubMed]
- Ouzzine, M.; Magdalou, J.; Burchell, B.; Fournel-Gigleux, S. Expression of a functionally active human hepatic UDP-glucuronosyltransferase (UGT1A6) lacking the N-terminal signal sequence in the endoplasmic reticulum. FEBS Lett. 1999, 454, 187–191. [Google Scholar] [PubMed]
- Ouzzine, M.; Magdalou, J.; Burchell, B.; Fournel-Gigleux, S. An internal signal sequence mediates the targeting and retention of the human UDP-glucuronosyltransferase 1A6 to the endoplasmic reticulum. J. Biol. Chem. 1999, 274, 31401–31409. [Google Scholar] [CrossRef] [PubMed]
- Vanstapel, F.; Blanckaert, N. Topology and regulation of bilirubin UDP-glucuronyltransferase in sealed native microsomes from rat liver. Arch. Biochem. Biophys. 1988, 263, 216–225. [Google Scholar] [CrossRef]
- Shepherd, S.R.; Baird, S.J.; Hallinan, T.; Burchell, B. An investigation of the transverse topology of bilirubin UDP-glucuronosyltransferase in rat hepatic endoplasmic reticulum. Biochem. J. 1989, 259, 617–620. [Google Scholar] [CrossRef] [PubMed]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef]
- Jackson, M.R.; Burchell, B. The full length coding sequence of rat liver androsterone UDP-glucuronyltransferase cDNA and comparison with other members of this gene family. Nucleic Acids Res. 1986, 14, 779–795. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, P.I. Rat liver UDP-glucuronosyltransferase. cDNA sequence and expression of a form glucuronidating 3-hydroxyandrogens. J. Biol. Chem. 1986, 261, 14112–14117. [Google Scholar] [PubMed]
- Iyanagi, T.; Haniu, M.; Sogawa, K.; Fujii-Kuriyama, Y.; Watanabe, S.; Shively, J.E.; Anan, K.F. Cloning and characterization of cDNA encoding 3-methylcholanthrene inducible rat mRNA for UDP-glucuronosyltransferase. J. Biol. Chem. 1986, 261, 15607–15614. [Google Scholar] [PubMed]
- Fujiwara, R.; Yokoi, T.; Nakajima, M. Structure and protein-protein interactions of human UDP-glucuronosyltransferases. Front. Pharmacol. 2016, 7, 388. [Google Scholar] [CrossRef] [PubMed]
- Miley, M.J.; Zielinska, A.K.; Keenan, J.E.; Bratton, S.M.; Radominska-Pandya, A.; Redinbo, M.R. Crystal structure of the cofactor-binding domain of the human phase II drug-metabolism enzyme UDP-glucuronosyltransferase 2B7. J. Mol. Biol. 2007, 369, 498–511. [Google Scholar] [CrossRef] [PubMed]
- Axelrod, J.; Kalckar, H.M.; Maxwell, E.S.; Strominger, J.L. Enzymatic formation of uridine diphosphoglucuronic acid. J. Biol. Chem. 1957, 224, 79–90. [Google Scholar] [PubMed]
- Banerjee, R.P.; Pennington, M.W.; Garza, A.; Owens, I.S. Mapping the UDP-glucuronic acid binding site in UDP-glucuronosyltransferase 1A10 by homology-based modeling: Confirmation with biochemical evidence. Biochemistry 2008, 47, 7385–7392. [Google Scholar] [CrossRef] [PubMed]
- Locuson, C.W.; Tracy, T.S. Comparative modelling of the human UDP-glucuronosyltransferases: Insights into structure and mechanism. Xenobiotica 2007, 37, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Revesz, K.; Toth, B.; Staines, A.G.; Coughtrie, M.W.; Mandl, J.; Csala, M. Luminal accumulation of newly synthesized morphine-3-glucuronide in rat liver microsomal vesicles. Biofactors 2013, 39, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Zakim, D.; Vessey, D.A. Membrane dependence of uridine diphosphate glucuronyltransferase: Effect of the membrane on kinetic properties. Biochem. Soc. Trans. 1974, 2, 1165–1167. [Google Scholar] [CrossRef]
- Berry, C.; Hallinan, T. ‘Coupled transglucuronidation’: A new tool for studying the latency of UDP-glucuronyl transferase. FEBS Lett. 1974, 42, 73–76. [Google Scholar] [CrossRef]
- Vessey, D.A.; Zakim, D. Stimulation of microsomal uridine diphosphate glucuronyltransferase by glucuronic acid derivatives. Biochem. J. 1974, 139, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Winsnes, A. Inhibition of hepatic UDP glucuronyltransferase by nucleotides. Biochim. Biophys. Acta 1972, 289, 88–96. [Google Scholar] [CrossRef]
- Winsnes, A. The effects of sulfhydryl reacting agents on hepatic UDP-glucuronyltransferase in vitro. Biochim. Biophys. Acta 1971, 242, 549–559. [Google Scholar] [CrossRef]
- Bossuyt, X.; Blanckaert, N. Carrier-mediated transport of intact UDP-glucuronic acid into the lumen of endoplasmic-reticulum-derived vesicles from rat liver. Biochem. J. 1994, 302, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Bossuyt, X.; Blanckaert, N. Mechanism of stimulation of microsomal UDP-glucuronosyltransferase by UDP-N-acetylglucosamine. Biochem J. 1995, 305, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.C.; Ziurys, J.C.; Gollan, J.L. A membrane transporter mediates access of uridine 5'-diphosphoglucuronic acid from the cytosol into the endoplasmic reticulum of rat hepatocytes: Implications for glucuronidation reactions. Biochim. Biophys. Acta 1988, 967, 149–157. [Google Scholar] [CrossRef]
- Nuwayhid, N.; Glaser, J.H.; Johnson, J.C.; Conrad, H.E.; Hauser, S.C.; Hirschberg, C.B. Xylosylation and glucuronosylation reactions in rat liver golgi apparatus and endoplasmic reticulum. J. Biol. Chem. 1986, 261, 12936–12941. [Google Scholar] [PubMed]
- Bossuyt, X.; Blanckaert, N. Functional characterization of carrier-mediated transport of uridine diphosphate N-acetylglucosamine across the endoplasmic reticulum membrane. Eur. J. Biochem. 1994, 223, 981–988. [Google Scholar] [CrossRef]
- Bossuyt, X.; Blanckaert, N. Carrier-mediated transport of uridine diphosphoglucuronic acid across the endoplasmic reticulum membrane is a prerequisite for UDP-glucuronosyltransferase activity in rat liver. Biochem. J. 1997, 323, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Muraoka, M.; Kawakita, M.; Ishida, N. Molecular characterization of human UDP-glucuronic acid/UDP-N-acetylgalactosamine transporter, a novel nucleotide sugar transporter with dual substrate specificity. FEBS Lett. 2001, 495, 87–93. [Google Scholar] [CrossRef]
- Kobayashi, T.; Sleeman, J.E.; Coughtrie, M.W.; Burchell, B. Molecular and functional characterization of microsomal UDP-glucuronic acid uptake by members of the nucleotide sugar transporter (NST) family. Biochem. J. 2006, 400, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Muraoka, M.; Miki, T.; Ishida, N.; Hara, T.; Kawakita, M. Variety of nucleotide sugar transporters with respect to the interaction with nucleoside mono- and diphosphates. J. Biol. Chem. 2007, 282, 24615–24622. [Google Scholar] [CrossRef] [PubMed]
- Dutton, G.J.; Storey, I.D.E. The isolation of a compound of uridine diphosphate and glucuronic acid from liver. Biochem. J. 1953, 53, xxxvii–xxxviii. [Google Scholar] [PubMed]
- Storey, I.D.E.; Dutton, G.J. Uridine compounds in glucuronic acid metabolism. 2. The isolation and structure of ‘uridine-diphosphate-glucuronic acid’. Biochem. J. 1955, 59, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Rowland, A.; Mackenzie, P.I.; Miners, J.O. Transporter-mediated uptake of UDP-glucuronic acid by human liver microsomes: Assay conditions, kinetics, and inhibition. Drug Metab. Dispos. 2015, 43, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Goto, S.; Taniguchi, M.; Muraoka, M.; Toyoda, H.; Sado, Y.; Kawakita, M.; Hayashi, S. UDP-sugar transporter implicated in glycosylation and processing of notch. Nat. Cell Biol. 2001, 3, 816–822. [Google Scholar] [CrossRef] [PubMed]
- Selva, E.M.; Hong, K.; Baeg, G.H.; Beverley, S.M.; Turco, S.J.; Perrimon, N.; Hacker, U. Dual role of the fringe connection gene in both heparan sulphate and fringe-dependent signalling events. Nat. Cell Biol. 2001, 3, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Suda, T.; Kamiyama, S.; Suzuki, M.; Kikuchi, N.; Nakayama, K.; Narimatsu, H.; Jigami, Y.; Aoki, T.; Nishihara, S. Molecular cloning and characterization of a human multisubstrate specific nucleotide-sugar transporter homologous to drosophila fringe connection. J. Biol. Chem. 2004, 279, 26469–26474. [Google Scholar] [CrossRef] [PubMed]
- Dutton, G.J. Commentary: Control of UDP-glucuronyltransferase activity. Biochem. Pharmacol. 1975, 24, 1835–1841. [Google Scholar] [CrossRef]
- Hochman, Y.; Zakim, D. Studies of the catalytic mechanism of microsomal UDP-glucuronyltransferase. Alpha-glucuronidase activity and its stimulation by phospholipids. J. Biol. Chem. 1984, 259, 5521–5525. [Google Scholar] [PubMed]
- Vessey, D.A.; Zakim, D. Regulation of microsomal enzymes by phospholipids. V. Kinetic studies of hepatic uridine diphosphate-glucuronyltransferase. J. Biol. Chem. 1972, 247, 3023–3028. [Google Scholar] [PubMed]
- Magdalou, J.; Hochman, Y.; Zakim, D. Factors modulating the catalytic specificity of a pure form of UDP-glucuronyltransferase. J. Biol. Chem. 1982, 257, 13624–13629. [Google Scholar] [PubMed]
- Dannenberg, A.; Rotenberg, M.; Zakim, D. Regulation of UDP-glucuronosyltransferase by lipid-protein interactions. Comparison of the thermotropic properties of pure reconstituted enzyme with microsomal enzyme. J. Biol. Chem. 1989, 264, 238–242. [Google Scholar] [PubMed]
- Hochman, Y.; Kelley, M.; Zakim, D. Modulation of the number of ligand binding sites of UDP-glucuronyltransferase by the gel to liquid-crystal phase transition of phosphatidylcholines. J. Biol. Chem. 1983, 258, 6509–6516. [Google Scholar] [PubMed]
- Erickson, R.H.; Zakim, D.; Vessey, D.A. Preparation and properties of a phospholipid-free form of microsomal UDP-glucuronyltransferase. Biochemistry 1978, 17, 3706–3711. [Google Scholar] [CrossRef] [PubMed]
- Hochman, Y.; Zakim, D.A. Comparison of the kinetic properties of two different forms of microsomal UDP-glucuronyltransferase. J. Biol. Chem. 1983, 258, 4143–4146. [Google Scholar] [PubMed]
- Rotenberg, M.; Zakim, D. Effect of phospholipids on the thermal stability of microsomal UDP-glucuronosyltransferase. Biochemistry 1989, 28, 8577–8582. [Google Scholar] [CrossRef] [PubMed]
- Hochman, Y.; Zakim, D.; Vessey, D.A. A kinetic mechanism for modulation of the activity of microsomal UDP-glucuronyltransferase by phospholipids. Effects of lysophosphatidylcholines. J. Biol. Chem. 1981, 256, 4783–4788. [Google Scholar] [PubMed]
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Ritchie, K.; Murakoshi, H.; Jacobson, K.; Kusumi, A. Phospholipids undergo hop diffusion in compartmentalized cell membrane. J. Cell Biol. 2002, 157, 1071–1081. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.A.; London, E. Functions of lipid rafts in biological membranes. Annu. Rev. Cell Dev. Biol. 1998, 14, 111–136. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.G.; Fujiwara, T.K.; Sanematsu, F.; Iino, R.; Edidin, M.; Kusumi, A. GPI-anchored receptor clusters transiently recruit Lyn and G alpha for temporary cluster immobilization and Lyn activation: Single-molecule tracking study 1. J. Cell Biol. 2007, 177, 717–730. [Google Scholar] [CrossRef] [PubMed]
- Castuma, C.E.; Brenner, R.R. Cholesterol-dependent modification of microsomal dynamics and UDP-glucuronyltransferase kinetics. Biochemistry 1986, 25, 4733–4738. [Google Scholar] [CrossRef] [PubMed]
- Castuma, C.E.; Brenner, R.R. Effect of dietary cholesterol on microsomal membrane composition, dynamics and kinetic properties of UDP-glucuronyltransferase. Biochim. Biophys. Acta 1986, 855, 231–242. [Google Scholar] [CrossRef]
- Rotenberg, M.; Zakim, D. Effects of cholesterol on the function and thermotropic properties of pure UDP-glucuronosyltransferase. J. Biol. Chem. 1991, 266, 4159–4161. [Google Scholar] [PubMed]
- Vessey, D.A.; Zakim, D. Regulation of microsomal enzymes by phospholipids. IV. Species differences in the properties of microsomal UDP-glucuronyltransferase. Biochim. Biophys. Acta 1972, 268, 61–69. [Google Scholar] [CrossRef]
- Peters, W.H.; te Morsche, R.H.; Roelofs, H.M. Combined polymorphisms in UDP-glucuronosyltransferases 1A1 and 1A6: Implications for patients with Gilbert’s syndrome. J. Hepatol. 2003, 38, 3–8. [Google Scholar] [CrossRef]
- Ishii, Y.; An, K.; Nishimura, Y.; Yamada, H. ATP serves as an endogenous inhibitor of UDP-glucuronosyltransferase (UGT): A new insight into the latency of UGT. Drug Metab. Dispos. 2012, 40, 2081–2089. [Google Scholar] [CrossRef] [PubMed]
- Ishii, Y.; Nurrochmad, A.; Yamada, H. Modulation of UDP-glucuronosyltransferase activity by endogenous compounds. Drug Metab. Pharmacokinet 2010, 25, 134–148. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.; Maeda, S.; Ikushiro, S.; Mackenzie, P.I.; Ishii, Y.; Yamada, H. Inhibitory effects of adenine nucleotides and related substances on UDP-glucuronosyltransferase: Structure-effect relationships and evidence for an allosteric mechanism. Biochim. Biophys. Acta 2007, 1770, 1557–1566. [Google Scholar] [CrossRef] [PubMed]
- Hallinan, T.; Pohl, K.R.; de Brito, R. Studies on the inhibition of hepatic microsomal glucuronidation by uridine nucleotides or adenosine triphosphate. Med. Biol. 1979, 57, 269–273. [Google Scholar] [PubMed]
- Fujiwara, R.; Nakajima, M.; Yamanaka, H.; Katoh, M.; Yokoi, T. Product inhibition of UDP-glucuronosyltransferase (UGT) enzymes by UDP obfuscates the inhibitory effects of UGT substrates. Drug Metab. Dispos. 2008, 36, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Yokota, H.; Ando, F.; Iwano, H.; Yuasa, A. Inhibitory effects of uridine diphosphate on UDP-glucuronosyltransferase. Life Sci. 1998, 63, 1693–1699. [Google Scholar] [CrossRef]
- Agarwal, A.K.; Monder, C.; Eckstein, B.; White, P.C. Cloning and expression of rat cDNA encoding corticosteroid 11 beta-dehydrogenase. J. Biol. Chem. 1989, 264, 18939–18943. [Google Scholar] [PubMed]
- Rostami-Hodjegan, A. Physiologically based pharmacokinetics joined with in vitro-in vivo extrapolation of ADME: A marriage under the arch of systems pharmacology. Clin. Pharmacol. Ther. 2012, 92, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Gill, K.L.; Houston, J.B.; Galetin, A. Characterization of in vitro glucuronidation clearance of a range of drugs in human kidney microsomes: Comparison with liver and intestinal glucuronidation and impact of albumin. Drug Metab. Dispos. 2012, 40, 825–835. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Coughtrie, M.W.H. Revisiting the Latency of Uridine Diphosphate-Glucuronosyltransferases (UGTs)—How Does the Endoplasmic Reticulum Membrane Influence Their Function? Pharmaceutics 2017, 9, 32. https://doi.org/10.3390/pharmaceutics9030032
Liu Y, Coughtrie MWH. Revisiting the Latency of Uridine Diphosphate-Glucuronosyltransferases (UGTs)—How Does the Endoplasmic Reticulum Membrane Influence Their Function? Pharmaceutics. 2017; 9(3):32. https://doi.org/10.3390/pharmaceutics9030032
Chicago/Turabian StyleLiu, Yuejian, and Michael W. H. Coughtrie. 2017. "Revisiting the Latency of Uridine Diphosphate-Glucuronosyltransferases (UGTs)—How Does the Endoplasmic Reticulum Membrane Influence Their Function?" Pharmaceutics 9, no. 3: 32. https://doi.org/10.3390/pharmaceutics9030032