
Nanostructured Microparticles Repolarize Macrophages and Induce Cell Death in an In Vitro Model of Tumour-Associated Macrophages
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.2.1. THP-1
2.2.2. Primary Peripheral Blood Human Monocyte-Derived Macrophages (HMDMs)
2.2.3. Reporter Cell Lines
2.2.4. Primary Murine Bone Marrow-Derived Macrophages (BMMs)
2.3. Particle Synthesis and Characterization
2.3.1. Synthesis of Aspherical Cylindrical Silica Microparticles (µRs)
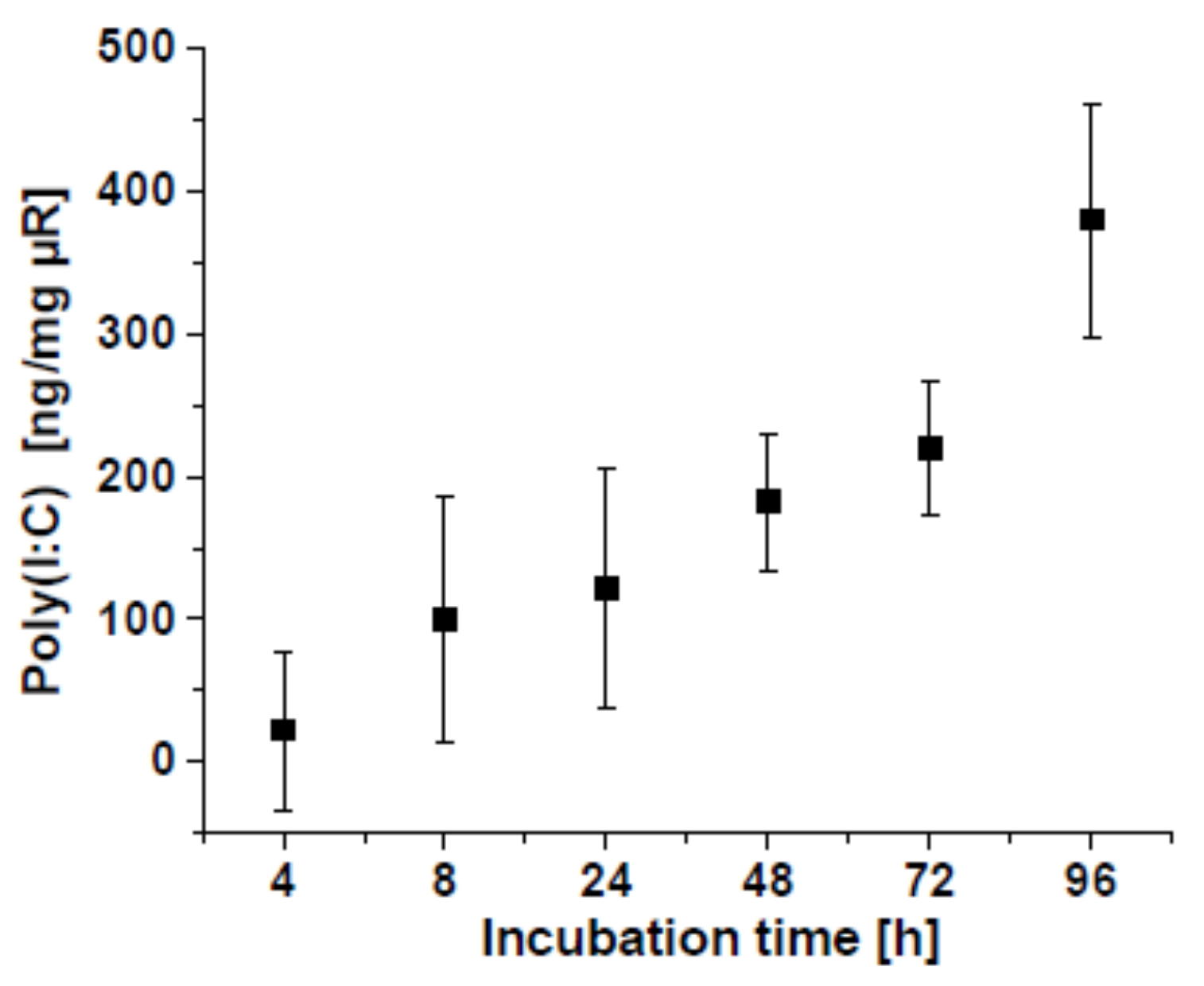
2.3.2. Poly(I:C)-Release from µRs
2.4. Analysis of Cellular µR Uptake
2.4.1. Flow Cytometry
2.4.2. Live-Cell Microscopy-Based Analysis
2.4.3. Confocal Laser Scanning Microscopy (CLSM)
2.5. Surface Protein Expression Analysis Employing Flow Cytometry
2.6. RNA Isolation, Reverse Transcription, and Quantitative RT-PCR (qPCR)
2.7. Endotoxin Assay
2.8. Reporter Cell Assay (HEK-Dual™ hTLR2, HEK-Blue™-hTLR2, HEK-Blue™ IL-1R and THP1-XBlue™)
2.9. Determination of Cell Viability
2.9.1. MTT Assay
2.9.2. Live-Cell Microscopy-Based Analysis
2.10. Statistics
3. Results
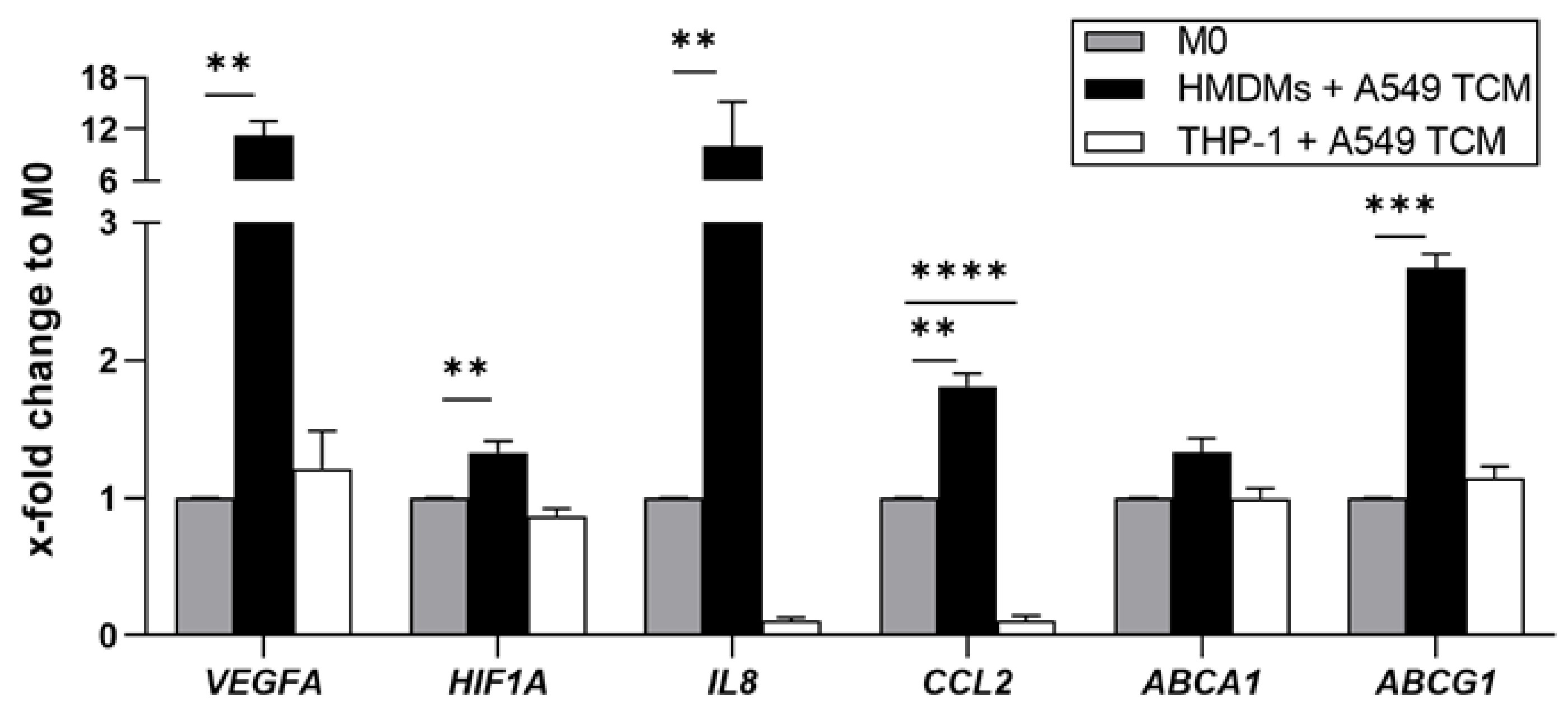
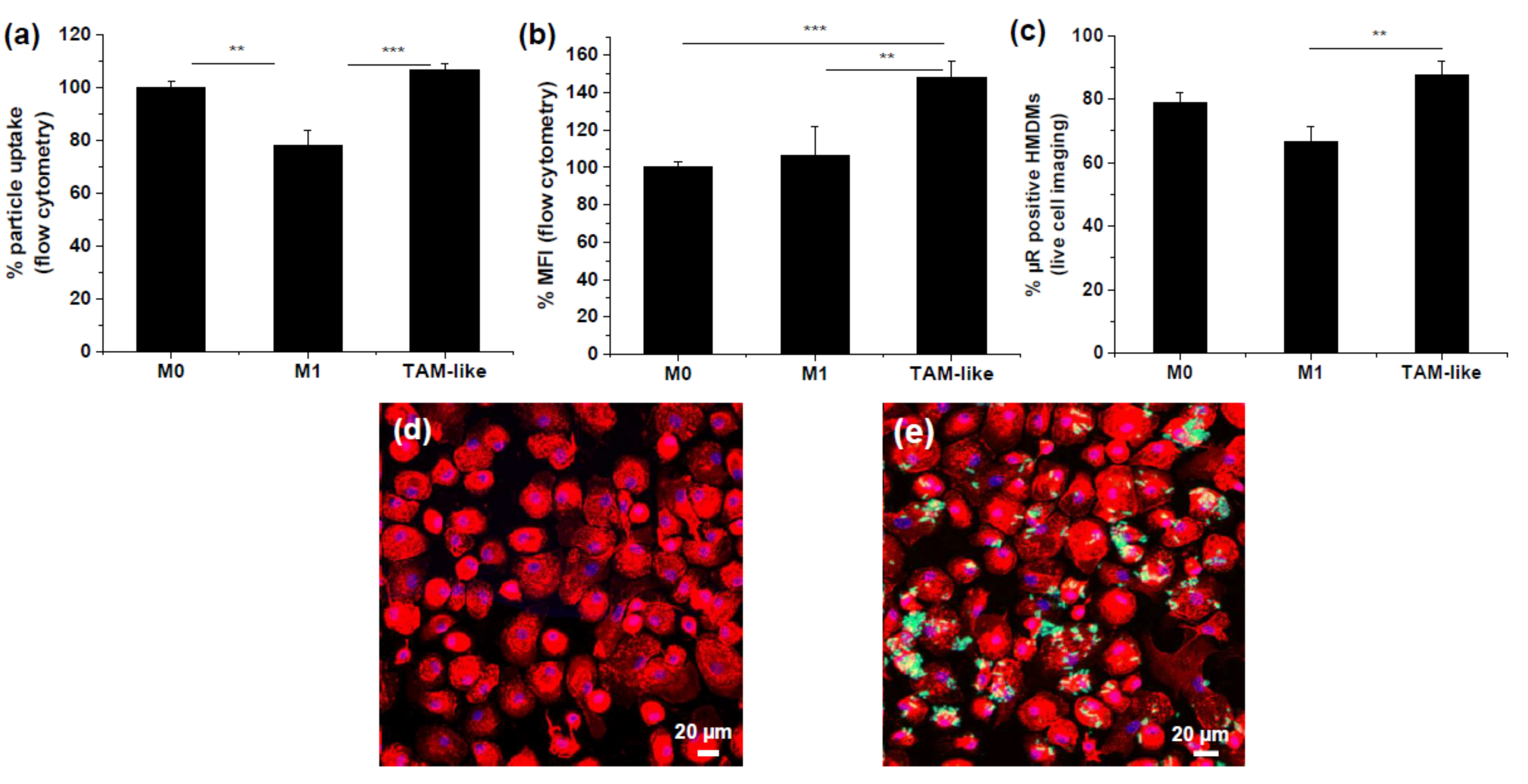
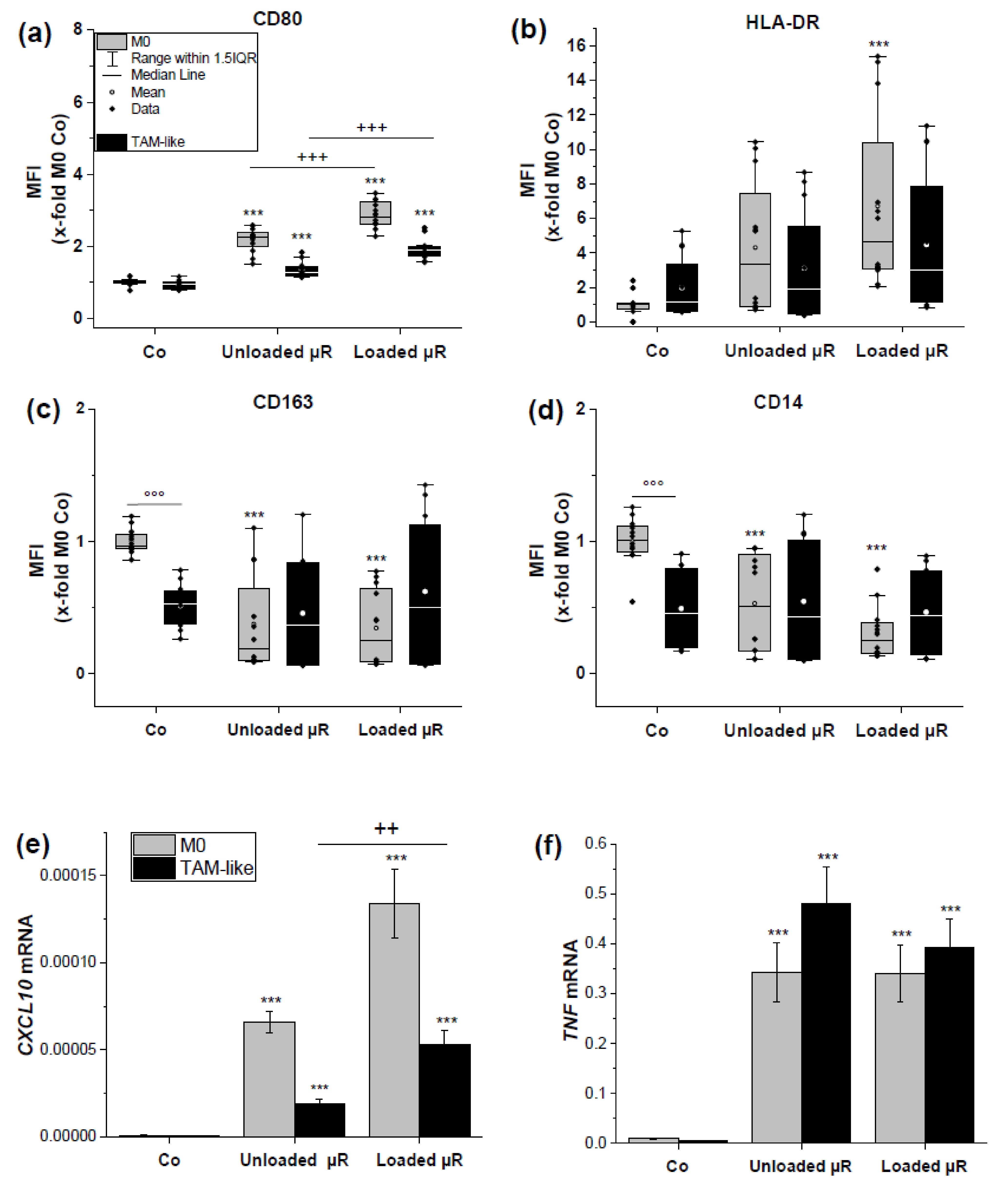
3.1. Uptake of Rod-Shaped Microparticles by TAMs
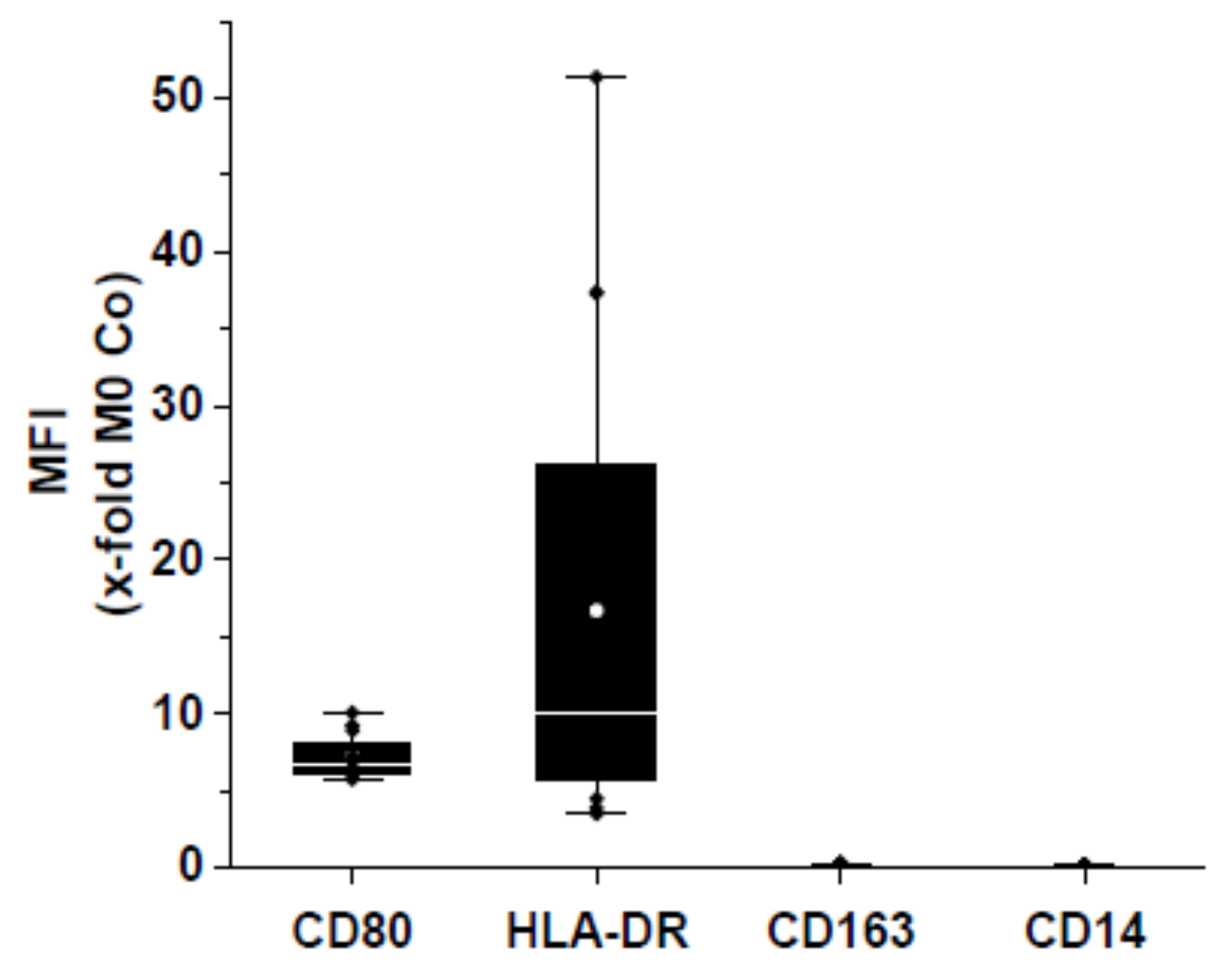
3.2. Loaded and Unloaded µRs Lead to an M1-Like Phenotype
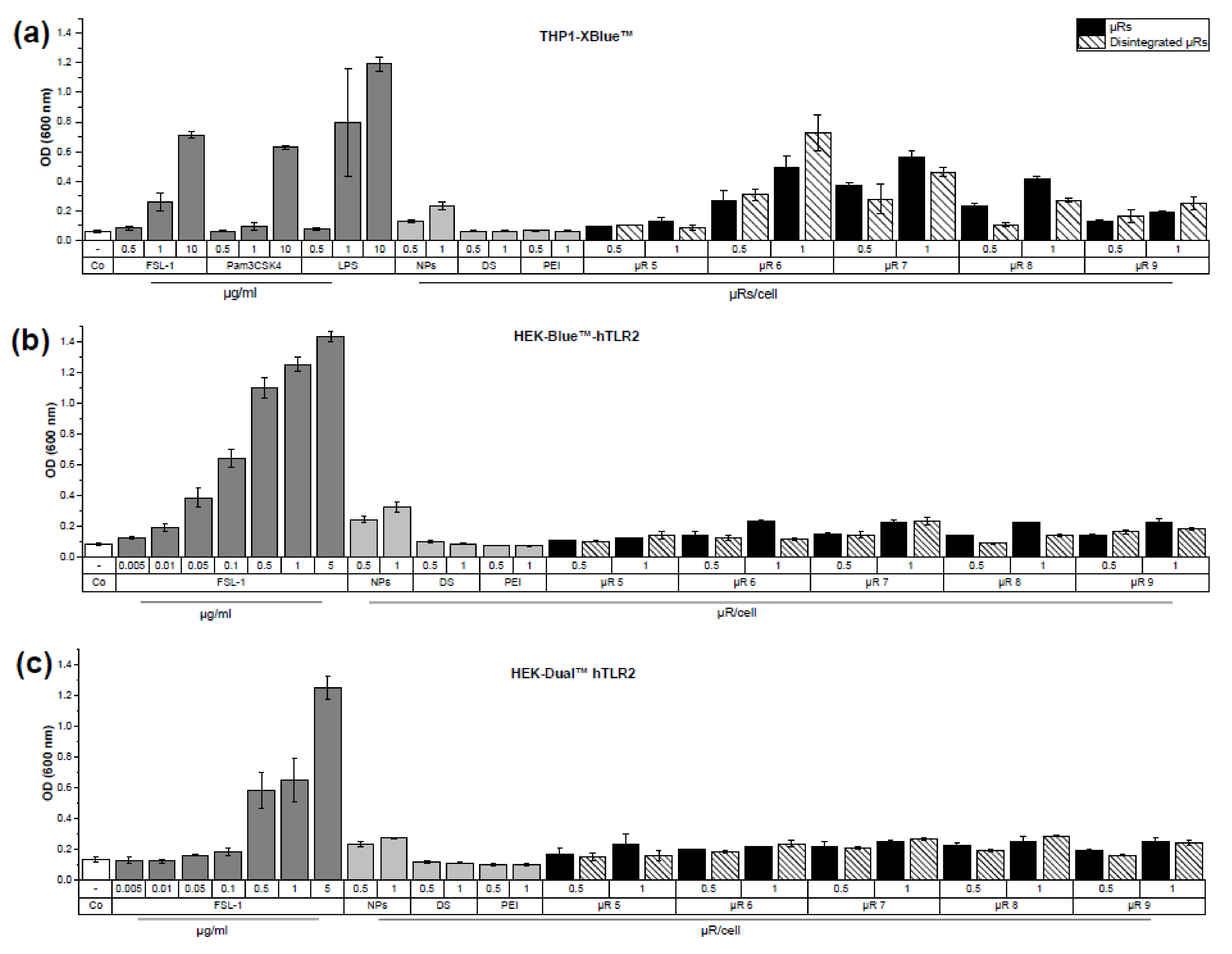
3.3. Evaluation of µRs’ Inflammatory Potential
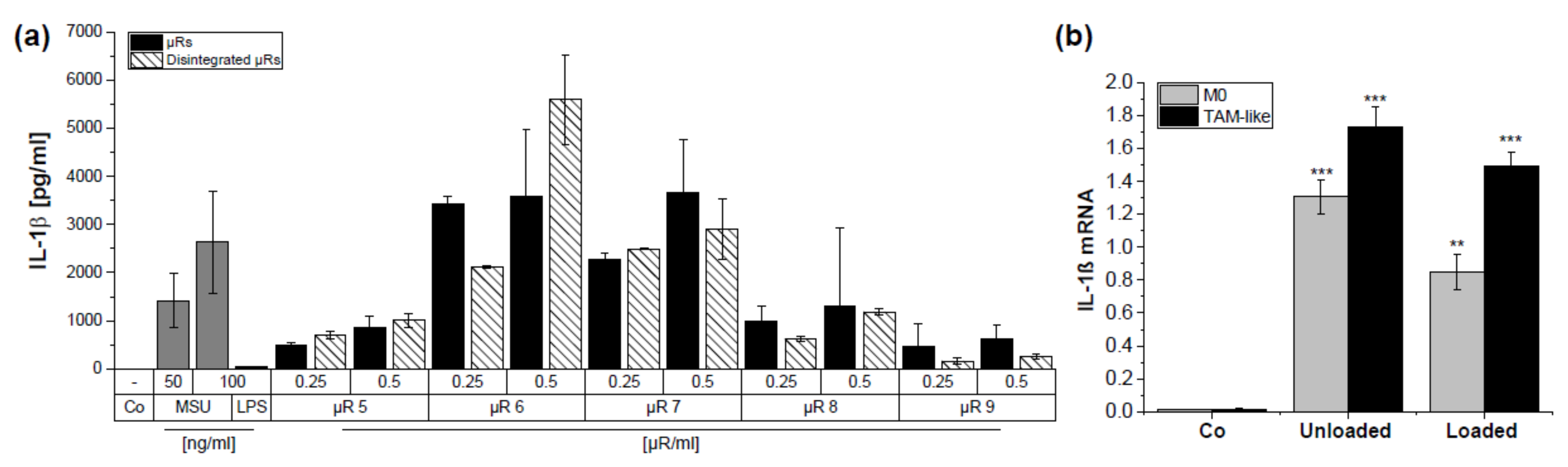
3.4. µRs Promote IL-1β Secretion in HMDMs
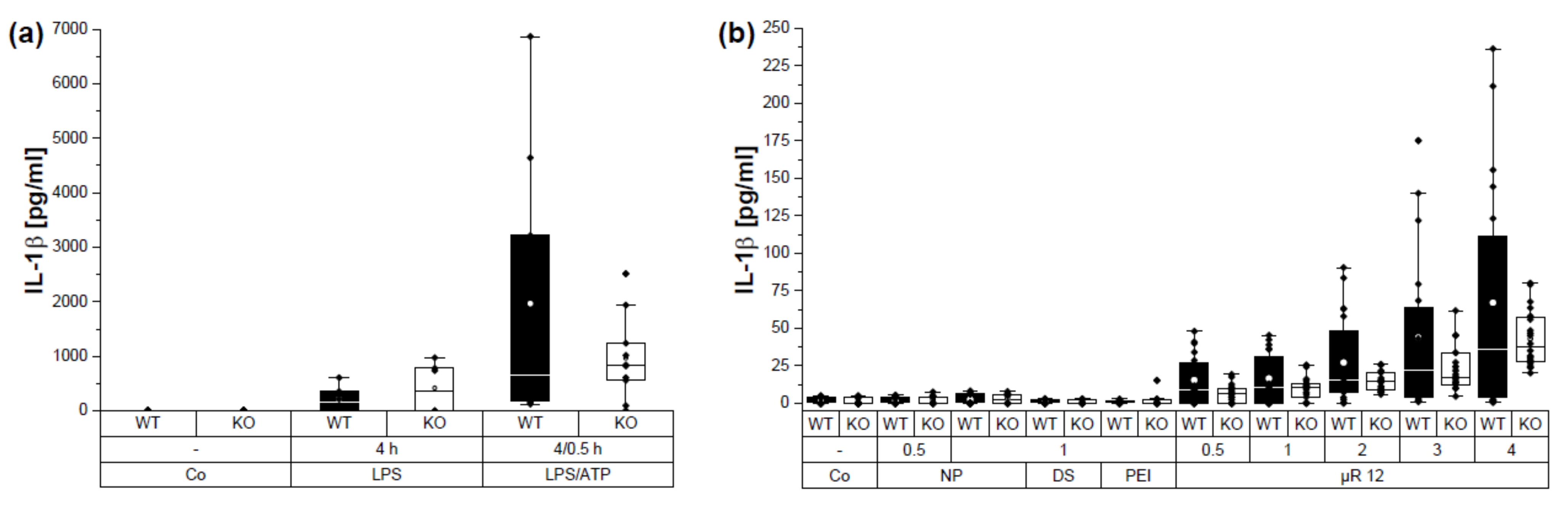
3.5. µRs Induced IL-1β Secretion Is NLRP3 Dependent
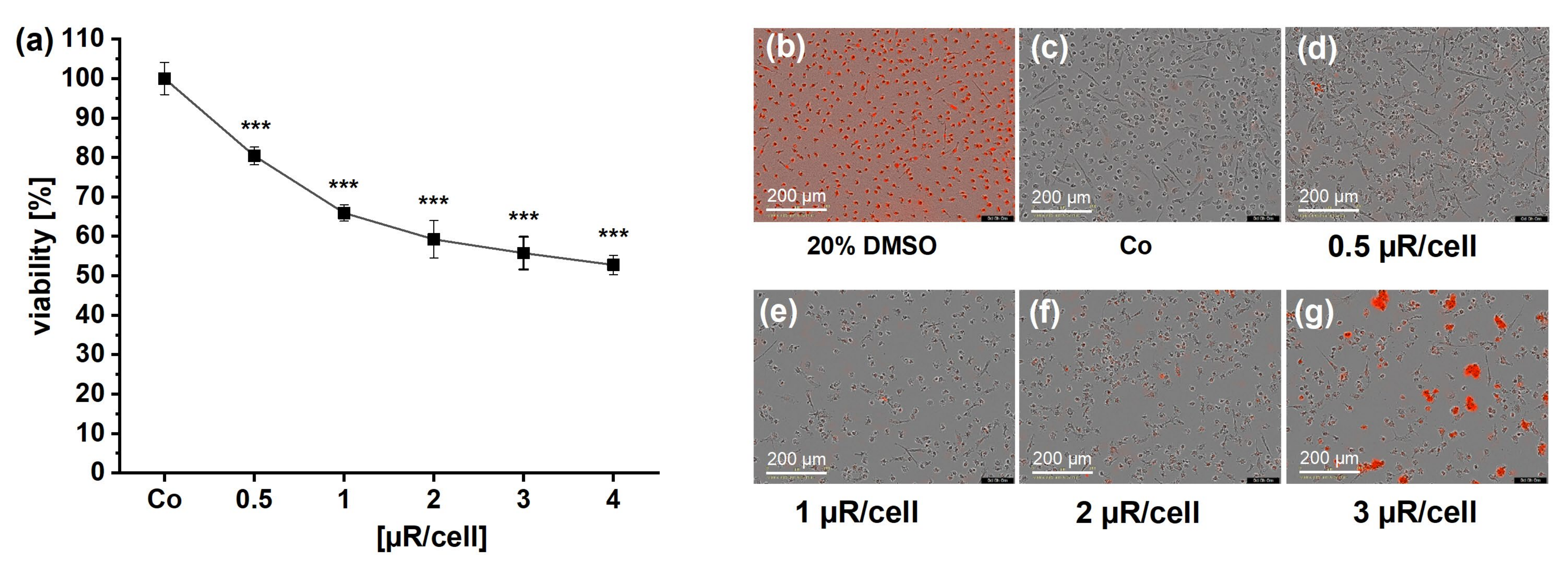
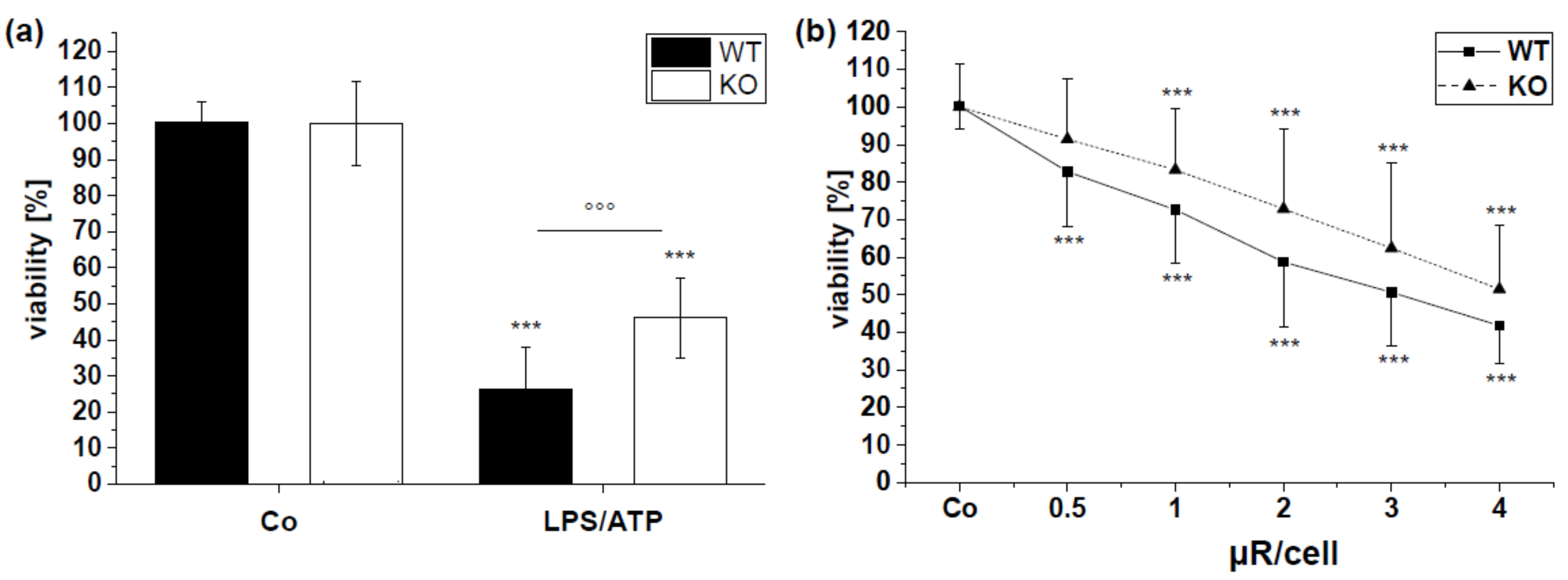
3.6. Cytotoxicity Is Involved in µR Effect on Viability
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, L.; Zhang, Y. Tumor-associated macrophages: From basic research to clinical application. J. Hematol. Oncol. 2017, 10, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedighzadeh, S.S.; Khoshbin, A.P.; Razi, S.; Keshavarz-Fathi, M.; Rezaei, N. A narrative review of tumor-associated macrophages in lung cancer: Regulation of macrophage polarization and therapeutic implications. Transl. Lung Cancer Res. 2021, 10, 1889–1916. [Google Scholar] [CrossRef] [PubMed]
- Dahlem, C.; Abuhaliema, A.; Kessler, S.M.; Kröhler, T.; Zoller, B.G.E.; Chanda, S.; Wu, Y.; Both, S.; Müller, F.; Lepikhov, K.; et al. First Small-Molecule Inhibitors Targeting the RNA-Binding Protein IGF2BP2/IMP2 for Cancer Therapy. ACS Chem. Biol. 2022, 17, 361–375. [Google Scholar] [CrossRef]
- Mantovani, A.; Sozzani, S.; Locati, M.; Allavena, P.; Sica, A. Macrophage polarization: Tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002, 23, 549–555. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Marchesi, F.; Garlanda, C. Macrophages as tools and targets in cancer therapy. Nat. Rev. Drug Discov. 2022, 21, 799–820. [Google Scholar] [CrossRef]
- Ostuni, R.; Kratochvill, F.; Murray, P.J.; Natoli, G. Macrophages and cancer: From mechanisms to therapeutic implications. Trends Immunol. 2015, 36, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Zheng, X.; Weigert, A.; Reu, S.; Guenther, S.; Mansouri, S.; Bassaly, B.; Gattenlöhner, S.; Grimminger, F.; Pullamsetti, S.; Seeger, W.; et al. Spatial Density and Distribution of Tumor-Associated Macrophages Predict Survival in Non-Small Cell Lung Carcinoma. Cancer Res. 2020, 80, 4414–4425. [Google Scholar] [CrossRef]
- Anfray, C.; Ummarino, A.; Andón, F.T.; Allavena, P. Current Strategies to Target Tumor-Associated-Macrophages to Improve Anti-Tumor Immune Responses. Cells 2019, 9, 46. [Google Scholar] [CrossRef] [Green Version]
- Hoppstädter, J.; Dembek, A.; Höring, M.; Schymik, H.S.; Dahlem, C.; Sultan, A.; Wirth, N.; Al-Fityan, S.; Diesel, B.; Gasparoni, G.; et al. Dysregulation of cholesterol homeostasis in human lung cancer tissue and tumour-associated macrophages. EBioMedicine 2021, 72, 103578. [Google Scholar] [CrossRef]
- Vidyarthi, A.; Khan, N.; Agnihotri, T.; Negi, S.; Das, D.K.; Aqdas, M.; Chatterjee, D.; Colegio, O.R.; Tewari, M.K.; Agrewala, J.N. TLR-3 Stimulation Skews M2 Macrophages to M1 through IFN-αβ Signaling and Restricts Tumor Progression. Front. Immunol. 2018, 9, 1650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, K.H.; Cha, S.B.; Lee, S.H.; Bae, H.S.; Ham, C.S.; Lee, M.G.; Kim, D.H.; Han, S.H. A novel defined TLR3 agonist as an effective vaccine adjuvant. Front. Immunol. 2023, 14, 1075291. [Google Scholar] [CrossRef] [PubMed]
- Hoppstädter, J.; Diesel, B.; Linnenberger, R.; Hachenthal, N.; Flamini, S.; Minet, M.; Leidinger, P.; Backes, C.; Grässer, F.; Meese, E.; et al. Amplified Host Defense by Toll-Like Receptor-Mediated Downregulation of the Glucocorticoid-Induced Leucine Zipper (GILZ) in Macrophages. Front. Immunol. 2019, 9, 3111. [Google Scholar] [CrossRef] [PubMed]
- Möhwald, M.; Pinnapireddy, S.R.; Wonnenberg, B.; Pourasghar, M.; Jurisic, M.; Jung, A.; Fink-Straube, C.; Tschernig, T.; Bakowsky, U.; Schneider, M. Aspherical, Nanostructured Microparticles for Targeted Gene Delivery to Alveolar Macrophages. Adv. Healthc. Mater. 2017, 6, 1700478. [Google Scholar] [CrossRef]
- Fischer, T.; Tschernig, T.; Drews, F.; Brix, K.; Meier, C.; Simon, M.; Kautenburger, R.; Schneider, M. siRNA delivery to macrophages using aspherical, nanostructured microparticles as delivery system for pulmonary administration. Eur. J. Pharm. Biopharm. 2021, 158, 284–293. [Google Scholar] [CrossRef]
- Tschernig, T.; Fischer, T.; Grissmer, A.; Beckmann, A.; Meier, C.; Lipp, P.; Schneider, M. Silica nanoparticles of microrods enter lung epithelial cells. Biomed. Rep. 2018, 9, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, X.; Wang, J.; Wang, J.; Zou, H.; Hu, X.; Yang, L.; Shen, P.; Wang, K.; Li, Y.; et al. Alveolar Macrophages-Mediated Translocation of Intratracheally Delivered Perfluorocarbon Nanoparticles to Achieve Lung Cancer 19F-MR Imaging. Nano Lett. 2023, 23, 2964–2973. [Google Scholar] [CrossRef] [PubMed]
- Hoppstädter, J.; Seif, M.; Dembek, A.; Cavelius, C.; Huwer, H.; Kraegeloh, A.; Kiemer, A.K. M2 polarization enhances silica nanoparticle uptake by macrophages. Front. Pharmacol. 2015, 6, 55. [Google Scholar] [CrossRef] [Green Version]
- Dembek, A.; Laggai, S.; Kessler, S.M.; Czepukojc, B.; Simon, Y.; Kiemer, A.K.; Hoppstädter, J. Hepatic interleukin-6 production is maintained during endotoxin tolerance and facilitates lipid accumulation. Immunobiology 2017, 222, 786–796. [Google Scholar] [CrossRef] [PubMed]
- Hoppstädter, J.; Kessler, S.M.; Bruscoli, S.; Huwer, H.; Riccardi, C.; Kiemer, A.K. Glucocorticoid-Induced Leucine Zipper: A Critical Factor in Macrophage Endotoxin Tolerance. J. Immunol. 2015, 194, 6057–6067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohler, D.; Schneider, M.; Krüger, M.; Lehr, C.M.; Möhwald, H.; Wang, D. Template-assisted polyelectrolyte encapsulation of nanoparticles into dispersible, hierarchically nanostructured microfibers. Adv. Mater. 2011, 23, 1376–1379. [Google Scholar] [CrossRef] [PubMed]
- Lamichhane, S.; Anderson, J.; Remund, T.; Kelly, P.; Mani, G. Dextran sulfate as a drug delivery platform for drug-coated balloons: Preparation, characterization, in vitro drug elution, and smooth muscle cell response. J. Biomed. Mater. Res. Part B Appl. Biomater. 2016, 104, 1416–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, D.; Huang, C.; Lin, Z.; Zhan, S.; Kong, L.; Fang, C.; Li, J. Macrophage polarization and function with emphasis on the evolving roles of coordinated regulation of cellular signaling pathways. Cell. Signal. 2014, 26, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Decher, G. Fuzzy Nanoassemblies: Toward Layered Polymeric Multicomposites. Science 1997, 277, 1232–1237. [Google Scholar] [CrossRef]
- Fischer, T.; Winter, I.; Drumm, R.; Schneider, M. Cylindrical Microparticles Composed of Mesoporous Silica Nanoparticles for the Targeted Delivery of a Small Molecule and a Macromolecular Drug to the Lungs: Exemplified with Curcumin and siRNA. Pharmaceutics 2021, 13, 844. [Google Scholar] [CrossRef] [PubMed]
- Lababidi, N.; Kissi, E.O.; Walid, A.M.E.; Sigal, V.; Haupenthal, J.; Schwarz, B.C.; Hirsch, A.K.H.; Rades, T.; Schneider, M. Spray-drying of inhalable, multifunctional formulations for the treatment of biofilms formed in cystic fibrosis. J. Control. Release 2019, 314, 62–71. [Google Scholar] [CrossRef]
- Torge, A.; Pavone, G.; Jurisic, M.; Lima-Engelmann, K.; Schneider, M. A comparison of spherical and cylindrical microparticles composed of nanoparticles for pulmonary application. Aerosol Sci. Technol. 2019, 53, 53–62. [Google Scholar] [CrossRef]
- Stefaniak, A.B.; Guilmette, R.A.; Day, G.A.; Hoover, M.D.; Breysse, P.N.; Scripsick, R.C. Characterization of phagolysosomal simulant fluid for study of beryllium aerosol particle dissolution. Toxicol. In Vitro 2005, 19, 123–134. [Google Scholar] [CrossRef]
- Linnenberger, R.; Hoppstädter, J.; Wrublewsky, S.; Ampofo, E.; Kiemer, A.K. Statins and Bempedoic Acid: Different Actions of Cholesterol Inhibitors on Macrophage Activation. Int. J. Mol. Sci. 2021, 22, 12480. [Google Scholar] [CrossRef]
- Hoppstädter, J.; Diesel, B.; Zarbock, R.; Breinig, T.; Monz, D.; Koch, M.; Meyerhans, A.; Gortner, L.; Lehr, C.M.; Huwer, H.; et al. Differential cell reaction upon Toll-like receptor 4 and 9 activation in human alveolar and lung interstitial macrophages. Respir. Res. 2010, 11, 124. [Google Scholar] [CrossRef] [Green Version]
- Astanina, K.; Simon, Y.; Cavelius, C.; Petry, S.; Kraegeloh, A.; Kiemer, A.K. Superparamagnetic iron oxide nanoparticles impair endothelial integrity and inhibit nitric oxide production. Acta Biomater. 2014, 10, 4896–4911. [Google Scholar] [CrossRef] [PubMed]
- Dahlem, C.; Siow, W.X.; Lopatniuk, M.; Tse, W.K.F.; Kessler, S.M.; Kirsch, S.H.; Hoppstädter, J.; Vollmar, A.M.; Müller, R.; Luzhetskyy, A.; et al. Thioholgamide A, a New Anti-Proliferative Anti-Tumor Agent, Modulates Macrophage Polarization and Metabolism. Cancers 2020, 12, 1288. [Google Scholar] [CrossRef]
- Hoppstädter, J.; Dembek, A.; Linnenberger, R.; Dahlem, C.; Barghash, A.; Fecher-Trost, C.; Fuhrmann, G.; Koch, M.; Kraegeloh, A.; Huwer, H.; et al. Toll-Like Receptor 2 Release by Macrophages: An Anti-inflammatory Program Induced by Glucocorticoids and Lipopolysaccharide. Front. Immunol. 2019, 10, 1634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tedesco, S.; De Majo, F.; Kim, J.; Trenti, A.; Trevisi, L.; Fadini, G.P.; Bolego, C.; Zandstra, P.W.; Cignarella, A.; Vitiello, L. Convenience versus Biological Significance: Are PMA-Differentiated THP-1 Cells a Reliable Substitute for Blood-Derived Macrophages When Studying In Vitro Polarization? Front. Pharmacol. 2018, 9, 71. [Google Scholar] [CrossRef] [Green Version]
- Kucki, M.; Cavelius, C.; Kraegeloh, A. Interference of silica nanoparticles with the traditional Limulus amebocyte lysate gel clot assay. J. Innate Immun. 2014, 20, 327–336. [Google Scholar] [CrossRef]
- Li, Y.; Boraschi, D. Endotoxin contamination: A key element in the interpretation of nanosafety studies. Nanomedicine 2016, 11, 269–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Fujita, M.; Boraschi, D. Endotoxin Contamination in Nanomaterials Leads to the Misinterpretation of Immunosafety Results. Front. Immunol. 2017, 8, 472. [Google Scholar] [CrossRef]
- Himly, M.; Geppert, M.; Hofer, S.; Hofstätter, N.; Horejs-Höck, J.; Duschl, A. When Would Immunologists Consider a Nanomaterial to be Safe? Recommendations for Planning Studies on Nanosafety. Small 2020, 16, 1907483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diesel, B.; Hoppstädter, J.; Hachenthal, N.; Zarbock, R.; Cavelius, C.; Wahl, B.; Thewes, N.; Jacobs, K.; Kraegeloh, A.; Kiemer, A.K. Activation of Rac1 GTPase by nanoparticulate structures in human macrophages. Eur. J. Pharm. Biopharm. 2013, 84, 315–324. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Leader, A.M.; Grout, J.A.; Maier, B.B.; Nabet, B.Y.; Park, M.D.; Tabachnikova, A.; Chang, C.; Walker, L.; Lansky, A.; Le Berichel, J.; et al. Single-cell analysis of human non-small cell lung cancer lesions refines tumor classification and patient stratification. Cancer Cell 2021, 39, 1594–1609.e1512. [Google Scholar] [CrossRef] [PubMed]
- Kapellos, T.S.; Taylor, L.; Lee, H.; Cowley, S.A.; James, W.S.; Iqbal, A.J.; Greaves, D.R. A novel real time imaging platform to quantify macrophage phagocytosis. Biochem. Pharmacol. 2016, 116, 107–119. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Katz, B.P.; Spinola, S.M. Haemophilus ducreyi-induced interleukin-10 promotes a mixed M1 and M2 activation program in human macrophages. Infect. Immun. 2012, 80, 4426–4434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangal, S.; Gao, W.; Li, T.; Zhou, Q.T. Pulmonary delivery of nanoparticle chemotherapy for the treatment of lung cancers: Challenges and opportunities. Acta Pharmacol. Sin. 2017, 38, 782–797. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Mendieta, S.; Guillén, D.; Vasquez-Martínez, N.; Hernández-Pando, R.; Sánchez, S.; Rodríguez-Sanoja, R. Understanding the Phagocytosis of Particles: The Key for Rational Design of Vaccines and Therapeutics. Pharm. Res. 2022, 39, 1823–1849. [Google Scholar] [CrossRef]
- Sumitomo, R.; Hirai, T.; Fujita, M.; Murakami, H.; Otake, Y.; Huang, C.L. M2 tumor-associated macrophages promote tumor progression in non-small-cell lung cancer. Exp. Ther. Med. 2019, 18, 4490–4498. [Google Scholar] [CrossRef]
- Reimer, T.; Brcic, M.; Schweizer, M.; Jungi, T.W. poly(I:C) and LPS induce distinct IRF3 and NF-κB signaling during type-I IFN and TNF responses in human macrophages. J. Leukoc. Biol. 2008, 83, 1249–1257. [Google Scholar] [CrossRef]
- Dacoba, T.G.; Anfray, C.; Mainini, F.; Allavena, P.; Alonso, M.J.; Torres Andón, F.; Crecente-Campo, J. Arginine-Based Poly(I:C)-Loaded Nanocomplexes for the Polarization of Macrophages Toward M1-Antitumoral Effectors. Front. Immunol. 2020, 11, 1412. [Google Scholar] [CrossRef]
- Márquez-Rodas, I.; Longo, F.; Rodriguez-Ruiz, M.E.; Calles, A.; Ponce, S.; Jove, M.; Rubio-Viqueira, B.; Perez-Gracia, J.L.; Gómez-Rueda, A.; López-Tarruella, S.; et al. Intratumoral nanoplexed poly I:C BO-112 in combination with systemic anti-PD-1 for patients with anti-PD-1-refractory tumors. Sci. Transl. Med. 2020, 12, eabb0391. [Google Scholar] [CrossRef]
- Hartmann, G.; Krieg, A.M. CpG DNA and LPS induce distinct patterns of activation in human monocytes. Gene Ther. 1999, 6, 893. [Google Scholar] [CrossRef] [Green Version]
- Smulders, S.; Kaiser, J.-P.; Zuin, S.; Van Landuyt, K.L.; Golanski, L.; Vanoirbeek, J.; Wick, P.; Hoet, P.H.M. Contamination of nanoparticles by endotoxin: Evaluation of different test methods. Part. Fibre Toxicol. 2012, 9, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baron, L.; Gombault, A.; Fanny, M.; Villeret, B.; Savigny, F.; Guillou, N.; Panek, C.; Le Bert, M.; Lagente, V.; Rassendren, F.; et al. The NLRP3 inflammasome is activated by nanoparticles through ATP, ADP and adenosine. Cell Death Dis. 2015, 6, e1629. [Google Scholar] [CrossRef] [Green Version]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Gómez, D.M.; Urcuqui-Inchima, S.; Hernandez, J.C. Silica nanoparticles induce NLRP3 inflammasome activation in human primary immune cells. Innate Immun. 2017, 23, 697–708. [Google Scholar] [CrossRef] [PubMed]
- Merget, R.; Bauer, T.; Küpper, H.U.; Philippou, S.; Bauer, H.D.; Breitstadt, R.; Bruening, T. Health hazards due to the inhalation of amorphous silica. Arch. Toxicol. 2002, 75, 625–634. [Google Scholar] [CrossRef]
- Kusaka, T.; Nakayama, M.; Nakamura, K.; Ishimiya, M.; Furusawa, E.; Ogasawara, K. Effect of silica particle size on macrophage inflammatory responses. PLoS ONE 2014, 9, e92634. [Google Scholar] [CrossRef]
- Wei, X.; Wang, J.; Liang, M.; Song, M. Development of functional nanomedicines for tumor associated macrophages-focused cancer immunotherapy. Theranostics 2022, 12, 7821–7852. [Google Scholar] [CrossRef]
- Mouasni, S.; Gonzalez, V.; Schmitt, A.; Bennana, E.; Guillonneau, F.; Mistou, S.; Avouac, J.; Ea, H.K.; Devauchelle, V.; Gottenberg, J.-E.; et al. The classical NLRP3 inflammasome controls FADD unconventional secretion through microvesicle shedding. Cell Death Dis. 2019, 10, 190. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, M. Macrophage Recognition of Crystals and Nanoparticles. Front. Immunol. 2018, 9, 103. [Google Scholar] [CrossRef]
- Challagundla, N.; Saha, B.; Agrawal-Rajput, R. Insights into inflammasome regulation: Cellular, molecular, and pathogenic control of inflammasome activation. Immunol. Res. 2022, 70, 578–606. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Analysis | Plate Format | Seeding Density [Cells/Well] | Differentiation + Polarization [d] | Treatment Time [h] |
|---|---|---|---|---|
| IL1-β secretion | 96-well | 20,000 | 5 | 24 |
| MTT-assay | 96-well | 50,000 | 5 | 24 |
| flow cytometry (µR uptake) | 24-well | 250,000 | 5 + 2 | 1/3 |
| flow cytometry (membrane expression) | 12-well | 244,000 | 5 + 1 | 48 |
| Microscopy | 24-well | 200,000 | 5 + 2 | 1/3 |
| live-cell microscopy (µR uptake) | 96-well | 40,000 | 5 + 2 | 1/3 |
| live-cell microscopy (cytotoxicity) | 96-well | 40,000 | 5 | 24 |
| mRNA expression | 6-well | 600,000 | 5 + 1 | 4 |
| mRNA expression (TAM models) | 12-well | 500,000 | 6 + 1 | / |
| µR [µg/mL] | 100 | 200 | 400 | 600 | 800 |
|---|---|---|---|---|---|
| Treatment [µRs/cell] | 0.5 | 1 | 2 | 3 | 4 |
| Gene * | Accession Number | Forward (5′-3′) | Reverse (5′-3′) |
|---|---|---|---|
| RNA18S5 | NR_003286.2 | AGGTCTGTGATGCCCTTAGA | GAATGGGGTTCAACGGGTTA |
| VEGFA | NM_001171623.1 | CGCTTACTCTCACCTGCTTCTG | GGTCAACCACTCACACACACAC |
| HIF1A | NM_181054.3 | CGGGGACCGATTCACCAT | TTTCGACGTTCAGAACTTATCTTTT |
| ABCA1 | NM_005502.4 | CATCTGGTTCTATGCCCGCT | TCTGCATTCCACCTGACAGC |
| ABCG1 | NM_016818.3 | GCGCCAAACTCTTCGAGCTG | CGGATGCAACCTCCATGACAAA |
| IL8 | NM_000584.4 | GAGAAGTTTTTGAAGAGGGCTGA | GCTTGAAGTTTCACTGGCATCT |
| CCL2 | NM_002982.3 | TTGATGTTTTAAGTTTATCTTTCATGG | CAGGGGTAGAACTGTGGTTCA |
| TNF | NM_000594.4 | CTCCACCCATGTGCTCCTCA | CTCTGGCAGGGGCTCTTGAT |
| CXCL10 | NM_001565.4 | GAGCCTACAGCAGAGGAACC | AAGGCAGCAAATCAGAATCG |
| Reporter Cell Line | Receptor Expression |
|---|---|
| THP1-XBlue™ | TLR1/2, TLR2/6, TLR4, TLR5, TLR8, NOD1, NOD2 |
| HEK-Blue™-hTLR2 | TLR1/2, TLR2/6, TLR3, TLR5, NOD1 |
| HEK-Dual™ hTLR2 (NF/IL8) cells | TLR1/2, TLR2/6, NOD1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Fityan, S.; Diesel, B.; Fischer, T.; Ampofo, E.; Schomisch, A.; Mashayekhi, V.; Schneider, M.; Kiemer, A.K. Nanostructured Microparticles Repolarize Macrophages and Induce Cell Death in an In Vitro Model of Tumour-Associated Macrophages. Pharmaceutics 2023, 15, 1895. https://doi.org/10.3390/pharmaceutics15071895
Al-Fityan S, Diesel B, Fischer T, Ampofo E, Schomisch A, Mashayekhi V, Schneider M, Kiemer AK. Nanostructured Microparticles Repolarize Macrophages and Induce Cell Death in an In Vitro Model of Tumour-Associated Macrophages. Pharmaceutics. 2023; 15(7):1895. https://doi.org/10.3390/pharmaceutics15071895
Chicago/Turabian StyleAl-Fityan, Salma, Britta Diesel, Thorben Fischer, Emmanuel Ampofo, Annika Schomisch, Vida Mashayekhi, Marc Schneider, and Alexandra K. Kiemer. 2023. "Nanostructured Microparticles Repolarize Macrophages and Induce Cell Death in an In Vitro Model of Tumour-Associated Macrophages" Pharmaceutics 15, no. 7: 1895. https://doi.org/10.3390/pharmaceutics15071895