
Interaction of ALK Inhibitors with Polyspecific Organic Cation Transporters and the Impact of Substrate-Dependent Inhibition on the Prediction of Drug–Drug Interactions

Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Culture
2.3. Inhibition Screening and Uptake Assays in HEK293 Cells
2.4. Quantification of mIBG by LC-MS/MS
2.5. Data Analysis
3. Results
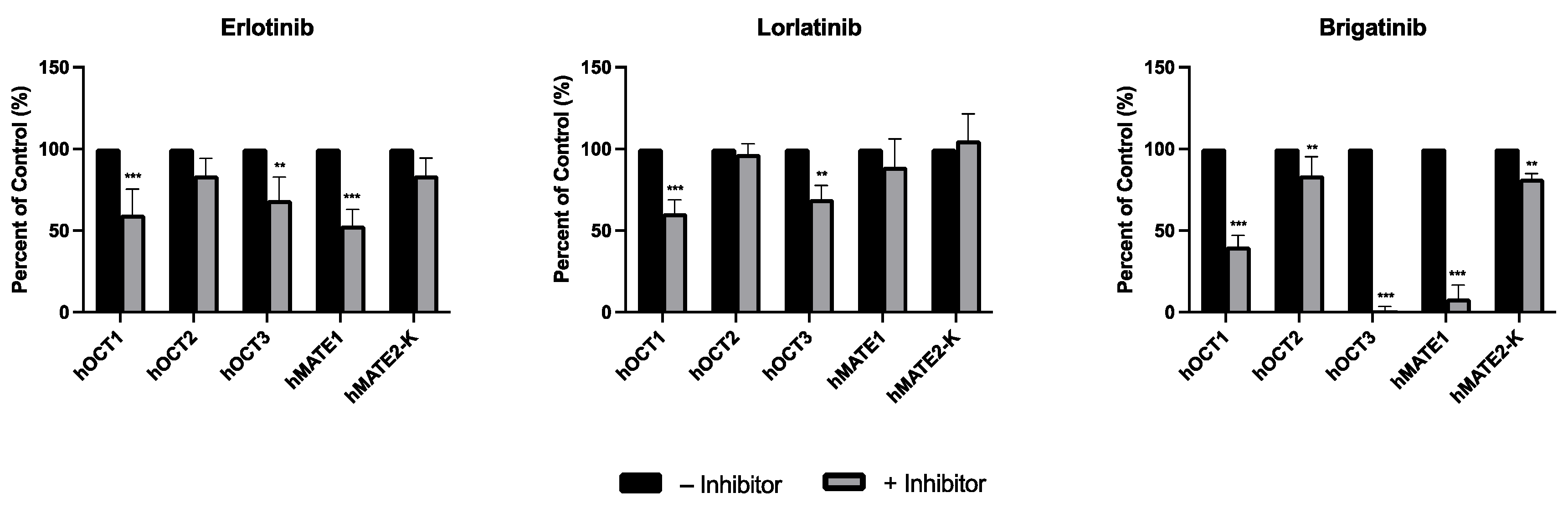
3.1. Inhibitory Screening of Selected TKIs on the Uptake of mIBG by hOCT1-3 and hMATE1/2-K
3.2. Dose-Dependent Inhibition Profile and IC50 Determinations of Selected TKIs on the Uptake of mIBG Mediated by hOCT1-3 and hMATE1/2-K
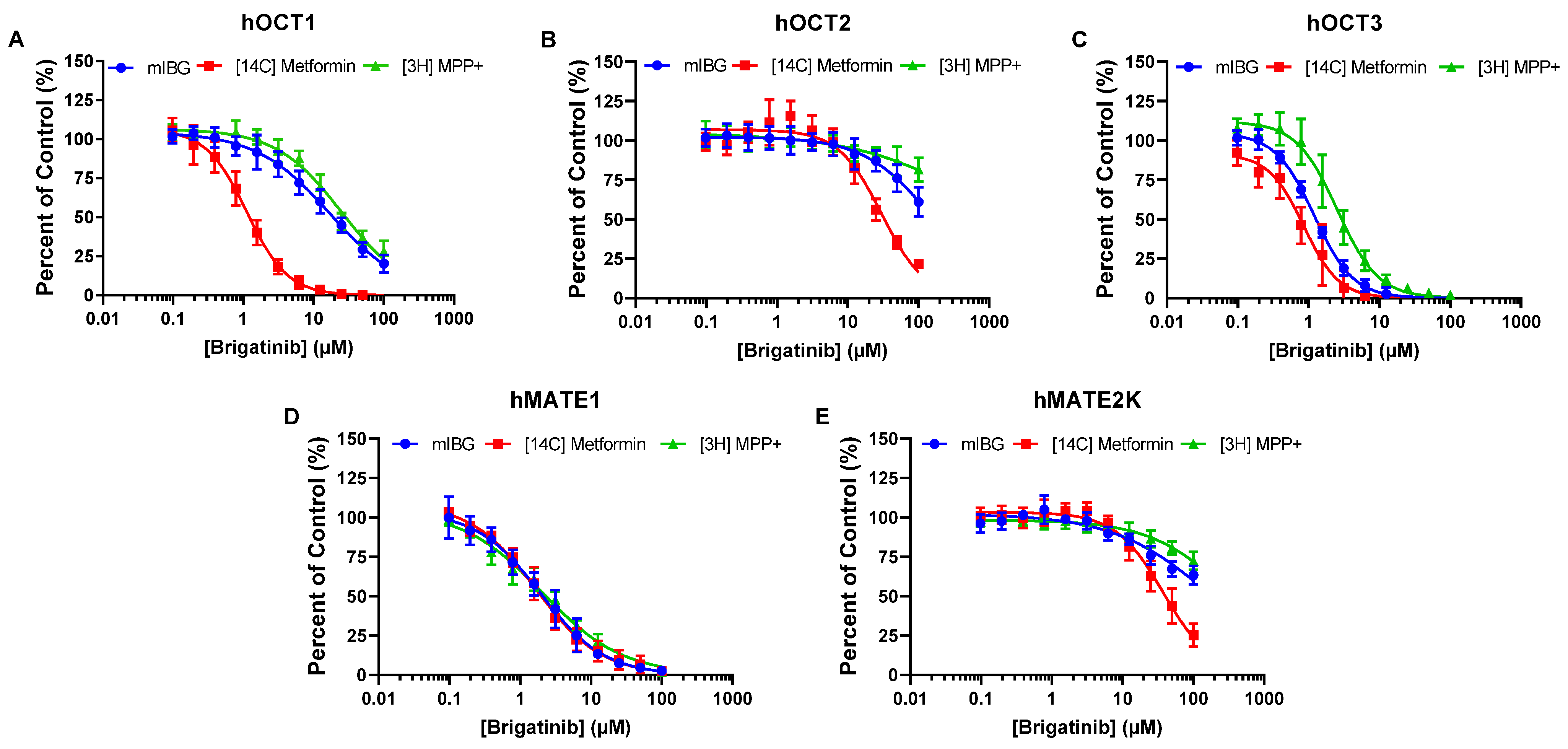
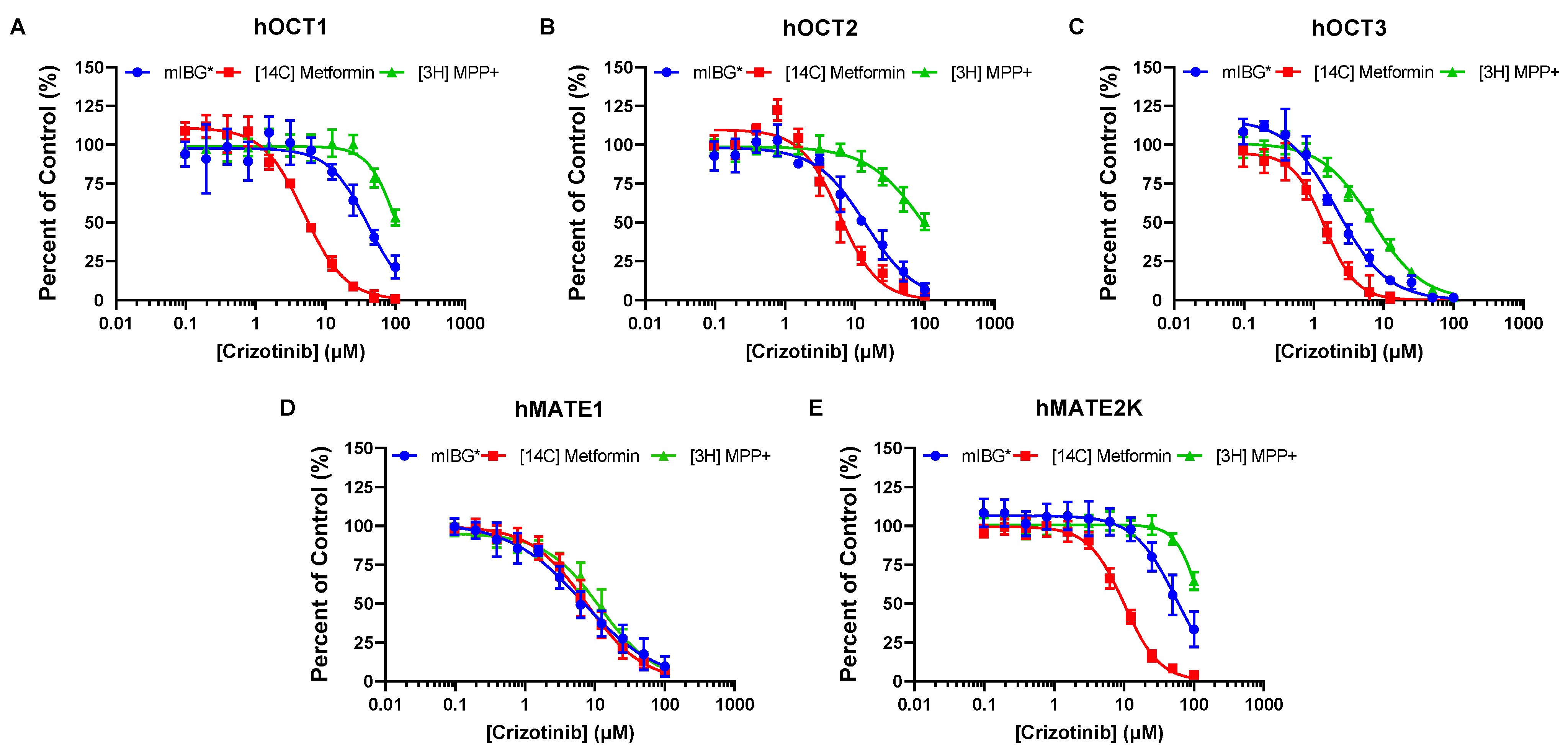
3.3. Brigatinib and Crizotinib Inhibition of hOCTs and hMATE2-K Is Highly Dependent on the Substrates Used
3.4. Impact of Substrate-Dependent Inhibition on the Prediction of DDI Potential for Brigatinib and Crizotinib
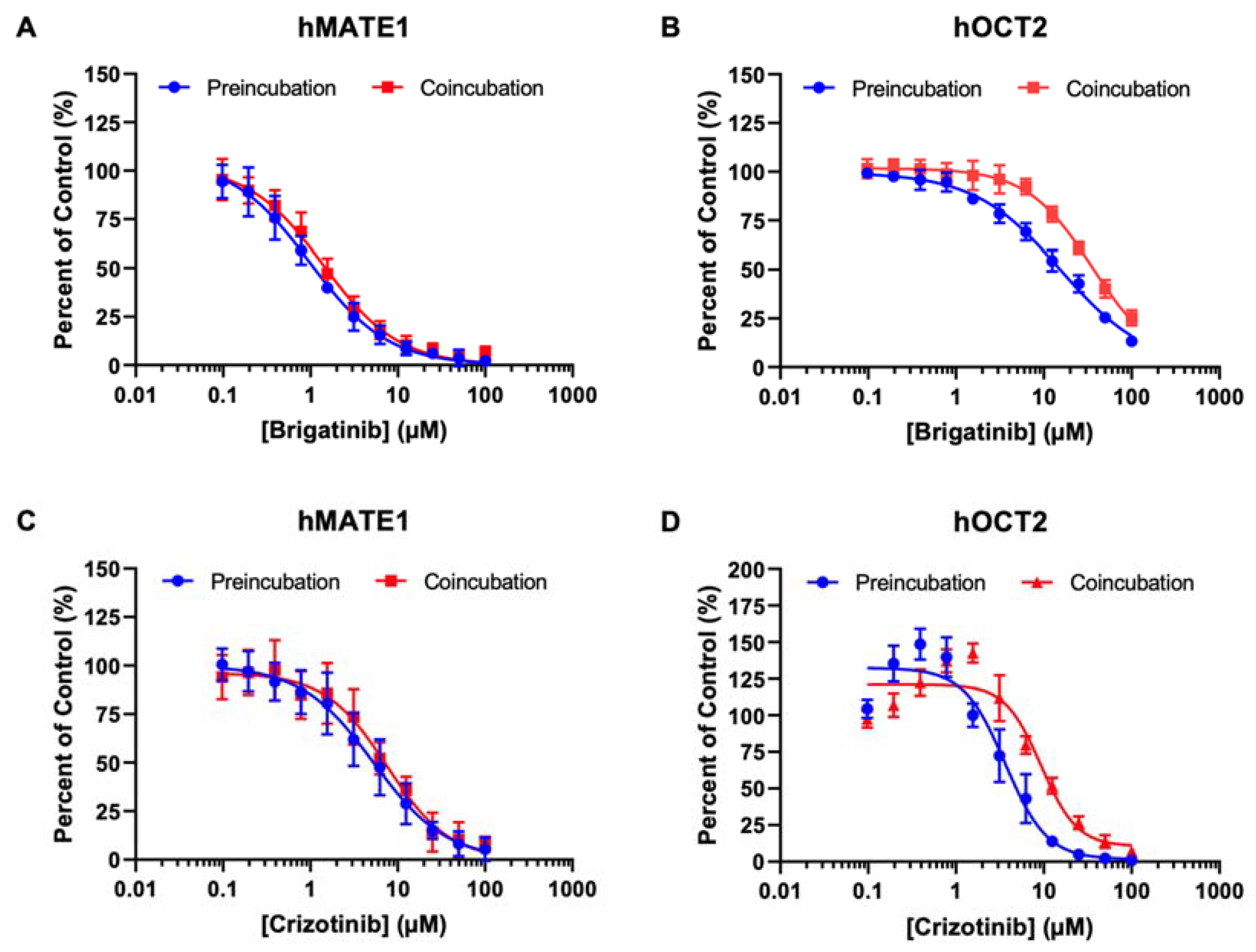
3.5. Effect of Preincubation on the Inhibitory Effects of Brigatinib and Crizotinib on hOCT2 and hMATE1
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Janoueix-Lerosey, I.; Lequin, D.; Brugières, L.; Ribeiro, A.; de Pontual, L.; Combaret, V.; Raynal, V.; Puisieux, A.; Schleiermacher, G.; Pierron, G.; et al. Somatic and Germline Activating Mutations of the ALK Kinase Receptor in Neuroblastoma. Nature 2008, 455, 967–970. [Google Scholar] [CrossRef] [PubMed]
- Bethune, G.; Bethune, D.; Ridgway, N.; Xu, Z. Epidermal Growth Factor Receptor (EGFR) in Lung Cancer: An Overview and Update. J. Thorac. Dis. 2010, 2, 48–51. [Google Scholar] [PubMed]
- Webb, T.R.; Slavish, J.; George, R.E.; Look, A.T.; Xue, L.; Jiang, Q.; Cui, X.; Rentrop, W.B.; Morris, S.W. Anaplastic Lymphoma Kinase: Role in Cancer Pathogenesis and Small-Molecule Inhibitor Development for Therapy. Expert Rev. Anticancer Ther. 2009, 9, 331–356. [Google Scholar] [CrossRef]
- Trigg, R.; Turner, S. ALK in Neuroblastoma: Biological and Therapeutic Implications. Cancers 2018, 10, 113. [Google Scholar] [CrossRef]
- Chiarle, R.; Voena, C.; Ambrogio, C.; Piva, R.; Inghirami, G. The Anaplastic Lymphoma Kinase in the Pathogenesis of Cancer. Nat. Rev. Cancer 2008, 8, 11–23. [Google Scholar] [CrossRef]
- Khan, M.; Lin, J.; Liao, G.; Tian, Y.; Liang, Y.; Li, R.; Liu, M.; Yuan, Y. ALK Inhibitors in the Treatment of ALK Positive NSCLC. Front. Oncol. 2019, 8, 557. [Google Scholar] [CrossRef] [PubMed]
- Chia, P.L.; John, T.; Dobrovic, A.; Mitchell, P. Prevalence and Natural History of ALK Positive Non-Small-Cell Lung Cancer and the Clinical Impact of Targeted Therapy with ALK Inhibitors. Clin. Epidemiol. 2014, 2014, 423–432. [Google Scholar] [CrossRef]
- Salido, M.; Pijuan, L.; Martínez-Avilés, L.; Galván, A.B.; Cañadas, I.; Rovira, A.; Zanui, M.; Martínez, A.; Longarón, R.; Sole, F.; et al. Increased ALK Gene Copy Number and Amplification Are Frequent in Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2011, 6, 21–27. [Google Scholar] [CrossRef]
- US Food and Drug Administration Crizotinib, Pharmacology Review. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/202570Orig1s000PharmR.pdf (accessed on 1 August 2023).
- Krytska, K.; Ryles, H.T.; Sano, R.; Raman, P.; Infarinato, N.R.; Hansel, T.D.; Makena, M.R.; Song, M.M.; Reynolds, C.P.; Mossé, Y.P. Crizotinib Synergizes with Chemotherapy in Preclinical Models of Neuroblastoma. Clin. Cancer Res. 2016, 22, 948–960. [Google Scholar] [CrossRef]
- US National Cancer Institute Drugs Approved for Non-Small Cell Lung Cancer. Available online: https://www.cancer.gov/about-cancer/treatment/drugs/lung (accessed on 7 February 2023).
- Goldsmith, K.C.; Park, J.R.; Kayser, K.; Malvar, J.; Chi, Y.-Y.; Groshen, S.G.; Villablanca, J.G.; Krytska, K.; Lai, L.M.; Acharya, P.T.; et al. Lorlatinib with or without Chemotherapy in ALK-Driven Refractory/Relapsed Neuroblastoma: Phase 1 Trial Results. Nat. Med. 2023, 29, 1092–1102. [Google Scholar] [CrossRef]
- Giacomini, K.M.; Huang, S.-M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.R.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; Hillgren, K.M.; et al. Membrane Transporters in Drug Development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar] [CrossRef] [PubMed]
- Wagner, D.J.; Hu, T.; Wang, J. Polyspecific Organic Cation Transporters and Their Impact on Drug Intracellular Levels and Pharmacodynamics. Pharmacol. Res. 2016, 111, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Li, L.P.; Song, F.F.; Weng, Y.Y.; Yang, X.; Wang, K.; Lei, H.M.; Ma, J.; Zhou, H.; Jiang, H.D. Role of OCT2 and MATE1 in Renal Disposition and Toxicity of Nitidine Chloride. Br. J. Pharmacol. 2016, 173, 2543–2554. [Google Scholar] [CrossRef]
- Sata, R.; Ohtani, H.; Tsujimoto, M.; Murakami, H.; Koyabu, N.; Nakamura, T.; Uchiumi, T.; Kuwano, M.; Nagata, H.; Tsukimori, K.; et al. Functional Analysis of Organic Cation Transporter 3 Expressed in Human Placenta. J. Pharmacol. Exp. Ther. 2005, 315, 888–895. [Google Scholar] [CrossRef]
- Lee, N.; Duan, H.; Hebert, M.F.; Liang, C.J.; Rice, K.M.; Wang, J. Taste of a Pill. J. Biol. Chem. 2014, 289, 27055–27064. [Google Scholar] [CrossRef]
- Koepsell, H.; Lips, K.; Volk, C. Polyspecific Organic Cation Transporters: Structure, Function, Physiological Roles, and Biopharmaceutical Implications. Pharm. Res. 2007, 24, 1227–1251. [Google Scholar] [CrossRef]
- Kekuda, R.; Prasad, P.D.; Wu, X.; Wang, H.; Fei, Y.-J.; Leibach, F.H.; Ganapathy, V. Cloning and Functional Characterization of a Potential-Sensitive, Polyspecific Organic Cation Transporter (OCT3) Most Abundantly Expressed in Placenta. J. Biol. Chem. 1998, 273, 15971–15979. [Google Scholar] [CrossRef]
- Duan, H.; Wang, J. Selective Transport of Monoamine Neurotransmitters by Human Plasma Membrane Monoamine Transporter and Organic Cation Transporter 3. J. Pharmacol. Exp. Ther. 2010, 335, 743–753. [Google Scholar] [CrossRef]
- Lee, N.; Hebert, M.F.; Wagner, D.J.; Easterling, T.R.; Liang, C.J.; Rice, K.; Wang, J. Organic Cation Transporter 3 Facilitates Fetal Exposure to Metformin during Pregnancy. Mol. Pharmacol. 2018, 94, 1125–1131. [Google Scholar] [CrossRef]
- Zhang, S.; Lovejoy, K.S.; Shima, J.E.; Lagpacan, L.L.; Shu, Y.; Lapuk, A.; Chen, Y.; Komori, T.; Gray, J.W.; Chen, X.; et al. Organic Cation Transporters Are Determinants of Oxaliplatin Cytotoxicity. Cancer Res. 2006, 66, 8847–8857. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.M.; Zavorka Thomas, M.; Magdy, T.; Eisenmann, E.D.; Uddin, M.E.; DiGiacomo, D.F.; Pan, A.; Keiser, M.; Otter, M.; Xia, S.H.; et al. Targeting OCT3 Attenuates Doxorubicin-Induced Cardiac Injury. Proc. Natl. Acad. Sci. USA 2021, 118, e2020168118. [Google Scholar] [CrossRef] [PubMed]
- Hucke, A.; Ciarimboli, G. The Role of Transporters in the Toxicity of Chemotherapeutic Drugs: Focus on Transporters for Organic Cations. J. Clin. Pharmacol. 2016, 56 (Suppl. 7), S157–S172. [Google Scholar] [CrossRef] [PubMed]
- López Quiñones, A.J.; Wagner, D.J.; Wang, J. Characterization of Meta -Iodobenzylguanidine (MIBG) Transport by Polyspecific Organic Cation Transporters: Implication for MIBG Therapy. Mol. Pharmacol. 2020, 98, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Masereeuw, R.; Russel, F.G.M. Mechanisms and Clinical Implications of Renal Drug Excretion. Drug Metab. Rev. 2001, 33, 299–351. [Google Scholar] [CrossRef]
- Morrissey, K.M.; Stocker, S.L.; Wittwer, M.B.; Xu, L.; Giacomini, K.M. Renal Transporters in Drug Development. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 503–529. [Google Scholar] [CrossRef]
- Koepsell, H. Role of Organic Cation Transporters in Drug–Drug Interaction. Expert Opin. Drug Metab. Toxicol. 2015, 11, 1619–1633. [Google Scholar] [CrossRef]
- US Food and Drug Adminstration In Vitro Drug Interaction Studies—Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions Guidance for Industry. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/in-vitro-drug-interaction-studies-cytochrome-p450-enzyme-and-transporter-mediated-drug-interactions (accessed on 7 February 2023).
- Koepsell, H. Multiple Binding Sites in Organic Cation Transporters Require Sophisticated Procedures to Identify Interactions of Novel Drugs. Biol. Chem. 2019, 400, 195–207. [Google Scholar] [CrossRef]
- Yin, J.; Duan, H.; Wang, J. Impact of Substrate-Dependent Inhibition on Renal Organic Cation Transporters HOCT2 and HMATE1/2-K-Mediated Drug Transport and Intracellular Accumulation. J. Pharmacol. Exp. Ther. 2016, 359, 401–410. [Google Scholar] [CrossRef]
- Martínez-Guerrero, L.J.; Wright, S.H. Substrate-Dependent Inhibition of Human MATE1 by Cationic Ionic Liquids. J. Pharmacol. Exp. Ther. 2013, 346, 495–503. [Google Scholar] [CrossRef]
- Martínez-Guerrero, L.J.; Morales, M.; Ekins, S.; Wright, S.H. Lack of Influence of Substrate on Ligand Interaction with the Human Multidrug and Toxin Extruder, MATE1. Mol. Pharmacol. 2016, 90, 254–264. [Google Scholar] [CrossRef]
- Uddin, M.E.; Garrison, D.A.; Kim, K.; Jin, Y.; Eisenmann, E.D.; Huang, K.M.; Gibson, A.A.; Hu, Z.; Sparreboom, A.; Hu, S. Influence of YES1 Kinase and Tyrosine Phosphorylation on the Activity of OCT1. Front. Pharmacol. 2021, 12, 644342. [Google Scholar] [CrossRef]
- Alim, K.; Moreau, A.; Bruyère, A.; Jouan, E.; Denizot, C.; Nies, A.T.; Parmentier, Y.; Fardel, O. Inhibition of Organic Cation Transporter 3 Activity by Tyrosine Kinase Inhibitors. Fundam. Clin. Pharmacol. 2021, 35, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Minematsu, T.; Giacomini, K.M. Interactions of Tyrosine Kinase Inhibitors with Organic Cation Transporters and Multidrug and Toxic Compound Extrusion Proteins. Mol. Cancer Ther. 2011, 10, 531–539. [Google Scholar] [CrossRef] [PubMed]
- Infarinato, N.R.; Park, J.H.; Krytska, K.; Ryles, H.T.; Sano, R.; Szigety, K.M.; Li, Y.; Zou, H.Y.; Lee, N.V.; Smeal, T.; et al. The ALK/ROS1 Inhibitor PF-06463922 Overcomes Primary Resistance to Crizotinib in ALK-Driven Neuroblastoma. Cancer Discov. 2016, 6, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Siaw, J.T.; Wan, H.; Pfeifer, K.; Rivera, V.M.; Guan, J.; Palmer, R.H.; Hallberg, B. Brigatinib, an Anaplastic Lymphoma Kinase Inhibitor, Abrogates Activity and Growth in ALK-Positive Neuroblastoma Cells, Drosophila and Mice. Oncotarget 2016, 7, 29011–29022. [Google Scholar] [CrossRef]
- Nalluri, S.; Peirce, S.K.; Tanos, R.; Abdella, H.A.; Karmali, D.; Hogarty, M.D.; Goldsmith, K.C. EGFR Signaling Defines Mcl−1 Survival Dependency in Neuroblastoma. Cancer Biol. Ther. 2015, 16, 276–286. [Google Scholar] [CrossRef]
- Amundsen, R.; Christensen, H.; Zabihyan, B.; Åsberg, A. Cyclosporine A, but Not Tacrolimus, Shows Relevant Inhibition of Organic Anion-Transporting Protein 1B1-Mediated Transport of Atorvastatin. Drug Metab. Dispos. 2010, 38, 1499–1504. [Google Scholar] [CrossRef]
- Gertz, M.; Cartwright, C.M.; Hobbs, M.J.; Kenworthy, K.E.; Rowland, M.; Houston, J.B.; Galetin, A. Cyclosporine Inhibition of Hepatic and Intestinal CYP3A4, Uptake and Efflux Transporters: Application of PBPK Modeling in the Assessment of Drug-Drug Interaction Potential. Pharm. Res. 2013, 30, 761–780. [Google Scholar] [CrossRef]
- Coleman, R.E.; Stubbs, J.B.; Barrett, J.A.; de la Guardia, M.; LaFrance, N.; Babich, J.W. Radiation Dosimetry, Pharmacokinetics, and Safety of UltratraceTM Iobenguane I-131 in Patients with Malignant Pheochromocytoma/Paraganglioma or Metastatic Carcinoid. Cancer Biother. Radiopharm. 2009, 24, 469–475. [Google Scholar] [CrossRef]
- Chin, B.B.; Kronauge, J.F.; Femia, F.J.; Chen, J.; Maresca, K.P.; Hillier, S.; Petry, N.A.; James, O.G.; Oldan, J.D.; Armor, T.; et al. Phase-1 Clinical Trial Results of High-Specific-Activity Carrier-Free123 I-Iobenguane. J. Nucl. Med. 2014, 55, 765–771. [Google Scholar] [CrossRef]
- Lopez Quinones, A.; Salvador-Vieira, L.; Wang, J. Organic Cation Transporter 3 Mediates Tissue Uptake and Accumulation of Meta-Iodobenzylguanidine (MIBG). FASEB J. 2021, 35, fasebj.2021.35.S1.02218. [Google Scholar] [CrossRef]
- Bayer, M.; Schmitt, J.; Dittmann, H.; Handgretinger, R.; Bruchelt, G.; Sauter, A.W. Improved Selectivity of MIBG Uptake into Neuroblastoma Cells in Vitro and in Vivo by Inhibition of Organic Cation Transporter 3 Uptake Using Clinically Approved Corticosteroids. Nucl. Med. Biol. 2016, 43, 543–551. [Google Scholar] [CrossRef] [PubMed]
- López Quiñones, A.J.; Vieira, L.S.; Wang, J. Clinical Applications and the Roles of Transporters in Disposition, Tumor Targeting, and Tissue Toxicity of meta-Iodobenzylguanidine. Drug Metab. Dispos. 2022, 50, 1218–1227. [Google Scholar] [CrossRef]
- Streby, K.A.; Shah, N.; Ranalli, M.A.; Kunkler, A.; Cripe, T.P. Nothing but NET: A Review of Norepinephrine Transporter Expression and Efficacy of 131 I-mIBG Therapy. Pediatr. Blood Cancer 2015, 62, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Hemminki, K.; Li, X.; Sundquist, J.; Sundquist, K. Risk of Cancer Following Hospitalization for Type 2 Diabetes. Oncologist 2010, 15, 548–555. [Google Scholar] [CrossRef]
- Chen, H.; Yao, W.; Chu, Q.; Han, R.; Wang, Y.; Sun, J.; Wang, D.; Wang, Y.; Cao, M.; He, Y. Synergistic Effects of Metformin in Combination with EGFR-TKI in the Treatment of Patients with Advanced Non-Small Cell Lung Cancer and Type 2 Diabetes. Cancer Lett. 2015, 369, 97–102. [Google Scholar] [CrossRef]
- Gong, L.; Goswami, S.; Giacomini, K.M.; Altman, R.B.; Klein, T.E. Metformin Pathways. Pharmacogenet. Genom. 2012, 22, 820–827. [Google Scholar] [CrossRef]
- Hundal, R.S.; Krssak, M.; Dufour, S.; Laurent, D.; Lebon, V.; Chandramouli, V.; Inzucchi, S.E.; Schumann, W.C.; Petersen, K.F.; Landau, B.R.; et al. Mechanism by Which Metformin Reduces Glucose Production in Type 2 Diabetes. Diabetes 2000, 49, 2063–2069. [Google Scholar] [CrossRef]
- Gorbunov, D.; Gorboulev, V.; Shatskaya, N.; Mueller, T.; Bamberg, E.; Friedrich, T.; Koepsell, H. High-Affinity Cation Binding to Organic Cation Transporter 1 Induces Movement of Helix 11 and Blocks Transport after Mutations in a Modeled Interaction Domain between Two Helices. Mol. Pharmacol. 2008, 73, 50–61. [Google Scholar] [CrossRef]
- Minuesa, G.; Volk, C.; Molina-Arcas, M.; Gorboulev, V.; Erkizia, I.; Arndt, P.; Clotet, B.; Pastor-Anglada, M.; Koepsell, H.; Martinez-Picado, J. Transport of Lamivudine [(-)-β-l-2′,3′-Dideoxy-3′-Thiacytidine] and High-Affinity Interaction of Nucleoside Reverse Transcriptase Inhibitors with Human Organic Cation Transporters 1, 2, and 3. J. Pharmacol. Exp. Ther. 2009, 329, 252–261. [Google Scholar] [CrossRef]
- Tátrai, P.; Schweigler, P.; Poller, B.; Domange, N.; de Wilde, R.; Hanna, I.; Gáborik, Z.; Huth, F. A Systematic In Vitro Investigation of the Inhibitor Preincubation Effect on Multiple Classes of Clinically Relevant Transporters. Drug Metab. Dispos. 2019, 47, 768–778. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 (µM) | ||||||
|---|---|---|---|---|---|---|
| Brigatinib | Crizotinib | |||||
| Metformin | mIBG | MPP+ | Metformin | mIBG | MPP+ | |
| hOCT1 | 1.12 ± 0.04 | 17.1 ± 0.6 | 25.1 ± 1.2 | 4.98 ± 0.15 | 40.3 ± 5.5 1 | 105 ± 3 |
| hOCT2 | 30.2 ± 1.5 | 157 ± 11 | >300 | 6.07 ± 0.34 | 14.3 ± 1.7 1 | 98.1 ± 4.8 |
| hOCT3 | 0.85 ± 0.07 | 1.20 ± 0.03 | 2.54 ± 0.14 | 1.43 ± 0.11 | 2.14 ± 0.25 1 | 6.86 ± 0.20 |
| hMATE1 | 1.67 ± 0.14 | 1.93 ± 0.15 | 2.03 ± 0.24 | 7.60 ± 0.26 | 6.73 ± 0.39 | 11.8 ± 0.4 |
| hMATE2-K | 38.3 ± 1.7 | 172 ± 17 | >300 | 10.1 ± 0.3 | 55.4 ± 2.3 | 125 ± 4 |
| Metformin | mIBG | MPP+ | |
|---|---|---|---|
| hOCT1 | |||
| IC50 (µM) | 1.12 ± 0.06 | 17.1 ± 1.8 | 25.1 ± 1.4 |
| Brigatinib unbound Cmax/IC50 1 | 0.75 | 0.05 | 0.03 |
| In vivo DDI study recommendation | Yes | No | No |
| hOCT2 | |||
| IC50 (µM) | 30.2 ± 1.8 | 157 ± 21 | >300 |
| Brigatinib unbound Cmax/IC50 1 | 0.03 | 0.005 | <0.002 |
| In vivo DDI study recommendation | No | No | No |
| hOCT3 | |||
| IC50 (µM) | 0.85 ± 0.08 | 1.20 ± 0.04 | 2.54 ± 0.15 |
| Brigatinib unbound Cmax/IC50 1 | 0.99 | 0.70 | 0.33 |
| In vivo DDI study recommendation | Yes | Yes | Yes |
| hMATE1 | |||
| IC50 (µM) | 1.67 ± 0.15 | 1.93 ± 0.21 | 2.03 ± 0.25 |
| Brigatinib unbound Cmax/IC50 1 | 0.51 | 0.44 | 0.42 |
| In vivo DDI study recommendation | Yes | Yes | Yes |
| hMATE2-K | |||
| IC50 (µM) | 38.3 ± 2.0 | 172 ± 23 | >300 |
| Brigatinib unbound Cmax/IC50 1 | 0.02 | 0.004 | <0.002 |
| In vivo DDI study recommendation | No | No | No |
| Metformin | mIBG | MPP+ | |
|---|---|---|---|
| hOCT1 | |||
| IC50 (µM) | 4.98 ± 0.20 | 40.3 ± 5.5 2 | 105 ± 5 |
| Crizotinib unbound Cmax/IC50 1 | 0.02 | 0.002 | <0.002 |
| In vivo DDI study recommendation | No | No | No |
| hOCT2 | |||
| IC50 (µM) | 6.07 ± 0.36 | 14.3 ± 1.7 2 | 98.1 ± 6.4 |
| Crizotinib unbound Cmax/IC50 1 | 0.02 | 0.007 | <0.002 |
| In vivo DDI study recommendation | No | No | No |
| hOCT3 | |||
| IC50 (µM) | 1.43 ± 0.11 | 2.14 ± 0.25 2 | 6.86 ± 0.27 |
| Crizotinib unbound Cmax/IC50 1 | 0.07 | 0.05 | 0.01 |
| In vivo DDI study recommendation | No | No | No |
| hMATE1 | |||
| IC50 (µM) | 7.60 ± 0.46 | 6.73 ± 0.68 | 11.8 ± 0.7 |
| Crizotinib unbound Cmax/IC50 1 | 0.01 | 0.01 | 0.01 |
| In vivo DDI study recommendation | No | No | No |
| hMATE2-K | |||
| IC50 (µM) | 10.1 ± 0.3 | 55.4 ± 3.2 | 125 ± 6 |
| Crizotinib unbound Cmax/IC50 1 | 0.01 | 0.002 | <0.002 |
| In vivo DDI study recommendation | No | No | No |
| IC50 (µM) | ||||||
|---|---|---|---|---|---|---|
| Brigatinib | Crizotinib | |||||
| Preincubation | Coincubation | Fold Difference | Preincubation | Coincubation | Fold Difference | |
| hOCT2 | 15.5 ± 0.5 | 36.0 ± 1.1 | 2.3 | 3.64 ± 0.29 | 9.30 ± 0.77 | 2.6 |
| hMATE1 | 0.97 ± 0.09 | 1.41 ± 0.06 | 1.4 | 5.28 ± 0.48 | 7.57 ± 0.59 | 1.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsang, Y.P.; López Quiñones, A.J.; Vieira, L.S.; Wang, J. Interaction of ALK Inhibitors with Polyspecific Organic Cation Transporters and the Impact of Substrate-Dependent Inhibition on the Prediction of Drug–Drug Interactions. Pharmaceutics 2023, 15, 2312. https://doi.org/10.3390/pharmaceutics15092312
Tsang YP, López Quiñones AJ, Vieira LS, Wang J. Interaction of ALK Inhibitors with Polyspecific Organic Cation Transporters and the Impact of Substrate-Dependent Inhibition on the Prediction of Drug–Drug Interactions. Pharmaceutics. 2023; 15(9):2312. https://doi.org/10.3390/pharmaceutics15092312
Chicago/Turabian StyleTsang, Yik Pui, Antonio Jesús López Quiñones, Letícia Salvador Vieira, and Joanne Wang. 2023. "Interaction of ALK Inhibitors with Polyspecific Organic Cation Transporters and the Impact of Substrate-Dependent Inhibition on the Prediction of Drug–Drug Interactions" Pharmaceutics 15, no. 9: 2312. https://doi.org/10.3390/pharmaceutics15092312