

Structure–Activity Relationship of PAD4 Inhibitors and Their Role in Tumor Immunotherapy

Abstract
:1. Introduction
2. The Structure and Function of PAD4
3. Reversible (Non-Covalent) PAD4 Inhibitors

{kind=link}
{kind=link}
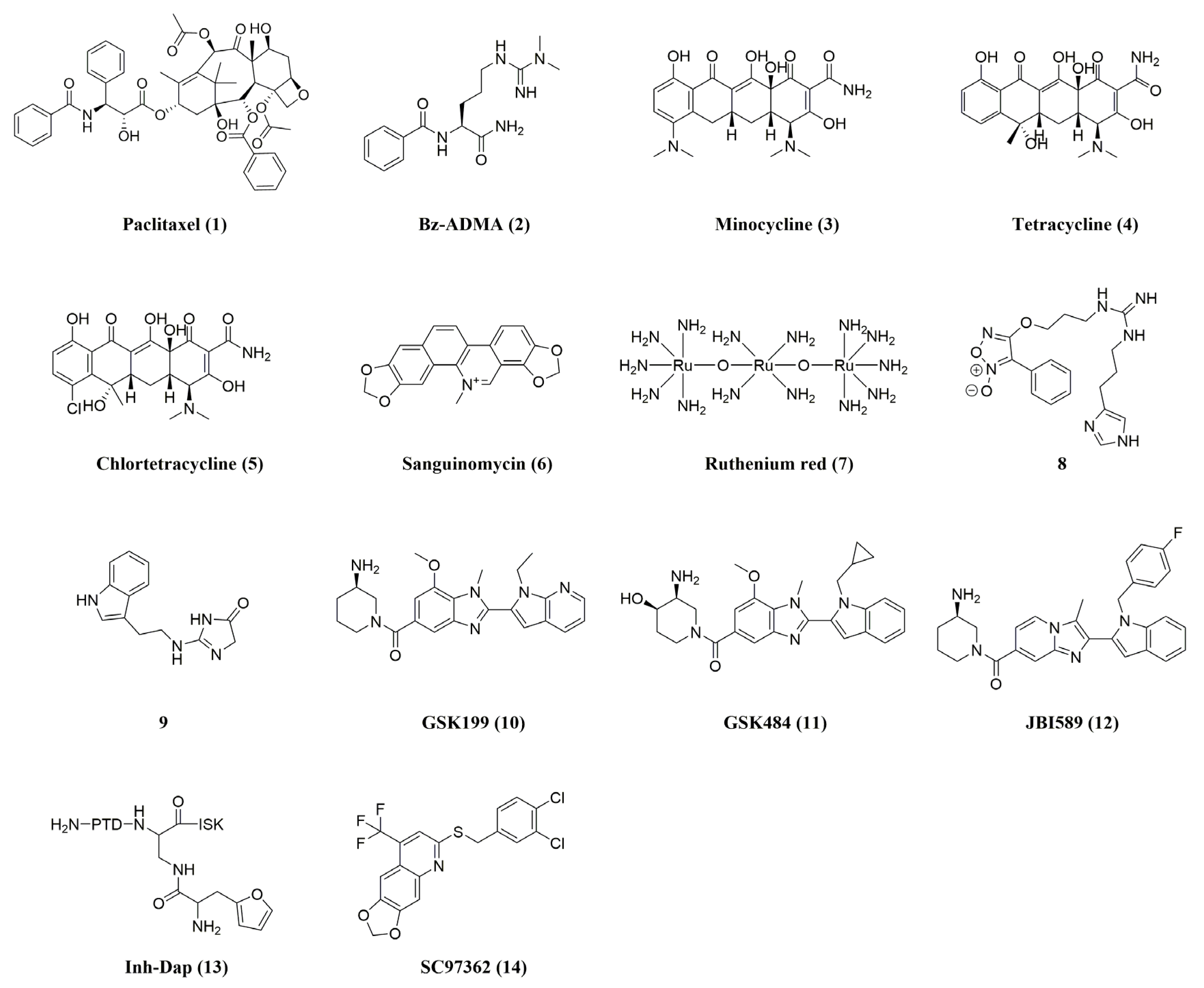{kind=link}
{kind=link}
{kind=link}
| Compound | Name | PAD4 Inhibition | In Vivo Activity (PAD4) | Ref. |
|---|---|---|---|---|
| 1 | Paclitaxel a | Ki= 4.5–10 mM | - | [21,42] |
| 2 | Bz-ADMA a | IC50 = 0.4 mM | - | [40,51] |
| 3 | Minocycline b | IC50 = 0.62 ± 0.01 mM | - | [21,40,41] |
| 4 | Tetracycline b | IC50 = 0.78 ± 0.14 mM | - | [43] |
| 5 | Chlortetracycline b | IC50 = 0.10 ± 0.01 mM | - | [40,43] |
| 6 | Sanguinomycin b | Ki PAD1 = 2000 µM PAD2 = 100 µM PAD3 = 60 µM PAD4 = 80 µM | - | [40] |
| 7 | Ruthenium red b | Ki PAD1 = 30 µM PAD2 = 17 µM PAD3 = 25 µM PAD4 = 10 µM | - | [40,41] |
| 8 | 8 a | 36% inhibition at 10 μM | - | [44] |
| 9 | 9 | IC50 (PAD3) = 100 nM | - | [45] |
| 10 | GSK199 a | IC50 (0 mM Ca2+) = 200 nM IC50 (2 mM Ca2+) = 1.0 μM | Collagen-induced arthritis mouse model | [40,52,53] |
| 11 | GSK484 a | IC50 (0 mM Ca2+) = 50 nM IC50 (2 mM Ca2+) = 250 nM | MMTV-PyMT mouse model for mammary carcinoma (FVB/n background); RIP1-Tag2 mouse model for pancreatic neuroendocrine carcinoma (C57BL/6 background); xenograft MDA-MB-231 mouse model | [40,52,54,55] |
| 12 | JBI589 a | IC50 PAD4 = 122 nM PAD1 > 30 µM PAD2 > 30 µM PAD3 > 30 µM | Collagen-induced arthritis (CIA) model in DBA/1 J mice; mouse LL2, B16F10, and EL4 tumor models | [47,48] |
| 13 | Inh-Dap a | IC50 = 243.2 ± 2.4 μM | - | [41,49] |
| 14 | SC97362 c | IC50 = 1.88 ± 0.26 μM | - | [41,50] |
4. Irreversible (Covalent) PAD4 Inhibitors
| Compound | Name | PAD4 Inhibition | In Vivo Activity (PAD4) | Ref. |
|---|---|---|---|---|
| 15 | NSC95397 b | kinact/KI (M−1min−1) PAD1: 175 PAD2: 1600 PAD3: 9150 PAD4: 4530 | - | [21,56,57] |
| 16 | Streptonigrin b | IC50 = 2.5 ± 0.4 μM kinact/KI (M−1min−1) PAD1: 3700 PAD2: 12,000 PAD3: 3500 PAD4: 440,000 | - | [21,56,57] |
| 17 | 2-chloroacetamidine | - | - | [58,59] |
| 18 | BAA | IC50 = 0.25 ± 0.06 mM | - | [60,62] |
| 19 | Cl-amidine a | IC50 = 5.9 ± 0.3 μM kinact/KI (M−1min−1) PAD1: 37,000 PAD2: 1200 PAD3: 2000 PAD4: 13,000 | Collagen-induced arthritis mouse model; DSS mouse model of colitis; murine sepsis model; mouse model of periodontitis | [46,60,62,80,81] |
| 20 | F-amidine a | IC50 = 22 ± 2.10 μM kinact/KI (M−1min−1) PAD1: 2800 PAD2: 380 PAD3: 170 PAD4: 3000 | - | [60,62] |
| 21 | o-Cl-amidine a | IC50 = 2.2 ± 0.31 μM kinact/KI (M−1min−1) PAD1: 106,400 PAD2: 14,100 PAD3: 10,345 PAD4: 38,000 | - | [65] |
| 22 | o-F-amidine a | IC50 = 1.9 ± 0.21 μM kinact/KI (M−1min−1) PAD1: 180,900 PAD2: 7500 PAD3: 6700 PAD4: 32,500 | - | [65] |
| 23–26 | X-n-amidine (X = F or Cl, n = 2 or 4) a | IC50 > 520 µM | - | [59,60,65] |
| 27 | D-Cl-amidine d | kinact/KI (M−1min−1) PAD4: 1400 | - | [66] |
| 28 | D-F-amidine d | kinact/KI (M−1min−1) PAD4: 130 | - | [66] |
| 29 | D-o-Cl-amidine d | kinact/KI (M−1min−1) PAD4: 30 | - | [66] |
| 30 | D-o-F-amidine d | kinact/KI (M−1min−1) PAD4: 50 | - | [66] |
| 31 | R1 = R3 = H, R2 = COOH, X = Cl a | poor cellular activity | - | [67] |
| 32 | R1 = tBu, R2 = H, R3 = Ph, X = Cl a | MTT assay in U2OS cells, EC50 = 10 ± 2.5 μM | - | [67] |
| 33 | BB-Cl-amidine a | kinact/KI (M−1min−1) PAD1: 16,100 PAD2: 4100 PAD3: 6800 PAD4: 13,300 | Lupus-prone MRL/lpr mice; canine and feline mammary cancer xenograft mice | [68,69,82] |
| 34 | BB-F-amidine a | kinact/KI (M−1min−1) PAD1: 900 PAD2: 1200 PAD3: 3400 PAD4: 3750 | - | [68,69] |
| 35 | YW356 a | IC50 = 1–5 μM | Mouse sarcoma S-180 xenograft model; mouse nasopharyngeal carcinoma model; A549 xenograft mouse model | [70,71,72,73] |
| 36 | ZD-E-1M a | IC50 = 2.39 μM | Mouse S180 sarcoma model; orthotopic 4T1 breast cancer model; Lewis mouse model of lung cancer metastasis | [74] |
| 37 | 5i a | IC50 = 1.9 ± 0.65 μM | Mouse S180 sarcoma model; orthotopic 4T1 breast cancer model | [75] |
| 38 | TDFA a | IC50 = 2.3 μM kinact/KI (M−1min−1) PAD1: 1700 PAD2: 500 PAD3: 400 PAD4: 26,000 | - | [78] |
| 39 | TDCA a | IC50 = 3.4 μM kinact/KI (M−1min−1) PAD1: 21,000 PAD2: 300 PAD3: 920 PAD4: 24,000 | - | [78] |
| 40 | 40T a 40C a | kinact/KI (40T) = 600 M−1min−1 kinact/KI (40C) = 5970 M−1min−1 | - | [79] |
| 41 | 41T a 41C a | kinact/KI (41T) = 4520 M−1min−1 kinact/KI (41C) < 100 M−1min−1 | - | [79] |
| 42 | 4B a | IC50 = 1.89 ± 0.33 μM | Mouse S180 sarcoma model; orthotopic 4T1 breast cancer model; Lewis mouse model of lung cancer metastasis | [75,77] |
| 43 | K-CRGDV-4B | - | Lewis mouse model of lung cancer metastasis | [77] |
5. Delivery Systems for PAD4 Inhibitors
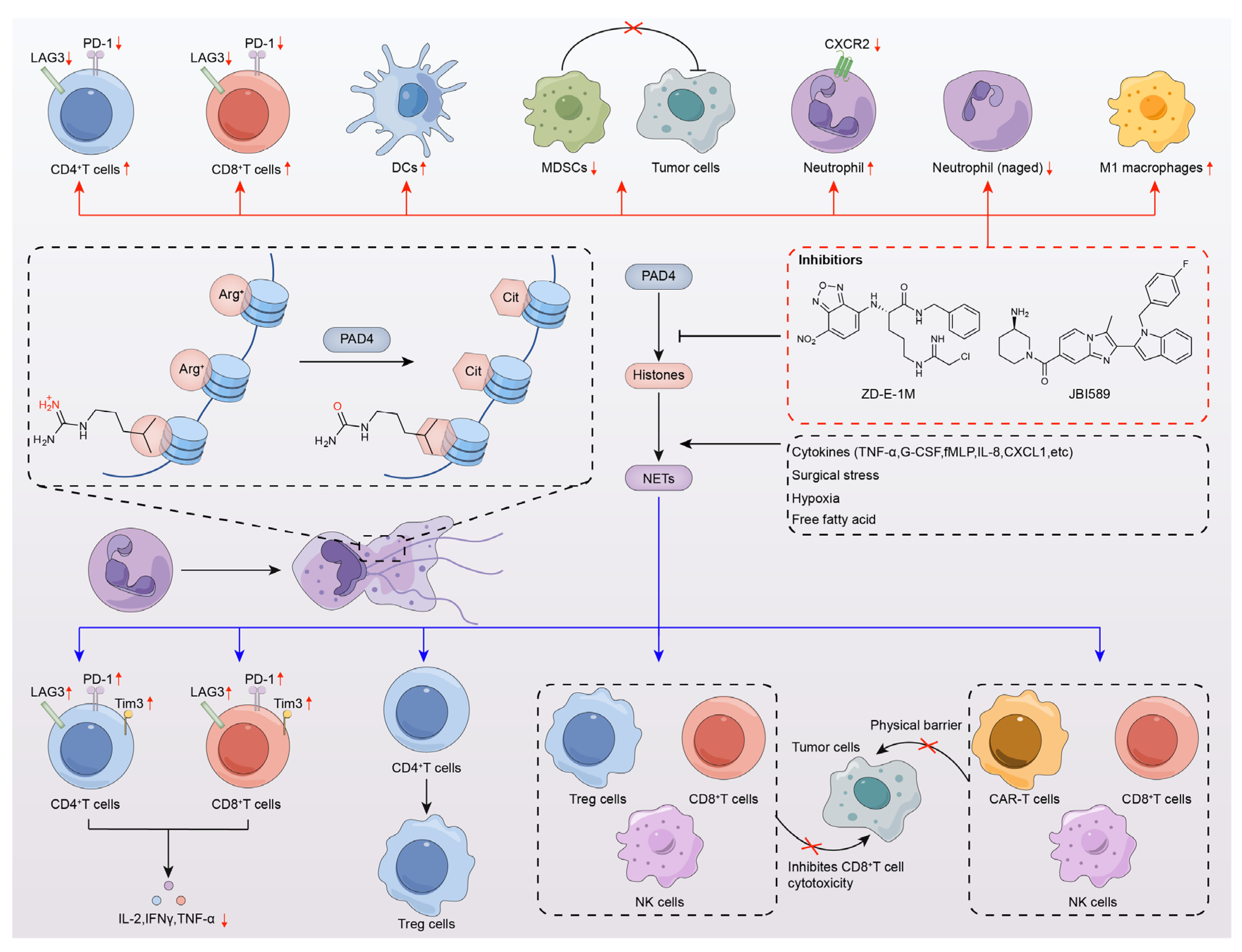
6. PAD4 Inhibitors Enhance Antitumor Immunotherapy
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alghamdi, M.; Al Ghamdi, K.A.; Khan, R.H.; Uversky, V.N.; Redwan, E.M. An interplay of structure and intrinsic disorder in the functionality of peptidylarginine deiminases, a family of key autoimmunity-related enzymes. Cell. Mol. Life Sci. 2019, 76, 4635–4662. [Google Scholar] [CrossRef]
- Nakashima, K.; Hagiwara, T.; Yamada, M. Nuclear localization of peptidylarginine deiminase V and histone deimination in granulocytes. J. Biol. Chem. 2002, 277, 49562–49568. [Google Scholar] [CrossRef]
- Knuckley, B.; Bhatia, M.; Thompson, P.R. Protein arginine deiminase 4: Evidence for a reverse protonation mechanism. Biochemistry 2007, 46, 6578–6587. [Google Scholar] [CrossRef]
- Darrah, E.; Giles, J.T.; Ols, M.L.; Bull, H.G.; Andrade, F.; Rosen, A. Erosive rheumatoid arthritis is associated with antibodies that activate PAD4 by increasing calcium sensitivity. Sci. Transl. Med. 2013, 5, 186ra65. [Google Scholar] [CrossRef] [PubMed]
- Rohrbach, A.S.; Slade, D.J.; Thompson, P.R.; Mowen, K.A. Activation of PAD4 in NET formation. Front. Immunol. 2012, 3, 360. [Google Scholar] [CrossRef]
- Delgado-Rizo, V.; Martínez-Guzmán, M.A.; Iñiguez-Gutierrez, L.; García-Orozco, A.; Alvarado-Navarro, A.; Fafutis-Morris, M. Neutrophil Extracellular Traps and Its Implications in Inflammation: An Overview. Front. Immunol. 2017, 8, 81. [Google Scholar] [CrossRef]
- Bronze-Da-Rocha, E.; Santos-Silva, A. Neutrophil Elastase Inhibitors and Chronic Kidney Disease. Int. J. Biol. Sci. 2018, 14, 1343–1360. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Lu, Y.; Wang, Y.; Wang, Y. PAD4 and Its Inhibitors in Cancer Progression and Prognosis. Pharmaceutics 2022, 14, 2414. [Google Scholar] [CrossRef]
- Griffin, G.K.; Wu, J.; Iracheta-Vellve, A.; Patti, J.C.; Hsu, J.; Davis, T.; Dele-Oni, D.; Du, P.P.; Halawi, A.G.; Ishizuka, J.J.; et al. Epigenetic silencing by SETDB1 suppresses tumour intrinsic immunogenicity. Nature 2021, 595, 309–314. [Google Scholar] [CrossRef]
- Zhang, S.-M.; Cai, W.L.; Liu, X.; Thakral, D.; Luo, J.; Chan, L.H.; McGeary, M.K.; Song, E.; Blenman, K.R.M.; Micevic, G.; et al. KDM5B promotes immune evasion by recruiting SETDB1 to silence retroelements. Nature 2021, 598, 682–687. [Google Scholar] [CrossRef] [PubMed]
- Musella, M.; Manduca, N.; Maccafeo, E.; Sistigu, A. Epigenetics behind tumor immunology: A mini review. Oncogene 2023, 42, 2932–2938. [Google Scholar] [CrossRef]
- Qiu, J.; Xu, B.; Ye, D.; Ren, D.; Wang, S.; Benci, J.L.; Xu, Y.; Ishwaran, H.; Beltra, J.-C.; Wherry, E.J.; et al. Cancer cells resistant to immune checkpoint blockade acquire interferon-associated epigenetic memory to sustain T cell dysfunction. Nat. Cancer 2023, 4, 43–61. [Google Scholar] [CrossRef]
- Esteller, M. Epigenetics in cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef]
- Jones, P.A.; Baylin, S.B. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002, 3, 415–428. [Google Scholar] [CrossRef]
- Yang, J.; Xu, J.; Wang, W.; Zhang, B.; Yu, X.; Shi, S. Epigenetic regulation in the tumor microenvironment: Molecular mechanisms and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 210. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the Histone Code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef]
- Hemmers, S.; Teijaro, J.R.; Arandjelovic, S.; Mowen, K.A. PAD4-mediated neutrophil extracellular trap formation is not required for immunity against influenza infection. PLoS ONE 2011, 6, e22043. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Gu, Y.; Sun, H.; Ge, Q. Neutrophil extracellular traps in tumor progression and immunotherapy. Front. Immunol. 2023, 14, 1135086. [Google Scholar] [CrossRef] [PubMed]
- Arita, K.; Hashimoto, H.; Shimizu, T.; Nakashima, K.; Yamada, M.; Sato, M. Structural Basis for Ca(2+)-Induced Activation of Human PAD4. Nat. Struct. Mol. Biol. 2004, 11, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Arfman, T.; Wichapong, K.; Reutelingsperger, C.P.; Voorberg, J.; Nicolaes, G.A. PAD4 takes charge during neutrophil activation: Impact of PAD4 mediated NET formation on immune-mediated disease. J. Thromb. Haemost. 2021, 19, 1607–1617. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Thompson, P.R. Protein Arginine Deiminases (PADs): Biochemistry and Chemical Biology of Protein Citrullination. Acc. Chem. Res. 2019, 52, 818–832. [Google Scholar] [CrossRef]
- Jones, J.E.; Causey, C.P.; Knuckley, B.; Slack-Noyes, J.L.; Thompson, P.R. Protein arginine deiminase 4 (PAD4): Current understanding and future therapeutic potential. Curr. Opin. Drug Discov. Dev. 2009, 12, 616–627. [Google Scholar]
- Liu, Y.-L.; Chiang, Y.-H.; Liu, G.-Y.; Hung, H.-C. Functional role of dimerization of human peptidylarginine deiminase 4 (PAD4). PLoS ONE 2011, 6, e21314. [Google Scholar] [CrossRef]
- Liu, Y.-L.; Lee, C.-Y.; Huang, Y.-N.; Chen, H.-Y.; Liu, G.-Y.; Hung, H.-C. Probing the Roles of Calcium-Binding Sites during the Folding of Human Peptidylarginine Deiminase 4. Sci. Rep. 2017, 7, 2429. [Google Scholar] [CrossRef] [PubMed]
- Vossenaar, E.R.; Zendman, A.J.; van Venrooij, W.J.; Pruijn, G.J. PAD, a growing family of citrullinating enzymes: Genes, features and involvement in disease. BioEssays 2003, 25, 1106–1118. [Google Scholar] [CrossRef] [PubMed]
- Vossenaar, E.R.; Radstake, T.R.D.; van der Heijden, A.; Mansum, M.A.M.v.; Dieteren, C.; de Rooij, D.-J.; Barrera, P.; Zendman, A.J.W.; van Venrooij, W.J. Expression and activity of citrullinating peptidylarginine deiminase enzymes in monocytes and macrophages. Ann. Rheum. Dis. 2004, 63, 373–381. [Google Scholar] [CrossRef]
- Asaga, H.; Nakashima, K.; Senshu, T.; Ishigami, A.; Yamada, M. Immunocytochemical localization of peptidylarginine deiminase in human eosinophils and neutrophils. J. Leukoc. Biol. 2001, 70, 46–51. [Google Scholar] [CrossRef]
- Yang, C.; Dong, Z.-Z.; Zhang, J.; Teng, D.; Luo, X.; Li, D.; Zhou, Y. Peptidylarginine deiminases 4 as a promising target in drug discovery. Eur. J. Med. Chem. 2021, 226, 113840. [Google Scholar] [CrossRef]
- Chang, X.; Han, J.; Pang, L.; Zhao, Y.; Yang, Y.; Shen, Z. Increased PADI4 expression in blood and tissues of patients with malignant tumors. BMC Cancer 2009, 9, 40. [Google Scholar] [CrossRef]
- Arelaki, S.; Arampatzioglou, A.; Kambas, K.; Papagoras, C.; Miltiades, P.; Angelidou, I.; Mitsios, A.; Kotsianidis, I.; Skendros, P.; Sivridis, E.; et al. Gradient Infiltration of Neutrophil Extracellular Traps in Colon Cancer and Evidence for Their Involvement in Tumour Growth. PLoS ONE 2016, 11, e0154484. [Google Scholar] [CrossRef]
- Nakashima, K.; Arai, S.; Suzuki, A.; Nariai, Y.; Urano, T.; Nakayama, M.; Ohara, O.; Yamamura, K.-I.; Yamamoto, K.; Miyazaki, T. PAD4 regulates proliferation of multipotent haematopoietic cells by controlling c-myc expression. Nat. Commun. 2013, 4, 1836–1838. [Google Scholar] [CrossRef]
- Wang, S.; Wang, Y. Peptidylarginine deiminases in citrullination, gene regulation, health and pathogenesis. Biochim. Biophys. Acta Bioenerg. 2013, 1829, 1126–1135. [Google Scholar] [CrossRef]
- György, B.; Tóth, E.; Tarcsa, E.; Falus, A.; Buzás, E.I. Citrullination: A posttranslational modification in health and disease. Int. J. Biochem. Cell Biol. 2006, 38, 1662–1677. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, H.; Tang, J.; Guo, Z.; Wang, Y. Peptidylarginine Deiminase and Alzheimer’s Disease. J. Alzheimer’s Dis. 2022, 85, 473–484. [Google Scholar] [CrossRef]
- de Souza, H.S.P.; Fiocchi, C. Immunopathogenesis of IBD: Current state of the art. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Gremese, E.; Ferraccioli, G. The pathogenesis of microthrombi in COVID-19 cannot be controlled by DOAC: NETosis should be the target. J. Intern. Med. 2021, 289, 420–421. [Google Scholar] [CrossRef]
- Chapman, E.A.; Lyon, M.; Simpson, D.; Mason, D.; Beynon, R.J.; Moots, R.J.; Wright, H.L. Caught in a Trap? Proteomic Analysis of Neutrophil Extracellular Traps in Rheumatoid Arthritis and Systemic Lupus Erythematosus. Front. Immunol. 2019, 10, 423. [Google Scholar] [CrossRef]
- Carmona-Rivera, C.; Zhao, W.; Yalavarthi, S.; Kaplan, M.J. Neutrophil extracellular traps induce endothelial dysfunction in systemic lupus erythematosus through the activation of matrix metalloproteinase-2. Ann. Rheum. Dis. 2015, 74, 1417–1424. [Google Scholar] [CrossRef]
- Nemmara, V.V.; Thompson, P.R. Development of Activity-Based Proteomic Probes for Protein Citrullination. Curr. Top Microbiol. Immunol. 2019, 420, 233–251. [Google Scholar] [CrossRef]
- Ciesielski, O.; Biesiekierska, M.; Panthu, B.; Soszyński, M.; Pirola, L.; Balcerczyk, A. Citrullination in the pathology of inflammatory and autoimmune disorders: Recent advances and future perspectives. Cell. Mol. Life Sci. 2022, 79, 5413–5461. [Google Scholar] [CrossRef]
- Fuhrmann, J.; Clancy, K.W.; Thompson, P.R. Chemical biology of protein arginine modifications in epigenetic regulation. Chem. Rev. 2015, 115, 5413–5461. [Google Scholar] [CrossRef] [PubMed]
- Pritzker, L.; Moscarello, M. A novel microtubule independent effect of paclitaxel: The inhibition of peptidylarginine deiminase from bovine brain. Biochim. Biophys. Acta 1998, 1388, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Knuckley, B.; Luo, Y.; Thompson, P.R. Profiling Protein Arginine Deiminase 4 (PAD4): A novel screen to identify PAD4 inhibitors. Bioorganic Med. Chem. 2008, 16, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Bozdag, M.; Dreker, T.; Henry, C.; Tosco, P.; Vallaro, M.; Fruttero, R.; Scozzafava, A.; Carta, F.; Supuran, C.T. Novel small molecule protein arginine deiminase 4 (PAD4) inhibitors. Bioorganic Med. Chem. Lett. 2013, 23, 715–719. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, P.; Vagaska, B.; Merchant, R.; Matthews, C.J.; Marson, C.M. Discovery of a structurally novel, drug-like and potent inhibitor of peptidylarginine deiminase. MedChemComm 2013, 4, 1109–1113. [Google Scholar] [CrossRef]
- Willis, V.C.; Gizinski, A.M.; Banda, N.K.; Causey, C.P.; Knuckley, B.; Cordova, K.N.; Luo, Y.; Levitt, B.; Glogowska, M.; Chandra, P.; et al. N-α-benzoyl-N5-(2-chloro-1-iminoethyl)-L-ornithine amide, a protein arginine deiminase inhibitor, reduces the severity of mu-rine collagen-induced arthritis. J. Immunol. 2011, 186, 4396–4404. [Google Scholar] [CrossRef] [PubMed]
- Gajendran, C.; Fukui, S.; Sadhu, N.M.; Zainuddin, M.; Rajagopal, S.; Gosu, R.; Gutch, S.; Fukui, S.; Sheehy, C.E.; Chu, L.; et al. Alleviation of arthritis through prevention of neutrophil extracellular traps by an orally available inhibitor of protein arginine deiminase 4. Sci. Rep. 2023, 13, 3189. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Lin, C.; Garcia-Gerique, L.; Fu, S.; Cruz, Z.; Bonner, E.E.; Rosenwasser, M.; Rajagopal, S.; Sadhu, M.N.; Gajendran, C.; et al. A Novel Selective Inhibitor JBI-589 Targets PAD4-Mediated Neutrophil Migration to Suppress Tumor Progression. Cancer Res. 2022, 82, 3561–3572. [Google Scholar] [CrossRef]
- Teo, C.Y.; Tejo, B.A.; Leow, A.T.C.; Salleh, A.B.; Rahman, M.B.A. Novel furan-containing peptide-based inhibitors of protein arginine deiminase type IV (PAD4). Chem. Biol. Drug Des. 2017, 90, 1134–1146. [Google Scholar] [CrossRef]
- Aliko, A.; Kamińska, M.; Falkowski, K.; Bielecka, E.; Benedyk-Machaczka, M.; Malicki, S.; Kozieł, J.; Wong, A.; Bryzek, D.; Kantyka, T.; et al. Discovery of Novel Potential Reversible Peptidyl Arginine Deiminase Inhibitor. Int. J. Mol. Sci. 2019, 20, 2174. [Google Scholar] [CrossRef]
- Hidaka, Y.; Hagiwara, T.; Yamada, M. Methylation of the guanidino group of arginine residues prevents citrullination by peptidylarginine deiminase IV. FEBS Lett. 2005, 579, 4088–4092. [Google Scholar] [CrossRef]
- Lewis, H.D.; Liddle, J.; Coote, J.E.; Atkinson, S.J.; Barker, M.D.; Bax, B.D.; Bicker, K.L.; Bingham, R.P.; Campbell, M.; Chen, Y.H.; et al. Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nat. Chem. Biol. 2015, 11, 189–191. [Google Scholar] [CrossRef]
- Willis, V.C.; Banda, N.K.; Cordova, K.N.; Chandra, P.E.; Robinson, W.H.; Cooper, D.C.; Lugo, D.; Mehta, G.; Taylor, S.; Tak, P.P.; et al. Protein arginine deiminase 4 inhibition is sufficient for the amelioration of collagen-induced arthritis. Clin. Exp. Immunol. 2017, 188, 263–274. [Google Scholar] [CrossRef]
- Du, M.; Yang, W.; Schmull, S.; Gu, J.; Xue, S. Inhibition of peptidyl arginine deiminase-4 protects against myocardial infarction induced cardiac dysfunction. Int. Immunopharmacol. 2020, 78, 106055. [Google Scholar] [CrossRef]
- Wei, L.; Wang, X.; Luo, M.; Wang, H.; Chen, H.; Huang, C. The PAD4 inhibitor GSK484 enhances the radiosensitivity of triple-negative breast cancer. Hum. Exp. Toxicol. 2021, 40, 1074–1083. [Google Scholar] [CrossRef]
- Lewallen, D.M.; Bicker, K.L.; Madoux, F.; Chase, P.; Anguish, L.; Coonrod, S.; Hodder, P.; Thompson, P.R. A FluoPol-ABPP PAD2 high-throughput screen identifies the first calcium site inhibitor targeting the PADs. ACS Chem. Biol. 2014, 9, 913–921. [Google Scholar] [CrossRef]
- Knuckley, B.; Jones, J.E.; Bachovchin, D.A.; Slack, J.; Causey, C.P.; Brown, S.J.; Rosen, H.; Cravatt, B.F.; Thompson, P.R. A fluopol-ABPP HTS assay to identify PAD inhibitors. Chem. Commun. 2010, 46, 7175–7177. [Google Scholar] [CrossRef]
- Dreyton, C.J.; Knuckley, B.; Jones, J.E.; Lewallen, D.M.; Thompson, P.R. Mechanistic studies of protein arginine deiminase 2: Evidence for a substrate-assisted mechanism. Biochemistry 2014, 53, 4426–4433. [Google Scholar] [CrossRef]
- Knuckley, B.; Causey, C.P.; Jones, J.E.; Bhatia, M.; Dreyton, C.J.; Osborne, T.C.; Takahara, H.; Thompson, P.R. Substrate specificity and kinetic studies of PADs 1, 3, and 4 identify potent and selective inhibitors of protein arginine deimi-nase 3. Biochemistry 2010, 49, 4852–4863. [Google Scholar] [CrossRef]
- Luo, Y.; Arita, K.; Bhatia, M.; Knuckley, B.; Lee, Y.-H.; Stallcup, M.R.; Sato, M.; Thompson, P.R. Inhibitors and inactivators of protein arginine deiminase 4: Functional and structural characterization. Biochemistry 2006, 45, 11727–11736. [Google Scholar] [CrossRef]
- Fang, Q.; Stehr, A.M.; Naschberger, E.; Knopf, J.; Herrmann, M.; Stürzl, M. No NETs no TIME: Crosstalk between neutrophil extracellular traps and the tumor immune microenvironment. Front. Immunol. 2022, 13, 1075260. [Google Scholar] [CrossRef]
- Luo, Y.; Knuckley, B.; Lee, Y.-H.; Stallcup, M.R.; Thompson, P.R. A fluoroacetamidine-based inactivator of protein arginine deiminase 4: Design, synthesis, and in vitro and in vivo evaluation. J. Am. Chem. Soc. 2006, 128, 1092–1093. [Google Scholar] [CrossRef] [PubMed]
- Biron, B.M.; Chung, C.-S.; O’Brien, X.M.; Chen, Y.; Reichner, J.S.; Ayala, A. Cl-Amidine Prevents Histone 3 Citrullination and Neutrophil Extracellular Trap Formation, and Improves Survival in a Murine Sepsis Model. J. Innate Immun. 2017, 9, 22–32. [Google Scholar] [CrossRef]
- Ham, A.; Kim, M.; Brown, K.M.; Ma, Z.; D’Agati, V.; Lee, H.T.; Rabadi, M.M.; Han, S.J.; Li, H.; Cho, A.; et al. Peptidyl arginine deiminase-4 activation exacerbates kidney ischemia-reperfusion injury. Am. J. Physiol. Physiol. 2014, 307, F1052–F1062. [Google Scholar] [CrossRef]
- Causey, C.P.; Jones, J.E.; Slack, J.L.; Kamei, D.; Jones, L.E.; Subramanian, V.; Knuckley, B.; Ebrahimi, P.; Chumanevich, A.A.; Luo, Y.; et al. The development of N-α-(2-carboxyl)benzoyl-N(5)-(2-fluoro-1-iminoethyl)-l-ornithine amide (o-F-amidine) and N-α-(2-carboxyl)benzoyl-N(5)-(2-chloro-1-iminoethyl)-l-ornithine amide (o-Cl-amidine) as second generation protein arginine deiminase (PAD) inhibitors. J. Med. Chem. 2011, 54, 6919–6935. [Google Scholar] [CrossRef]
- Bicker, K.L.; Anguish, L.; Chumanevich, A.A.; Cameron, M.D.; Cui, X.; Witalison, E.; Subramanian, V.; Zhang, X.; Chumanevich, A.P.; Hofseth, L.J.; et al. D-amino acid based protein arginine deiminase inhibitors: Synthesis, pharmacokinetics, and in cellulo efficacy. ACS Med. Chem. Lett. 2012, 3, 1081–1085. [Google Scholar] [CrossRef]
- Subramanian, V.; Knight, J.S.; Parelkar, S.; Anguish, L.; Coonrod, S.A.; Kaplan, M.J.; Thompson, P.R. Design, synthesis, and biological evaluation of tetrazole analogs of Cl-amidine as protein arginine deiminase inhibitors. J. Med. Chem. 2015, 58, 1337–1344. [Google Scholar] [CrossRef]
- Knight, J.S.; Subramanian, V.; O’Dell, A.A.; Yalavarthi, S.; Zhao, W.; Smith, C.K.; Hodgin, J.B.; Thompson, P.R.; Kaplan, M.J. Peptidylarginine deiminase inhibition disrupts NET formation and protects against kidney, skin and vascular disease in lupus-prone MRL/lprmice. Ann. Rheum. Dis. 2015, 74, 2199–2206. [Google Scholar] [CrossRef]
- Muth, A.; Subramanian, V.; Beaumont, E.; Nagar, M.; Kerry, P.; McEwan, P.; Srinath, H.; Clancy, K.; Parelkar, S.; Thompson, P.R. Development of a Selective Inhibitor of Protein Arginine Deiminase 2. J. Med. Chem. 2017, 60, 3198–3211. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, P.; Wang, S.; Hu, J.; Chen, X.A.; Wu, J.; Fisher, M.; Oshaben, K.; Zhao, N.; Gu, Y.; et al. Anticancer peptidylarginine deiminase (PAD) inhibitors regulate the autophagy flux and the mammalian target of rapamycin complex 1 activity. J. Biol. Chem. 2012, 287, 25941–25953. [Google Scholar] [CrossRef]
- Chen, H.; Wei, L.; Luo, M.; Wang, X.; Zhan, Y.; Mao, Y.; Huang, C.; Li, J.; Lu, H. PAD4 inhibitor promotes DNA damage and radiosensitivity of nasopharyngeal carcinoma cells. Environ. Toxicol. 2021, 36, 2291–2301. [Google Scholar] [CrossRef]
- Zhou, Y.; An, L.-L.; Chaerkady, R.; Mittereder, N.; Clarke, L.; Cohen, T.S.; Chen, B.; Hess, S.; Sims, G.P.; Mustelin, T. Evidence for a direct link between PAD4-mediated citrullination and the oxidative burst in human neutrophils. Sci. Rep. 2018, 8, 15228. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Peng, Z.; Zhu, D.; Jia, Y.; Taledaohan, A.; Li, Y.; Liu, J.; Wang, Y.; Wang, Y. RGD Peptide and PAD4 Inhibitor-Loaded Gold Nanorods for Chemo-Photothermal Combined Therapy to Inhibit Tumor Growth, Prevent Lung Metastasis and Improve Biosafety. Int. J. Nanomed. 2021, 16, 5565–5580. [Google Scholar] [CrossRef]
- Zhu, D.; Lu, Y.; Gui, L.; Wang, W.; Hu, X.; Chen, S.; Wang, Y.; Wang, Y. Self-assembling, pH-responsive nanoflowers for inhibiting PAD4 and neutrophil extracellular trap formation and improving the tumor immune microenvironment. Acta Pharm. Sin. B 2022, 12, 2592–2608. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Lu, Y.; Hu, B.; Pang, Y.; Liu, B.; Zhang, M.; Wang, W.; Wang, Y. Highly-tumor-targeted PAD4 inhibitors with PBA modification inhibit tumors in vivo by specifically inhibiting the PAD4-H3cit-NETs pathway in neutrophils. Eur. J. Med. Chem. 2023, 258, 115619. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Xu, Z.; Hu, X.; Lai, Y.; Xu, J.; Hou, B.; Wang, Y.; Yu, H.; Tian, Y.; Zhang, W. Bioinspired Multivalent Peptide Nanotubes for Sialic Acid Targeting and Imaging-Guided Treatment of Metastatic Melanoma. Small 2019, 15, e1900157. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Taledaohan, A.; Jia, R.; Wang, X.; Jia, Y.; Liu, J.; Wang, Y. Chitosan nanomedicine containing RGD peptide and PAD4 inhibitor based on phenyl boronate coupling inhibition of primary tumor growth and lung metastasis. Biomed. Pharmacother. 2023, 168, 115826. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.E.; Slack, J.L.; Fang, P.; Zhang, X.; Subramanian, V.; Causey, C.P.; Coonrod, S.A.; Guo, M.; Thompson, P.R. Synthesis and screening of a haloacetamidine containing library to identify PAD4 selective inhibitors. ACS Chem. Biol. 2011, 7, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Mondal, S.; Parelkar, S.S.; Nagar, M.; Thompson, P.R. Photochemical Control of Protein Arginine Deiminase (PAD) Activity. ACS Chem. Biol. 2018, 13, 1057–1065. [Google Scholar] [CrossRef]
- Chumanevich, A.A.; Causey, C.P.; Knuckley, B.A.; Jones, J.E.; Poudyal, D.; Chumanevich, A.P.; Davis, T.; Matesic, L.E.; Thompson, P.R.; Hofseth, L.J. Suppression of colitis in mice by Cl-amidine: A novel peptidylarginine deiminase inhibitor. Am. J. Physiol. Liver Physiol. 2011, 300, G929–G938. [Google Scholar] [CrossRef]
- Shindo, S.; Pierrelus, R.; Ikeda, A.; Nakamura, S.; Heidari, A.; Pastore, M.R.; Leon, E.; Ruiz, S.; Chheda, H.; Khatiwala, R.; et al. Extracellular Release of Citrullinated Vimentin Directly Acts on Osteoclasts to Promote Bone Resorption in a Mouse Model of Periodontitis. Cells 2023, 12, 1109. [Google Scholar] [CrossRef]
- Ledet, M.M.; Anderson, R.; Harman, R.; Muth, A.; Thompson, P.R.; Coonrod, S.A.; Van de Walle, G.R. BB-Cl-Amidine as a novel therapeutic for canine and feline mammary cancer via activation of the endoplasmic reticulum stress pathway. BMC Cancer 2018, 18, 412. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, A.; Maji, A.; Potdar, P.D.; Singh, N.; Parikh, P.; Bisht, B.; Mukherjee, A.; Paul, M.K. Lung cancer immunotherapy: Progress, pitfalls, and promises. Mol. Cancer 2023, 22, 40. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Gui, L.; Feng, Q.; Taledaohan, A.; Li, Y.; Wang, W.; Wang, Y.; Wang, Y. TAT-Modified Gold Nanoparticles Enhance the Antitumor Activity of PAD4 Inhibitors. Int. J. Nanomed. 2020, 15, 6659–6671. [Google Scholar] [CrossRef]
- Liu, Y.; Bhattarai, P.; Dai, Z.; Chen, X. Photothermal therapy and photoacoustic imaging via nanotheranostics in fighting cancer. Chem. Soc. Rev. 2019, 48, 2053–2108. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Lv, W.; Li, S.; Zhang, Q.; He, W.; Min, Z.; Teng, C.; Chen, Y.; Liu, L.; Yin, J.; et al. Smart Liposomal Nanocarrier Enhanced the Treatment of Ischemic Stroke through Neutrophil Extracellular Traps and Cyclic Guanosine Monophosphate-Adenosine Monophosphate Synthase-Stimulator of Interferon Genes (cGAS-STING) Pathway Inhibition of Ischemic Penumbra. ACS Nano 2023, 17, 17845–17857. [Google Scholar] [CrossRef]
- Kakaradov, B.; Arsenio, J.; Widjaja, C.E.; He, Z.; Aigner, S.; Metz, P.J.; Yu, B.; Wehrens, E.J.; Lopez, J.; Kim, S.H.; et al. Early transcriptional and epigenetic regulation of CD8+ T cell differentiation revealed by single-cell RNA sequencing. Nat. Immunol. 2017, 18, 422–432. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Kaltenmeier, C.; Yazdani, H.O.; Morder, K.; Geller, D.A.; Simmons, R.L.; Tohme, S. Neutrophil Extracellular Traps Promote T Cell Exhaustion in the Tumor Microenvironment. Front. Immunol. 2021, 12, 785222. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, H.; Wang, Y.; Brown, Z.J.; Xia, Y.; Huang, Z.; Shen, C.; Hu, Z.; Beane, J.; Ansa-Addo, E.A.; et al. Regulatory T-cell and neutrophil extracellular trap interaction contributes to carcinogenesis in non-alcoholic steatohepatitis. J. Hepatol. 2021, 75, 1271–1283. [Google Scholar] [CrossRef]
- Que, H.; Fu, Q.; Lan, T.; Tian, X.; Wei, X. Tumor-associated neutrophils and neutrophil-targeted cancer therapies. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188762. [Google Scholar] [CrossRef] [PubMed]
- Biron, B.M.; Chung, C.-S.; Chen, Y.; Wilson, Z.; Fallon, E.A.; Reichner, J.S.; Ayala, A. PAD4 Deficiency Leads to Decreased Organ Dysfunction and Improved Survival in a Dual Insult Model of Hemorrhagic Shock and Sepsis. J. Immunol. 2018, 200, 1817–1828. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, Y.; Jia, R.; Taledaohan, A.; Wang, Y.; Wang, Y. Structure–Activity Relationship of PAD4 Inhibitors and Their Role in Tumor Immunotherapy. Pharmaceutics 2024, 16, 335. https://doi.org/10.3390/pharmaceutics16030335
Jia Y, Jia R, Taledaohan A, Wang Y, Wang Y. Structure–Activity Relationship of PAD4 Inhibitors and Their Role in Tumor Immunotherapy. Pharmaceutics. 2024; 16(3):335. https://doi.org/10.3390/pharmaceutics16030335
Chicago/Turabian StyleJia, Yijiang, Renbo Jia, Ayijiang Taledaohan, Yanming Wang, and Yuji Wang. 2024. "Structure–Activity Relationship of PAD4 Inhibitors and Their Role in Tumor Immunotherapy" Pharmaceutics 16, no. 3: 335. https://doi.org/10.3390/pharmaceutics16030335