
The Nanotechnology-Based Approaches against Kirsten Rat Sarcoma-Mutated Cancers
 , , , , , and
, , , , , and
Abstract
:1. Introduction
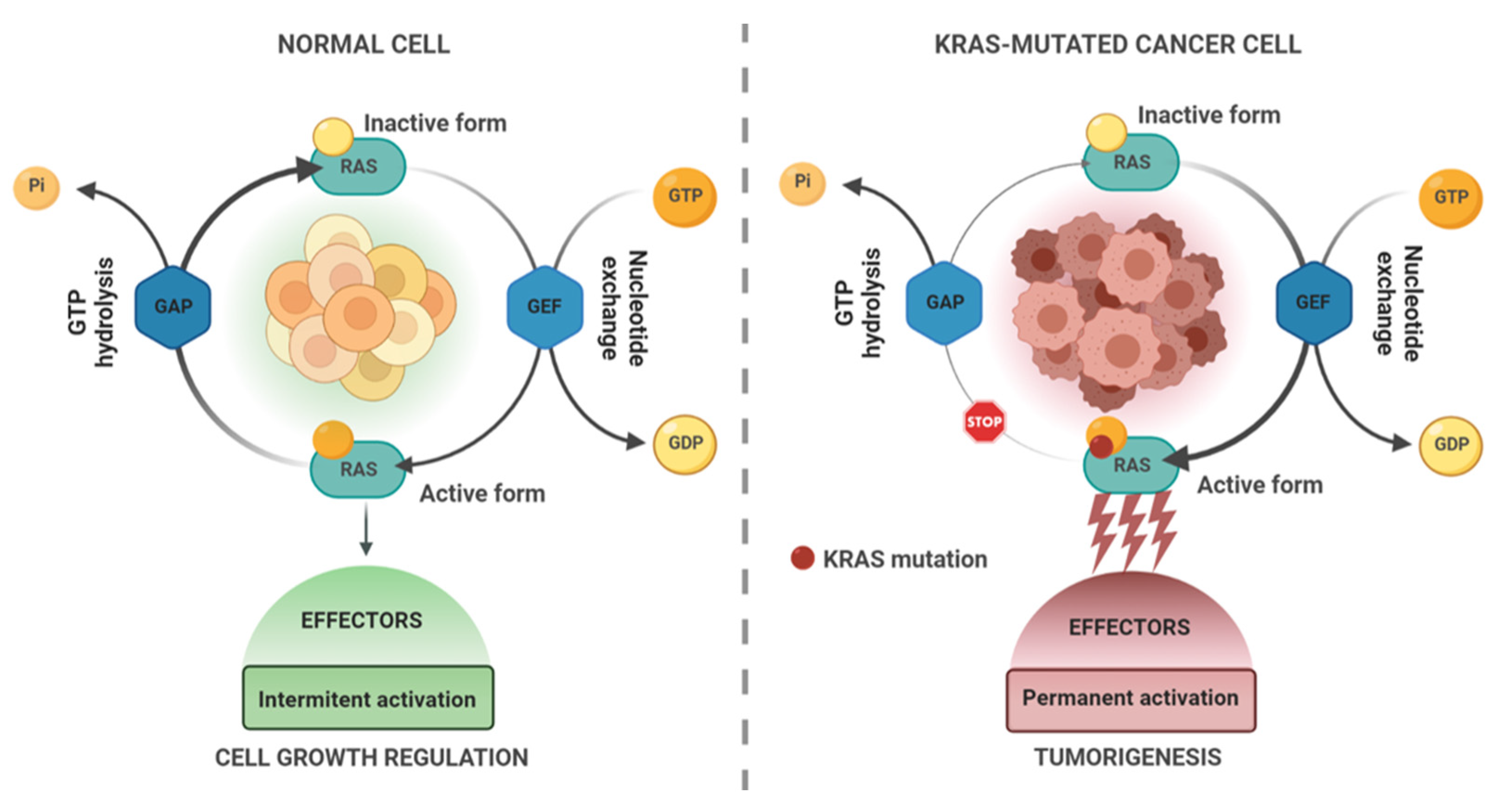
KRAS in Cancer
2. Actual Therapeutic Strategies against KRAS
3. The Importance of Nanotechnology
4. Nanotechnology-Based Anti-KRAS Therapies: The State of the Art
4.1. Chemical Therapy
4.2. Biotechnological/Biopharmaceutical Therapy
4.2.1. Peptide/Protein-Based Compounds
4.2.2. Gene Therapy
Gene Silencing
Gene Editing

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Vehicle | Cargo | Application | Development Stage | Reference |
|---|---|---|---|---|---|
| Chemotherapy | Amphiphilic hydroxyethyl starch-conjugated lauric acid and L-leucine NP | Camptothecin | Hepatic cancer | In vivo | [76] |
| PLGA NP | Afatinib | NSCLC | In vitro | [61] | |
| Gold NP | Doxorubicin | CRC | In vivo | [75] | |
| Liposomes (DOPA, DOTAP, Chol, DSPE-PEG) with calcium phosphate | Gemcitabine | PDAC | In vivo | [80] | |
| Silicasomes (DSPC/Chol/DSPE-PEG liposomes with mesoporous silica) | Irinotecan and an anti-PD-1 compound | PDAC | In vivo | [81] | |
| Silicasomes (DSPC/Chol/DSPE-PEG liposomes with mesoporous silica) | Platinum-based compounds and an anti-PD-1 compound | PDAC | In vivo | [82] | |
| PLGA-coated gold NP | 5-Fluorouracil | Lung cancer | In vitro | [116] | |
| PEG-b-PDPA micelles | Triptolide | PDAC | In vivo | [77] | |
| Cetuximab-conjugated PEG-PLGA NP | Camptothecin | Pancreatic cancer | In vivo | [117] | |
| Albumin NP | β-lapachone | PDAC | In vivo | [60] | |
| PpIX-C6-PEG8-KKKKKKSKTKC-OMe peptidic micelles | Protoporphyrin IX | Breast cancer | In vivo | [118] | |
| Avidin–nucleic acid nanoassemblies | Doxorubicin | Breast cancer | In vivo | [119] | |
| Peptide/protein-based therapy | Pluronic-based micelles | Anti-KRAS antibody | CRC and pancreatic cancer | In vivo | [87] |
| Cremophor EL-based micelles | Bicyclic peptide KS-58 | CRC and pancreatic cancer | In vivo | [88] | |
| MPN technology nanocapsules | Anti-KRAS antibody | PDAC | In vivo | [94] | |
| Gene therapy | Albumin NP | siKRAS | Lung cancer therapy | In vitro | [107,114] |
| p5RHH NP | siKRAS | Pancreatic Cancer | In vivo | [108] | |
| EVs | siKRAS | Pancreatic Cancer | Phase I | [45] | |
| Lipid NP | siKRAS and gemcitabine | Pancreatic cancer | In vivo | [97,107] | |
| PEI-modified hydroxyapatite NP | siKRAS | Pancreatic cancer | In vitro | [108,120] | |
| Cationic poly (cyclohexene carbonate) NP | siKRAS | Pancreatic cancer | In vitro | [45,121] | |
| Antibody-cationized gelatin NP | siKRAS | NSCLC | In vitro | [97,122] | |
| HA layer-by-layer liposomes | siKRAS miR-532-3p 5-Fluorouracil (5-FU) | CRC | In vivo | [99,120] | |
| Polyionic copolymer nanocarrier | miR-143#12 | Bladder cancer and RCC | In vivo | [110] | |
| PAMAM dendrimer | miRNA Mimic let-7b chloroquine | NSCLC | In vitro | [98] | |
| HA-decorated HAPD | Cas9 RNP sgRNAs targeting mutant APC and KRAS | CRC | In vivo | [114] | |
| Disulfide-bridged biguanidyl adamantine with β-cyclodextrin-conjugated low-molecular-weight polyethyleneimime nanocomplex | Cas9 RNP sgRNAs targeting mutant KRAS | CRC | In vivo | [112] | |
| EVs | CRISPR/Cas9 vector (LentiCRISPR V2 and pSpCas9(BB)-2A-GFP (PX458)) | Pancreatic cancer | In vivo | [115] | |
| Thiol-modified glycol chitosan NP | siKRAS and GDC-0941 | Ovarian cancer | In vivo | [102] | |
| Lipid NP | siGSTP | NSCLC, CRC, and pancreatic cancer | Phase I clinical trial | [103] |
5. Nanomedicine Challenges
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braicu, C.; Buse, M.; Busuioc, C.; Drula, R.; Gulei, D.; Raduly, L.; Rusu, A.; Irimie, A.; Atanasov, A.G.; Slaby, O.; et al. A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer. Cancers 2019, 11, 1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Liu, W.Z.; Liu, T.; Feng, X.; Yang, N.; Zhou, H.F. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J. Recept. Signal Transduct. Res. 2015, 35, 600–604. [Google Scholar] [CrossRef]
- Rodriguez-Viciana, P.; Warne, P.H.; Dhand, R.; Vanhaesebroeck, B.; Gout, I.; Fry, M.J.; Waterfield, M.D.; Downward, J. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature 1994, 370, 527–532. [Google Scholar] [CrossRef]
- Pacold, M.E.; Suire, S.; Perisic, O.; Lara-Gonzalez, S.; Davis, C.T.; Walker, E.H.; Hawkins, P.T.; Stephens, L.; Eccleston, J.F.; Williams, R.L. Crystal structure and functional analysis of Ras binding to its effector phosphoinositide 3-kinase gamma. Cell 2000, 103, 931–943. [Google Scholar] [CrossRef] [Green Version]
- Hofer, F.; Fields, S.; Schneider, C.; Martin, G.S. Activated Ras interacts with the Ral guanine nucleotide dissociation stimulator. Proc. Natl. Acad. Sci. USA 1994, 91, 11089–11093. [Google Scholar] [CrossRef] [Green Version]
- Neel, N.F.; Martin, T.D.; Stratford, J.K.; Zand, T.P.; Reiner, D.J.; Der, C.J. The RalGEF-Ral Effector Signaling Network: The Road Less Traveled for Anti-Ras Drug Discovery. Genes Cancer 2011, 2, 275–287. [Google Scholar] [CrossRef]
- Jancík, S.; Drábek, J.; Radzioch, D.; Hajdúch, M. Clinical relevance of KRAS in human cancers. J. Biomed. Biotechnol. 2010, 2010, 150960. [Google Scholar] [CrossRef] [Green Version]
- Fang, B. RAS signaling and anti-RAS therapy: Lessons learned from genetically engineered mouse models, human cancer cells, and patient-related studies. Acta Biochim. Biophys. Sin. 2016, 48, 27–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Maldonado, C.; Zimmer, Y.; Medová, M. A Comparative Analysis of Individual RAS Mutations in Cancer Biology. Front Oncol 2019, 9, 1088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.Q.; Kuttikrishnan, S.; Siveen, K.S.; Prabhu, K.S.; Shanmugakonar, M.; Al-Naemi, H.A.; Haris, M.; Dermime, S.; Uddin, S. RAS-mediated oncogenic signaling pathways in human malignancies. Semin. Cancer Biol. 2019, 54, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Karnoub, A.E.; Weinberg, R.A. Ras oncogenes: Split personalities. Nat. Rev. Mol. Cell Biol. 2008, 9, 517–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, M.; Zhong, K.; Jiang, T.; Liu, Z.; Kwan, H.Y.; Su, T. The current understanding on the impact of KRAS on colorectal cancer. Biomed. Pharmacother. 2021, 140, 111717. [Google Scholar] [CrossRef] [PubMed]
- Stolze, B.; Reinhart, S.; Bulllinger, L.; Fröhling, S.; Scholl, C. Comparative analysis of KRAS codon 12, 13, 18, 61, and 117 mutations using human MCF10A isogenic cell lines. Sci. Rep. 2015, 5, 8535. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Guo, Z.; Wang, F.; Fu, L. KRAS mutation: From undruggable to druggable in cancer. Signal. Transduct. Target. Ther. 2021, 6, 386. [Google Scholar] [CrossRef]
- Pantsar, T. The current understanding of KRAS protein structure and dynamics. Comput. Struct. Biotechnol. J. 2020, 18, 189–198. [Google Scholar] [CrossRef]
- Karimi, N.; Moghaddam, S.J. KRAS-Mutant Lung Cancer: Targeting Molecular and Immunologic Pathways, Therapeutic Advantages and Restrictions. Cells 2023, 12, 749. [Google Scholar] [CrossRef]
- Tang, D.; Kroemer, G.; Kang, R. Oncogenic KRAS blockade therapy: Renewed enthusiasm and persistent challenges. Mol. Cancer 2021, 20, 128. [Google Scholar] [CrossRef] [PubMed]
- Ghimessy, A.; Radeczky, P.; Laszlo, V.; Hegedus, B.; Renyi-Vamos, F.; Fillinger, J.; Klepetko, W.; Lang, C.; Dome, B.; Megyesfalvi, Z. Current therapy of KRAS-mutant lung cancer. Cancer Metastasis Rev. 2020, 39, 1159–1177. [Google Scholar] [CrossRef] [PubMed]
- Uprety, D.; Adjei, A.A. KRAS: From undruggable to a druggable Cancer Target. Cancer Treat. Rev. 2020, 89, 102070. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, E.; Drezner, N.; Li, X.; Mishra-Kalyani, P.; Liu, Y.; Zhao, H.; Bi, Y.; Liu, J.; Rahman, A.; Wearne, E.; et al. FDA Approval Summary: Sotorasib for KRAS G12C-Mutated Metastatic NSCLC. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2022, 28, 1482–1486. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Lin, J.; Li, C.; Ryan, M.; Zhang, J.; Kiedrowski, L.; Michel, A.; Syed, M.; Fella, K.; Sakhi, M.; et al. Clinical Acquired Resistance to KRASG12C Inhibition through a Novel KRAS Switch-II Pocket Mutation and Polyclonal Alterations Converging on RAS-MAPK Reactivation. Cancer Discov. 2021, 11, 1913–1922. [Google Scholar] [CrossRef]
- Lee, A. Sotorasib: A Review in KRAS G12C Mutation-Positive Non-small Cell Lung Cancer. Target. Oncol. 2022, 17, 727–733. [Google Scholar] [CrossRef]
- Jänne, P.A.; Riely, G.J.; Gadgeel, S.M.; Heist, R.S.; Ou, S.I.; Pacheco, J.M.; Johnson, M.L.; Sabari, J.K.; Leventakos, K.; Yau, E.; et al. Adagrasib in Non-Small-Cell Lung Cancer Harboring a KRAS G12C Mutation. N. Engl. J. Med. 2022, 387, 120–131. [Google Scholar] [CrossRef]
- Dhillon, S. Adagrasib: First Approval. Drugs 2023, 83, 275–285. [Google Scholar] [CrossRef]
- Khan, H.Y.; Nagasaka, M.; Li, Y.; Aboukameel, A.; Uddin, M.H.; Sexton, R.; Bannoura, S.; Mzannar, Y.; Al-Hallak, M.N.; Kim, S.; et al. Inhibitor of the Nuclear Transport Protein XPO1 Enhances the Anticancer Efficacy of KRAS G12C Inhibitors in Preclinical Models of KRAS G12C-Mutant Cancers. Cancer Res. Commun. 2022, 2, 342–352. [Google Scholar] [CrossRef]
- Patricelli, M.P.; Janes, M.R.; Li, L.S.; Hansen, R.; Peters, U.; Kessler, L.V.; Chen, Y.; Kucharski, J.M.; Feng, J.; Ely, T.; et al. Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discov. 2016, 6, 316–329. [Google Scholar] [CrossRef] [Green Version]
- Janes, M.R.; Zhang, J.; Li, L.S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589.e517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Zhao, H.; Liao, H.; Chen, J.; Liu, J. Discovery of novel quinazoline-based covalent inhibitors of KRAS G12C with various cysteine-targeting warheads as potential anticancer agents. Bioorg. Chem. 2021, 110, 104825. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhao, H.; Peng, X.; Liu, J.; Mai, R.; Chen, J.; Lin, L.; Chen, T.; Yan, J.; Shi, J. Discovery of novel Quinazoline-based KRAS G12C inhibitors as potential anticancer agents. Bioorg. Med. Chem. 2022, 71, 116962. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Li, L.; Liu, J.; Mai, R.; Chen, J. Discovery of ARS-1620 analogs as KRas G12C inhibitors with high in vivo antitumor activity. Bioorg. Chem. 2022, 121, 105652. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Chen, W.; Wang, J.; Zhao, G.; Chen, L.; Wan, Y.; Luo, Q.; Huang, H.; Yang, Y.; Chen, D.; et al. A PDX model combined with CD-DST assay to evaluate the antitumor properties of KRpep-2d and oxaliplatin in KRAS (G12D) mutant colorectal cancer. Heliyon 2022, 8, e12518. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Lin, B.; Nunomura, K.; Izawa, T.; Nakagawa, S. The K-Ras(G12D)-inhibitory peptide KS-58 suppresses growth of murine CT26 colorectal cancer cell-derived tumors. Sci. Rep. 2022, 12, 8121. [Google Scholar] [CrossRef]
- Sakamoto, K.; Masutani, T.; Hirokawa, T. Generation of KS-58 as the first K-Ras(G12D)-inhibitory peptide presenting anti-cancer activity in vivo. Sci. Rep. 2020, 10, 21671. [Google Scholar] [CrossRef]
- Teng, K.W.; Tsai, S.T.; Hattori, T.; Fedele, C.; Koide, A.; Yang, C.; Hou, X.; Zhang, Y.; Neel, B.G.; O’Bryan, J.P.; et al. Selective and noncovalent targeting of RAS mutants for inhibition and degradation. Nat. Commun. 2021, 12, 2656. [Google Scholar] [CrossRef]
- Guo, C.; Banerji, U. Searching for treatments for non-G12C-KRAS mutant cancers. Br. J. Cancer 2021, 125, 625–626. [Google Scholar] [CrossRef]
- Zhou, C.; Fan, Z.; Zhou, Z.; Li, Y.; Cui, R.; Liu, C.; Zhou, G.; Diao, X.; Jiang, H.; Zheng, M.; et al. Discovery of the First-in-Class Agonist-Based SOS1 PROTACs Effective in Human Cancer Cells Harboring Various KRAS Mutations. J. Med. Chem. 2022, 65, 3923–3942. [Google Scholar] [CrossRef]
- Salgia, R.; Pharaon, R.; Mambetsariev, I.; Nam, A.; Sattler, M. The improbable targeted therapy: KRAS as an emerging target in non-small cell lung cancer (NSCLC). Cell. Rep. Med. 2021, 2, 100186. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.J.; Revenko, A.S.; Hanson, L.L.; Ellston, R.; Staniszewska, A.; Whalley, N.; Pandey, S.K.; Revill, M.; Rooney, C.; Buckett, L.K.; et al. Targeting KRAS-dependent tumors with AZD4785, a high-affinity therapeutic antisense oligonucleotide inhibitor of KRAS. Sci. Transl. Med. 2017, 9, eaal5253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gort, E.; Johnson, M.L.; Hwang, J.J.; Pant, S.; Dünzinger, U.; Riemann, K.; Kitzing, T.; Janne, P.A. A phase I, open-label, dose-escalation trial of BI 1701963 as monotherapy and in combination with trametinib in patients with KRAS mutated advanced or metastatic solid tumors. J. Clin. Oncol. 2020, 38, TPS3651. [Google Scholar] [CrossRef]
- Chen, K.; Zhang, Y.; Qian, L.; Wang, P. Emerging strategies to target RAS signaling in human cancer therapy. J. Hematol. Oncol. 2021, 14, 116. [Google Scholar] [CrossRef]
- Surana, R.; LeBleu, V.S.; Lee, J.J.; Smaglo, B.G.; Zhao, D.; Lee, M.S.; Wolff, R.A.; Overman, M.J.; Mendt, M.C.; McAndrews, K.M.; et al. Phase I study of mesenchymal stem cell (MSC)-derived exosomes with KRAS G12D siRNA in patients with metastatic pancreatic cancer harboring a KRAS G12D mutation. In Proceedings of the ASCO Gastrointestinal Cancers Symposium, San Francisco, CA, USA, 20–22 January 2022; p. TPS633. [Google Scholar]
- Wang, J.; Martin-Romano, P.; Cassier, P.; Johnson, M.; Haura, E.; Lenox, L.; Guo, Y.; Bandyopadhyay, N.; Russell, M.; Shearin, E.; et al. Phase I Study of JNJ-74699157 in Patients with Advanced Solid Tumors Harboring the KRAS G12C Mutation. Oncologist 2022, 27, 536-e553. [Google Scholar] [CrossRef]
- Blaquier, J.B.; Cardona, A.F.; Recondo, G. Resistance to KRAS G12C Inhibitors in Non-Small Cell Lung Cancer. Front. Oncol. 2021, 11, 787585. [Google Scholar] [CrossRef]
- Liu, J.; Kang, R.; Tang, D. The KRAS-G12C inhibitor: Activity and resistance. Cancer Gene Ther. 2022, 29, 875–878. [Google Scholar] [CrossRef]
- Rasool, M.; Malik, A.; Waquar, S.; Arooj, M.; Zahid, S.; Asif, M.; Shaheen, S.; Hussain, A.; Ullah, H.; Gan, S.H. New challenges in the use of nanomedicine in cancer therapy. Bioengineered 2022, 13, 759–773. [Google Scholar] [CrossRef]
- Germain, M.; Caputo, F.; Metcalfe, S.; Tosi, G.; Spring, K.; Åslund, A.K.O.; Pottier, A.; Schiffelers, R.; Ceccaldi, A.; Schmid, R. Delivering the power of nanomedicine to patients today. J. Control. Release 2020, 326, 164–171. [Google Scholar] [CrossRef]
- Bhatia, S.N.; Chen, X.; Dobrovolskaia, M.A.; Lammers, T. Cancer nanomedicine. Nat. Rev. Cancer 2022, 22, 550–556. [Google Scholar] [CrossRef]
- Wu, J. The Enhanced Permeability and Retention (EPR) Effect: The Significance of the Concept and Methods to Enhance Its Application. J. Pers. Med. 2021, 11, 771. [Google Scholar] [CrossRef] [PubMed]
- Waheed, S.; Li, Z.; Zhang, F.; Chiarini, A.; Armato, U.; Wu, J. Engineering nano-drug biointerface to overcome biological barriers toward precision drug delivery. J. Nanobiotechnology 2022, 20, 395. [Google Scholar] [CrossRef]
- Pearce, A.K.; O’Reilly, R.K. Insights into Active Targeting of Nanoparticles in Drug Delivery: Advances in Clinical Studies and Design Considerations for Cancer Nanomedicine. Bioconjug. Chem. 2019, 30, 2300–2311. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Zhang, J.; Zhao, M.; Tang, S.; Cheng, X.; Zhang, W.; Li, W.; Liu, X.; Peng, H.; Wang, Q. Effects of polyethylene glycol on the surface of nanoparticles for targeted drug delivery. Nanoscale 2021, 13, 10748–10764. [Google Scholar] [CrossRef] [PubMed]
- Suk, J.S.; Xu, Q.; Kim, N.; Hanes, J.; Ensign, L.M. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Adv. Drug. Deliv. Rev. 2016, 99, 28–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Ghosh, D. Intracellular nanoparticle delivery by oncogenic KRAS-mediated macropinocytosis. Int. J. Nanomed. 2019, 14, 6589–6600. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Ng, T.S.C.; Wang, S.J.; Prytyskach, M.; Rodell, C.B.; Mikula, H.; Kohler, R.H.; Garlin, M.A.; Lauffenburger, D.A.; Parangi, S.; et al. Therapeutically reprogrammed nutrient signalling enhances nanoparticulate albumin bound drug uptake and efficacy in KRAS-mutant cancer. Nat. Nanotechnol. 2021, 16, 830–839. [Google Scholar] [CrossRef]
- Dou, L.; Liu, H.; Wang, K.; Liu, J.; Liu, L.; Ye, J.; Wang, R.; Deng, H.; Qian, F. Albumin binding revitalizes NQO1 bioactivatable drugs as novel therapeutics for pancreatic cancer. J. Control. Release 2022, 349, 876–889. [Google Scholar] [CrossRef]
- Elbatanony, R.S.; Parvathaneni, V.; Kulkarni, N.S.; Shukla, S.K.; Chauhan, G.; Kunda, N.K.; Gupta, V. Afatinib-loaded inhalable PLGA nanoparticles for localized therapy of non-small cell lung cancer (NSCLC)-development and in-vitro efficacy. Drug Deliv. Transl. Res. 2021, 11, 927–943. [Google Scholar] [CrossRef]
- Maiti, A.; Naqvi, K.; Kadia, T.M.; Borthakur, G.; Takahashi, K.; Bose, P.; Daver, N.G.; Patel, A.; Alvarado, Y.; Ohanian, M.; et al. Phase II Trial of MEK Inhibitor Binimetinib (MEK162) in RAS-mutant Acute Myeloid Leukemia. Clin. Lymphoma Myeloma Leuk. 2019, 19, 142–148.e141. [Google Scholar] [CrossRef] [PubMed]
- Sorokin, A.V.; Kanikarla Marie, P.; Bitner, L.; Syed, M.; Woods, M.; Manyam, G.; Kwong, L.N.; Johnson, B.; Morris, V.K.; Jones, P.; et al. Targeting RAS Mutant Colorectal Cancer with Dual Inhibition of MEK and CDK4/6. Cancer Res. 2022, 82, 3335–3344. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Schadendorf, D.; Ascierto, P.A.; Arance, A.; Dutriaux, C.; Di Giacomo, A.M.; Rutkowski, P.; Del Vecchio, M.; Gutzmer, R.; Mandala, M.; et al. Binimetinib versus dacarbazine in patients with advanced NRAS-mutant melanoma (NEMO): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Bikhezar, F.; de Kruijff, R.M.; van der Meer, A.J.G.M.; Torrelo Villa, G.; van der Pol, S.M.A.; Becerril Aragon, G.; Gasol Garcia, A.; Narayan, R.S.; de Vries, H.E.; Slotman, B.J.; et al. Preclinical evaluation of binimetinib (MEK162) delivered via polymeric nanocarriers in combination with radiation and temozolomide in glioma. J. Neurooncol. 2020, 146, 239–246. [Google Scholar] [CrossRef] [Green Version]
- Dingemans, A.M.; Mellema, W.W.; Groen, H.J.; van Wijk, A.; Burgers, S.A.; Kunst, P.W.; Thunnissen, E.; Heideman, D.A.; Smit, E.F. A phase II study of sorafenib in patients with platinum-pretreated, advanced (Stage IIIb or IV) non-small cell lung cancer with a KRAS mutation. Clin. Cancer Res. 2013, 19, 743–751. [Google Scholar] [CrossRef] [Green Version]
- Nogova, L.; Mattonet, C.; Scheffler, M.; Taubert, M.; Gardizi, M.; Sos, M.L.; Michels, S.; Fischer, R.N.; Limburg, M.; Abdulla, D.S.Y.; et al. Sorafenib and everolimus in patients with advanced solid tumors and KRAS-mutated NSCLC: A phase I trial with early pharmacodynamic FDG-PET assessment. Cancer Med. 2020, 9, 4991–5007. [Google Scholar] [CrossRef]
- Kong, F.H.; Ye, Q.F.; Miao, X.Y.; Liu, X.; Huang, S.Q.; Xiong, L.; Wen, Y.; Zhang, Z.J. Current status of sorafenib nanoparticle delivery systems in the treatment of hepatocellular carcinoma. Theranostics 2021, 11, 5464–5490. [Google Scholar] [CrossRef]
- Lai, H.; Zhong, L.; Huang, Y.; Zhao, Y.; Qian, Z. Progress in Application of Nanotechnology in Sorafenib. J. Biomed. Nanotechnol. 2021, 17, 529–557. [Google Scholar] [CrossRef]
- Caputo, T.M.; Cusano, A.M.; Ruvo, M.; Aliberti, A.; Cusano, A. Human Serum Albumin Nanoparticles as a Carrier for On-Demand Sorafenib Delivery. Curr. Pharm. Biotechnol. 2022, 23, 1214–1225. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Hirsh, V.; Zhang, L.; de Marinis, F.; Yang, J.C.; Wakelee, H.A.; Seto, T.; Wu, Y.L.; Novello, S.; Juhász, E.; et al. Monotherapy Administration of Sorafenib in Patients With Non-Small Cell Lung Cancer (MISSION) Trial: A Phase III, Multicenter, Placebo-Controlled Trial of Sorafenib in Patients with Relapsed or Refractory Predominantly Nonsquamous Non-Small-Cell Lung Cancer after 2 or 3 Previous Treatment Regimens. J. Thorac. Oncol. 2015, 10, 1745–1753. [Google Scholar] [CrossRef] [Green Version]
- Zheng, N.; Liu, W.; Li, B.; Nie, H.; Liu, J.; Cheng, Y.; Wang, J.; Dong, H.; Jia, L. Co-delivery of sorafenib and metapristone encapsulated by CXCR4-targeted PLGA-PEG nanoparticles overcomes hepatocellular carcinoma resistance to sorafenib. J. Exp. Clin. Cancer Res. 2019, 38, 232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, T.; Liu, X.; Li, H.; Zhang, J. Co-delivery of sorafenib and crizotinib encapsulated with polymeric nanoparticles for the treatment of. Drug Deliv. 2021, 28, 2108–2118. [Google Scholar] [CrossRef]
- Taha, A.M.; Aboulwafa, M.M.; Zedan, H.; Helmy, O.M. Ramucirumab combination with sorafenib enhances the inhibitory effect of sorafenib on HepG2 cancer cells. Sci. Rep. 2022, 12, 17889. [Google Scholar] [CrossRef] [PubMed]
- Hung, W.H.; Zheng, J.H.; Lee, K.C.; Cho, E.C. Doxorubicin conjugated AuNP/biopolymer composites facilitate cell cycle regulation and exhibit superior tumor suppression potential in KRAS mutant colorectal cancer. J. Biotechnol. 2019, 306, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, X.; Zhang, C.; Chen, X.; Sheng, W.; Li, P.; Qin, D.; Wang, F. Novel amphiphilic hydroxyethyl starch-based nanoparticles loading camptothecin exhibit high anticancer activity in HepG2 cells and zebrafish. Colloids Surf. B Biointerfaces 2023, 224, 113215. [Google Scholar] [CrossRef] [PubMed]
- Kong, C.; Li, Y.; Liu, Z.; Ye, J.; Wang, Z.; Zhang, L.; Kong, W.; Liu, H.; Liu, C.; Pang, H.; et al. Targeting the Oncogene KRAS Mutant Pancreatic Cancer by Synergistic Blocking of Lysosomal Acidification and Rapid Drug Release. ACS Nano 2019, 13, 4049–4063. [Google Scholar] [CrossRef]
- Thummuri, D.; Khan, S.; Underwood, P.W.; Zhang, P.; Wiegand, J.; Zhang, X.; Budamagunta, V.; Sobh, A.; Tagmount, A.; Loguinov, A.; et al. Overcoming Gemcitabine Resistance in Pancreatic Cancer Using the BCL-X. Mol. Cancer Ther. 2022, 21, 184–192. [Google Scholar] [CrossRef]
- Fanchon, L.M.; Russell, J.; Pillarsetty, N.; O’Donoghue, I.; Gangangari, K.; Yu, K.H.; Humm, J.L. Comparing the intra-tumoral distribution of Gemcitabine, 5-Fluorouracil, and Capecitabine in a murine model of pancreatic ductal adenocarcinoma. PLoS ONE 2020, 15, e0231745. [Google Scholar] [CrossRef] [Green Version]
- Das, M.; Li, J.; Bao, M.; Huang, L. Nano-delivery of Gemcitabine Derivative as a Therapeutic Strategy in a Desmoplastic KRAS Mutant Pancreatic Cancer. AAPS J. 2020, 22, 88. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, J.; Liao, Y.P.; Tang, I.; Zheng, E.; Qiu, W.; Lin, M.; Wang, X.; Ji, Y.; Mei, K.C.; et al. Combination Chemo-Immunotherapy for Pancreatic Cancer Using the Immunogenic Effects of an Irinotecan Silicasome Nanocarrier Plus Anti-PD-1. Adv. Sci. 2021, 8, 2002147. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, J.; Chang, C.H.; Liao, Y.P.; Lodico, J.J.; Tang, I.; Zheng, E.; Qiu, W.; Lin, M.; Wang, X.; et al. Development of Facile and Versatile Platinum Drug Delivering Silicasome Nanocarriers for Efficient Pancreatic Cancer Chemo-Immunotherapy. Small 2021, 17, e2005993. [Google Scholar] [CrossRef] [PubMed]
- Kesik-Brodacka, M. Progress in biopharmaceutical development. Biotechnol. Appl. Biochem. 2018, 65, 306–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moorkens, E.; Meuwissen, N.; Huys, I.; Declerck, P.; Vulto, A.G.; Simoens, S. The Market of Biopharmaceutical Medicines: A Snapshot of a Diverse Industrial Landscape. Front. Pharmacol. 2017, 8, 314. [Google Scholar] [CrossRef] [Green Version]
- Lagassé, H.A.; Alexaki, A.; Simhadri, V.L.; Katagiri, N.H.; Jankowski, W.; Sauna, Z.E.; Kimchi-Sarfaty, C. Recent advances in (therapeutic protein) drug development. F1000Res 2017, 6, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahlich, J.; Desai, A.; Greco, F.; Hill, K.; Jones, A.T.; Mrsny, R.J.; Pasut, G.; Perrie, Y.; Seib, F.P.; Seymour, L.W.; et al. Nanomedicines for the Delivery of Biologics. Pharmaceutics 2019, 11, 210. [Google Scholar] [CrossRef] [Green Version]
- Rafael, D.; Montero, S.; Carcavilla, P.; Andrade, F.; German-Cortés, J.; Diaz-Riascos, Z.V.; Seras-Franzoso, J.; Llaguno, M.; Fernández, B.; Pereira, A.; et al. Intracellular Delivery of Anti-Kirsten Rat Sarcoma Antibodies Mediated by Polymeric Micelles Exerts Strong. ACS Appl. Mater. Interfaces 2023, 15, 10398–10413. [Google Scholar] [CrossRef]
- Sakamoto, K.; Qi, Y.; Miyako, E. Nanoformulation of the K-Ras(G12D)-inhibitory peptide KS-58 suppresses colorectal and pancreatic cancer-derived tumors. Sci. Rep. 2023, 13, 518. [Google Scholar] [CrossRef]
- Singh, S.; Murillo, G.; Richner, J.; Singh, S.P.; Berleth, E.; Kumar, V.; Mehta, R.; Ramiya, V.; Parihar, A.S. A Broad-Based Characterization of a Cell-Penetrating, Single Domain Camelid Bi-Specific Antibody Monomer That Targets STAT3 and KRAS Dependent Cancers. Int. J. Mol. Sci. 2022, 23, 7565. [Google Scholar] [CrossRef]
- Haza, K.Z.; Martin, H.L.; Rao, A.; Turner, A.L.; Saunders, S.E.; Petersen, B.; Tiede, C.; Tipping, K.; Tang, A.A.; Ajayi, M.; et al. RAS-inhibiting biologics identify and probe druggable pockets including an SII-α3 allosteric site. Nat. Commun. 2021, 12, 4045. [Google Scholar] [CrossRef]
- Röth, S.; Macartney, T.J.; Konopacka, A.; Chan, K.H.; Zhou, H.; Queisser, M.A.; Sapkota, G.P. Targeting Endogenous K-RAS for Degradation through the Affinity-Directed Protein Missile System. Cell Chem. Biol. 2020, 27, 1151–1163.e1156. [Google Scholar] [CrossRef]
- Ajmal, A.; Ali, Y.; Khan, A.; Wadood, A.; Rehman, A.U. Identification of novel peptide inhibitors for the KRas-G12C variant to prevent oncogenic signaling. J. Biomol. Struct. Dyn. 2022, 40, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Samad, A.; Khurshid, B.; Mahmood, A.; Rehman, A.U.; Khalid, A.; Abdalla, A.N.; Algarni, A.S.; Wadood, A. Identification of novel peptide inhibitors for oncogenic KRAS G12D as therapeutic options using mutagenesis-based remodeling and MD simulations. J. Biomol. Struct. Dyn. 2023, 41, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Alonso Fernandez, M.J.; Desireé, T.O. Intracellular Delivery of Anti-KRAS Antibodies Formulated into Nanocapsules. 2020. Available online: https://patents.google.com/patent/EP3962491A1/en (accessed on 4 April 2023).
- Bio, L. Enabling Intracellular Antibody Delivery to Unlock High-Value Targets. 2022. Available online: https://www.nature.com/articles/d43747-022-00073-x (accessed on 4 April 2023).
- Pei, Y.; Chen, L.; Huang, Y.; Wang, J.; Feng, J.; Xu, M.; Chen, Y.; Song, Q.; Jiang, G.; Gu, X.; et al. Sequential Targeting TGF-β Signaling and KRAS Mutation Increases Therapeutic Efficacy in Pancreatic Cancer. Small 2019, 15, e1900631. [Google Scholar] [CrossRef] [PubMed]
- Anthiya, S.; Öztürk, S.C.; Yanik, H.; Tavukcuoglu, E.; Şahin, A.; Datta, D.; Charisse, K.; Álvarez, D.M.; Loza, M.I.; Calvo, A.; et al. Targeted siRNA lipid nanoparticles for the treatment of KRAS-mutant tumors. J. Control. Release 2023, 357, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Maghsoudnia, N.; Eftekhari, R.B.; Sohi, A.N.; Dorkoosh, F.A. Chloroquine Assisted Delivery of microRNA Mimic Let-7b to NSCLC Cell Line by PAMAM (G5) - HA Nano-Carrier. Curr. Drug Deliv. 2021, 18, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Shahidi, M.; Abazari, O.; Dayati, P.; Haghiralsadat, B.F.; Oroojalian, F.; Tofighi, D. Targeted delivery of 5-fluorouracil, miR-532-3p, and si-KRAS to the colorectal tumor using layer-by-layer liposomes. Front. Bioeng. Biotechnol. 2022, 10, 1013541. [Google Scholar] [CrossRef]
- Xue, W.; Dahlman, J.E.; Tammela, T.; Khan, O.F.; Sood, S.; Dave, A.; Cai, W.; Chirino, L.M.; Yang, G.R.; Bronson, R.; et al. Small RNA combination therapy for lung cancer. Proc. Natl. Acad. Sci. USA 2014, 111, E3553-3561. [Google Scholar] [CrossRef] [Green Version]
- Pecot, C.V.; Wu, S.Y.; Bellister, S.; Filant, J.; Rupaimoole, R.; Hisamatsu, T.; Bhattacharya, R.; Maharaj, A.; Azam, S.; Rodriguez-Aguayo, C.; et al. Therapeutic silencing of KRAS using systemically delivered siRNAs. Mol. Cancer Ther. 2014, 13, 2876–2885. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.J.; Lee, S.J.; Ryu, J.H.; Kim, S.H.; Kwon, I.C.; Roberts, T.M. Combination of KRAS gene silencing and PI3K inhibition for ovarian cancer treatment. J. Control. Release 2020, 318, 98–108. [Google Scholar] [CrossRef]
- O’Brien, Z.; Wang, L.; Majeti, B.; Clamme, J.; Baclig, R.; Chu, J.; Fong, S.; Harborth, J.; Ibarra, J.; Yin, H.; et al. A novel lipid nanoparticle (NBF-006) encapsulating glutathione S-transferase P (GSTP) siRNA for the treatment of KRAS-driven non-small cell lung cancer. In Proceedings of the AACR Annual Meeting, Washington, DC, USA, 27–30 September 2018; p. 5917. [Google Scholar]
- Cecchin, R.; Troyer, Z.; Witwer, K.; Morris, K.V. Extracellular vesicles: The next generation in gene therapy delivery. Mol. Ther. 2023, 31, 1225–1230. [Google Scholar] [CrossRef]
- Mendt, M.; Kamerkar, S.; Sugimoto, H.; McAndrews, K.M.; Wu, C.C.; Gagea, M.; Yang, S.; Blanko, E.V.R.; Peng, Q.; Ma, X.; et al. Generation and testing of clinical-grade exosomes for pancreatic cancer. JCI Insight 2018, 3, e99263. [Google Scholar] [CrossRef] [PubMed]
- Kamerkar, S.; LeBleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, A.; Dalle Vedove, E.; Isert, L.; Merkel, O.M. Targeting KRAS Mutant Lung Cancer Cells with siRNA-Loaded Bovine Serum Albumin Nanoparticles. Pharm. Res. 2019, 36, 133. [Google Scholar] [CrossRef] [PubMed]
- Strand, M.S.; Krasnick, B.A.; Pan, H.; Zhang, X.; Bi, Y.; Brooks, C.; Wetzel, C.; Sankpal, N.; Fleming, T.; Goedegebuure, S.P.; et al. Precision delivery of RAS-inhibiting siRNA to KRAS driven cancer via peptide-based nanoparticles. Oncotarget 2019, 10, 4761–4775. [Google Scholar] [CrossRef] [Green Version]
- Pekow, J.; Meckel, K.; Dougherty, U.; Butun, F.; Mustafi, R.; Lim, J.; Crofton, C.; Chen, X.; Joseph, L.; Bissonnette, M. Tumor suppressors miR-143 and miR-145 and predicted target proteins API5, ERK5, K-RAS, and IRS-1 are differentially expressed in proximal and distal colon. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G179-187. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, Y.; Taniguchi, K.; Tsujino, T.; Heishima, K.; Inamoto, T.; Takai, T.; Minami, K.; Azuma, H.; Miyata, K.; Hayashi, K.; et al. Anti-cancer Effects of a Chemically Modified miR-143 on Bladder Cancer by Either Systemic or Intravesical Treatment. Mol. Ther. Methods Clin. Dev. 2019, 13, 290–302. [Google Scholar] [CrossRef] [Green Version]
- Takai, T.; Tsujino, T.; Yoshikawa, Y.; Inamoto, T.; Sugito, N.; Kuranaga, Y.; Heishima, K.; Soga, T.; Hayashi, K.; Miyata, K.; et al. Synthetic miR-143 Exhibited an Anti-Cancer Effect via the Downregulation of K-RAS Networks of Renal Cell Cancer Cells In Vitro and In Vivo. Mol. Ther. 2019, 27, 1017–1027. [Google Scholar] [CrossRef] [Green Version]
- Wan, T.; Chen, Y.; Pan, Q.; Xu, X.; Kang, Y.; Gao, X.; Huang, F.; Wu, C.; Ping, Y. Genome editing of mutant KRAS through supramolecular polymer-mediated delivery of Cas9 ribonucleoprotein for colorectal cancer therapy. J. Control Release 2020, 322, 236–247. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, L.; Liu, H.; Cheng, K. Delivery strategies of the CRISPR-Cas9 gene-editing system for therapeutic applications. J. Control. Release 2017, 266, 17–26. [Google Scholar] [CrossRef]
- Wan, T.; Pan, Q.; Liu, C.; Guo, J.; Li, B.; Yan, X.; Cheng, Y.; Ping, Y. A Duplex CRISPR-Cas9 Ribonucleoprotein Nanomedicine for Colorectal Cancer Gene Therapy. Nano Lett. 2021, 21, 9761–9771. [Google Scholar] [CrossRef]
- McAndrews, K.M.; Xiao, F.; Chronopoulos, A.; LeBleu, V.S.; Kugeratski, F.G.; Kalluri, R. Exosome-mediated delivery of CRISPR/Cas9 for targeting of oncogenic Kras. Life Sci. Alliance 2021, 4, e202000875. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.; Vishwakarma, L.; Guleri, S.K.; Kumar, G. 5-Fluorouracil-Impregnated PLGA Coated Gold Nanoparticles for Augmented Delivery to Lung Cancer: In Vitro Investigations. Anticancer Agents Med. Chem. 2022, 22, 2292–2302. [Google Scholar] [CrossRef] [PubMed]
- McDaid, W.J.; Greene, M.K.; Johnston, M.C.; Pollheimer, E.; Smyth, P.; McLaughlin, K.; Van Schaeybroeck, S.; Straubinger, R.M.; Longley, D.B.; Scott, C.J. Repurposing of Cetuximab in antibody-directed chemotherapy-loaded nanoparticles in EGFR therapy-resistant pancreatic tumours. Nanoscale 2019, 11, 20261–20273. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Gao, F.; Wu, W.; Qiu, W.X.; Zhang, L.; Li, R.; Zhuang, Z.N.; Yu, W.; Cheng, H.; Zhang, X.Z. Enzyme-Driven Membrane-Targeted Chimeric Peptide for Enhanced Tumor Photodynamic Immunotherapy. ACS Nano 2019, 13, 11249–11262. [Google Scholar] [CrossRef]
- Roncato, F.; Rruga, F.; Porcù, E.; Casarin, E.; Ronca, R.; Maccarinelli, F.; Realdon, N.; Basso, G.; Alon, R.; Viola, G.; et al. Improvement and extension of anti-EGFR targeting in breast cancer therapy by integration with the Avidin-Nucleic-Acid-Nano-Assemblies. Nat. Commun. 2018, 9, 4070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, D.; Xu, X.; Iqbal, M.Z.; Zhao, Q.; Zhao, R.; Farheen, J.; Zhang, Q.; Zhang, P.; Kong, X. siRNA-Loaded Hydroxyapatite Nanoparticles for KRAS Gene Silencing in Anti-Pancreatic Cancer Therapy. Pharmaceutics 2021, 13, 1428. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lin, Z.I.; Yang, J.; Liu, G.L.; Hu, Z.; Huang, H.; Li, X.; Liu, Q.; Ma, M.; Xu, Z.; et al. Carbon Dioxide-Derived Biodegradable and Cationic Polycarbonates as a New siRNA Carrier for Gene Therapy in Pancreatic Cancer. Nanomaterials 2021, 11, 2312. [Google Scholar] [CrossRef]
- Sreedurgalakshmi, K.; Srikar, R.; Harikrishnan, K.; Srinivasan, L.; Rajkumari, R. Cetuximab-siRNA Conjugate Linked Through Cationized Gelatin Knocks Down KRAS G12C Mutation in NSCLC Sensitizing the Cells Toward Gefitinib. Technol. Cancer Res. Treat. 2021, 20, 15330338211041453. [Google Scholar] [CrossRef]
- Wu, L.P.; Wang, D.; Li, Z. Grand challenges in nanomedicine. Mater. Sci. Eng. C Mater. Biol. Appl. 2020, 106, 110302. [Google Scholar] [CrossRef]
- Yang, C.; Merlin, D. Challenges to Safe Nanomedicine Treatment. Nanomaterials 2023, 13, 1171. [Google Scholar] [CrossRef]
- Metselaar, J.M.; Lammers, T. Challenges in nanomedicine clinical translation. Drug Deliv. Transl. Res. 2020, 10, 721–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]







| Type | Advantages | Disadvantages | Diagram |
|---|---|---|---|
| Liposomes | Biocompatibility. Biodegradability. Co-loading of drugs with different polarities. | Difficult and high production costs. Storage stability. Leakage of drugs. |  |
| Solid Lipid Nanoparticles | Biocompatibility. Biodegradability. Simultaneous loading of drugs with different polarities. | Low loading of hydrophilic drugs. Long-term stability (crystallization). |  |
| Polymeric Nanoparticles | Controlled release properties. High stability. | Difficulties in scale-up. Potentially more toxic. Low loading capacity. |  |
| Polymeric Micelles | Co-loading of drugs with different polarities. Easy and cheap preparation. | Loading limitations to some drugs. |  |
| Dendrimers | High drug loading. Versatility of surface functionalization. | High toxicity. Hemolytic properties. Non-biodegradability. |  |
| Extracellular Vesicles | Biocompatibility. Biodegradability. Stability. Versatility of drug loading. Versatility of surface functionalization. | Difficult and high production costs. Heterogeneity in production. Difficulties in scale-up. |  |
| Protein and Peptide Nanoparticles | Biocompatibility. Biodegradability. Versatility of functionalization. | High production cost. Stability. |  |
| Inorganic Nanoparticles | Versatility of surface functionalization. Stimuli-responsiveness. | Long-term toxicity. Stability. Non-biodegradability. |  |
| Nucleic acid Nanoparticles | Biocompatibility. Biodegradability. Versatility of functionalization. | Stability. High production cost. |  |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andrade, F.; German-Cortés, J.; Montero, S.; Carcavilla, P.; Baranda-Martínez-Abascal, D.; Moltó-Abad, M.; Seras-Franzoso, J.; Díaz-Riascos, Z.V.; Rafael, D.; Abasolo, I. The Nanotechnology-Based Approaches against Kirsten Rat Sarcoma-Mutated Cancers. Pharmaceutics 2023, 15, 1686. https://doi.org/10.3390/pharmaceutics15061686
Andrade F, German-Cortés J, Montero S, Carcavilla P, Baranda-Martínez-Abascal D, Moltó-Abad M, Seras-Franzoso J, Díaz-Riascos ZV, Rafael D, Abasolo I. The Nanotechnology-Based Approaches against Kirsten Rat Sarcoma-Mutated Cancers. Pharmaceutics. 2023; 15(6):1686. https://doi.org/10.3390/pharmaceutics15061686
Chicago/Turabian StyleAndrade, Fernanda, Júlia German-Cortés, Sara Montero, Pilar Carcavilla, Diego Baranda-Martínez-Abascal, Marc Moltó-Abad, Joaquín Seras-Franzoso, Zamira Vanessa Díaz-Riascos, Diana Rafael, and Ibane Abasolo. 2023. "The Nanotechnology-Based Approaches against Kirsten Rat Sarcoma-Mutated Cancers" Pharmaceutics 15, no. 6: 1686. https://doi.org/10.3390/pharmaceutics15061686