
Mesenchymal Stem Cell Membrane-Coated TPCS2a-Loaded Nanoparticles for Breast Cancer Photodynamic Therapy
 , , and
, , and
Abstract
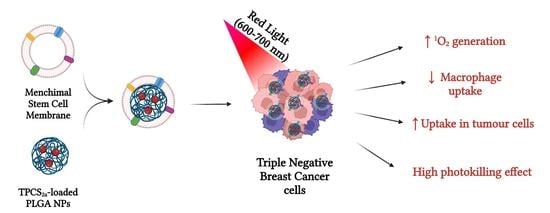
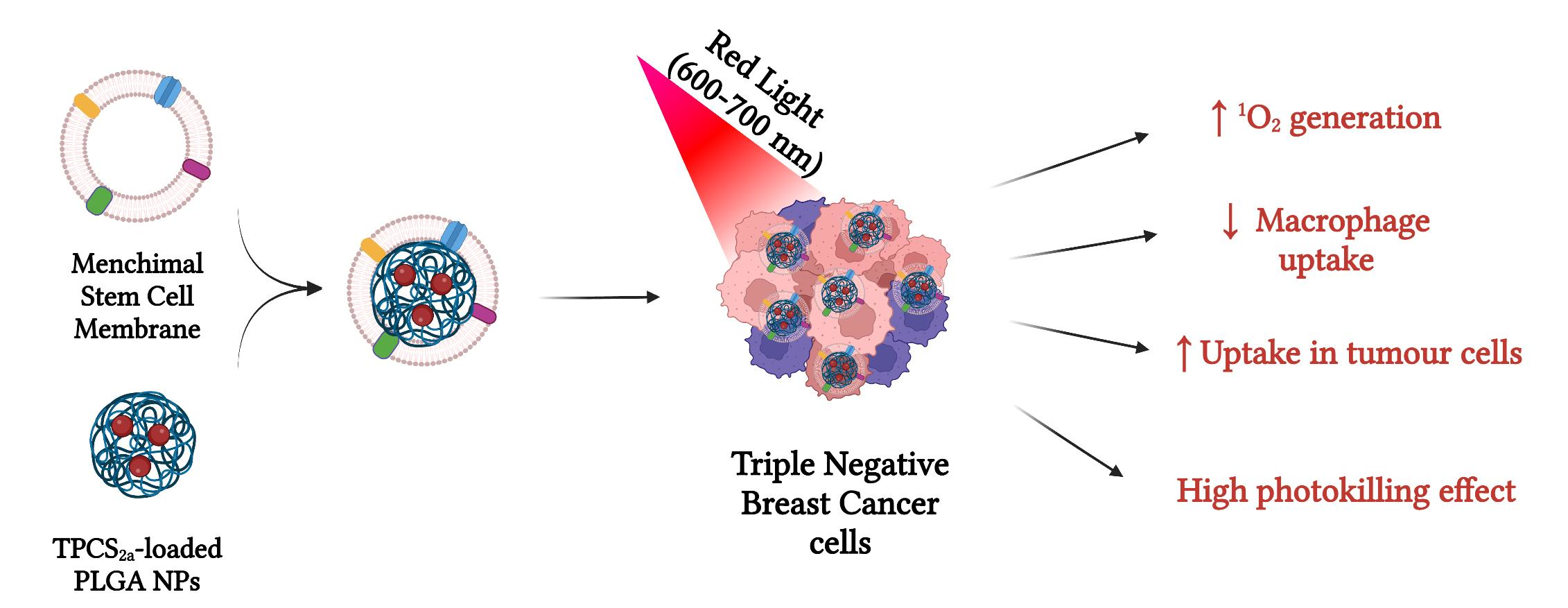
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Cell Lines
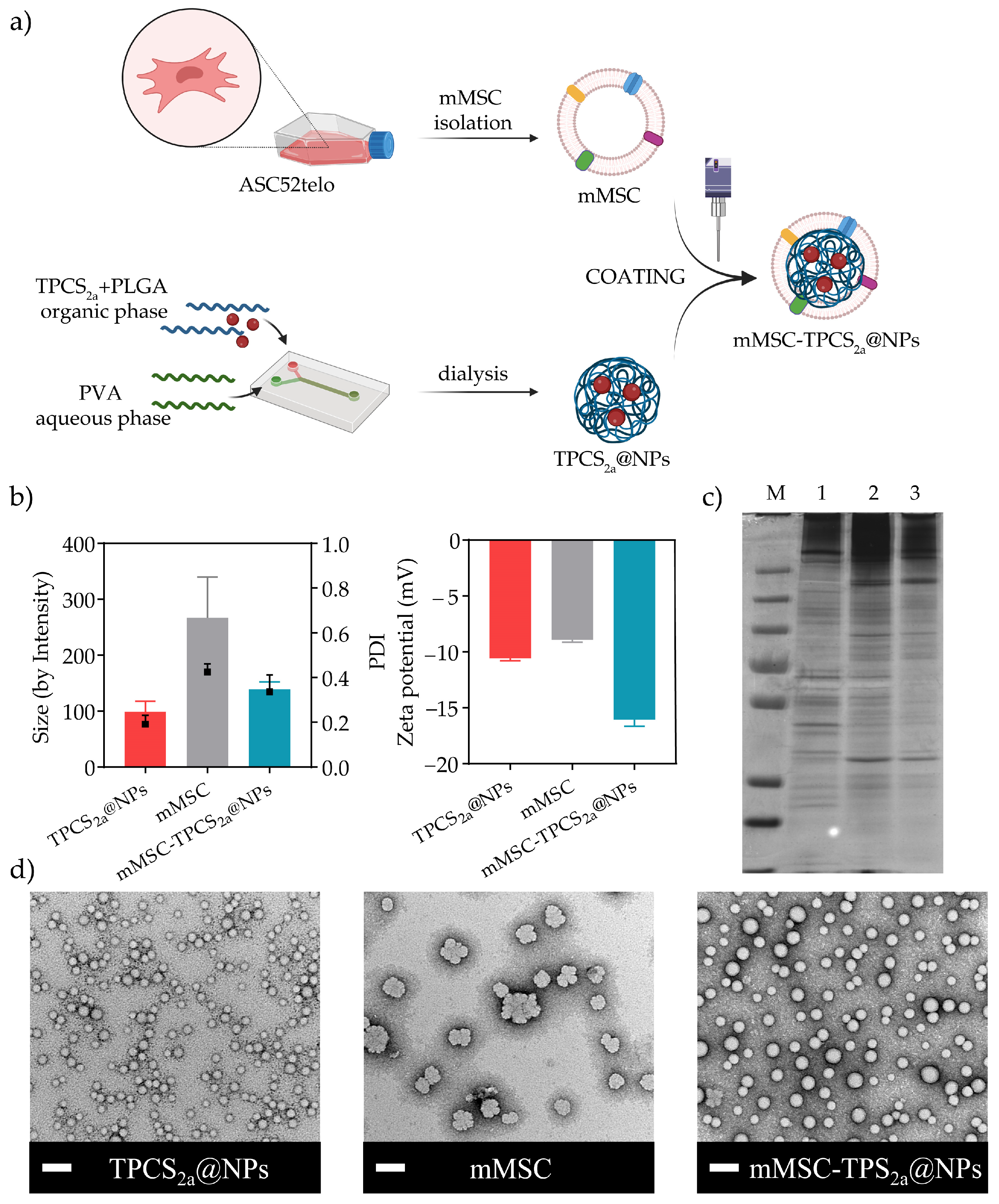
2.3. Synthesis of NPs
2.3.1. Synthesis of TPCS2a-Loaded PLGA NPs (TPCS2a@NPs)
2.3.2. Isolation of Mesenchymal Stem Cell-Derived Plasma Membranes (mMSC)
2.3.3. Preparation of Biomimetic TPCS2a NPs (mMSC-TPCS2a@NPs)
2.4. Characterization of TPCS2a@NPs and mMSC-TPCS2a@NPs
2.4.1. Size Distribution and Zeta Potential Analyses
2.4.2. Freeze-Drying Studies
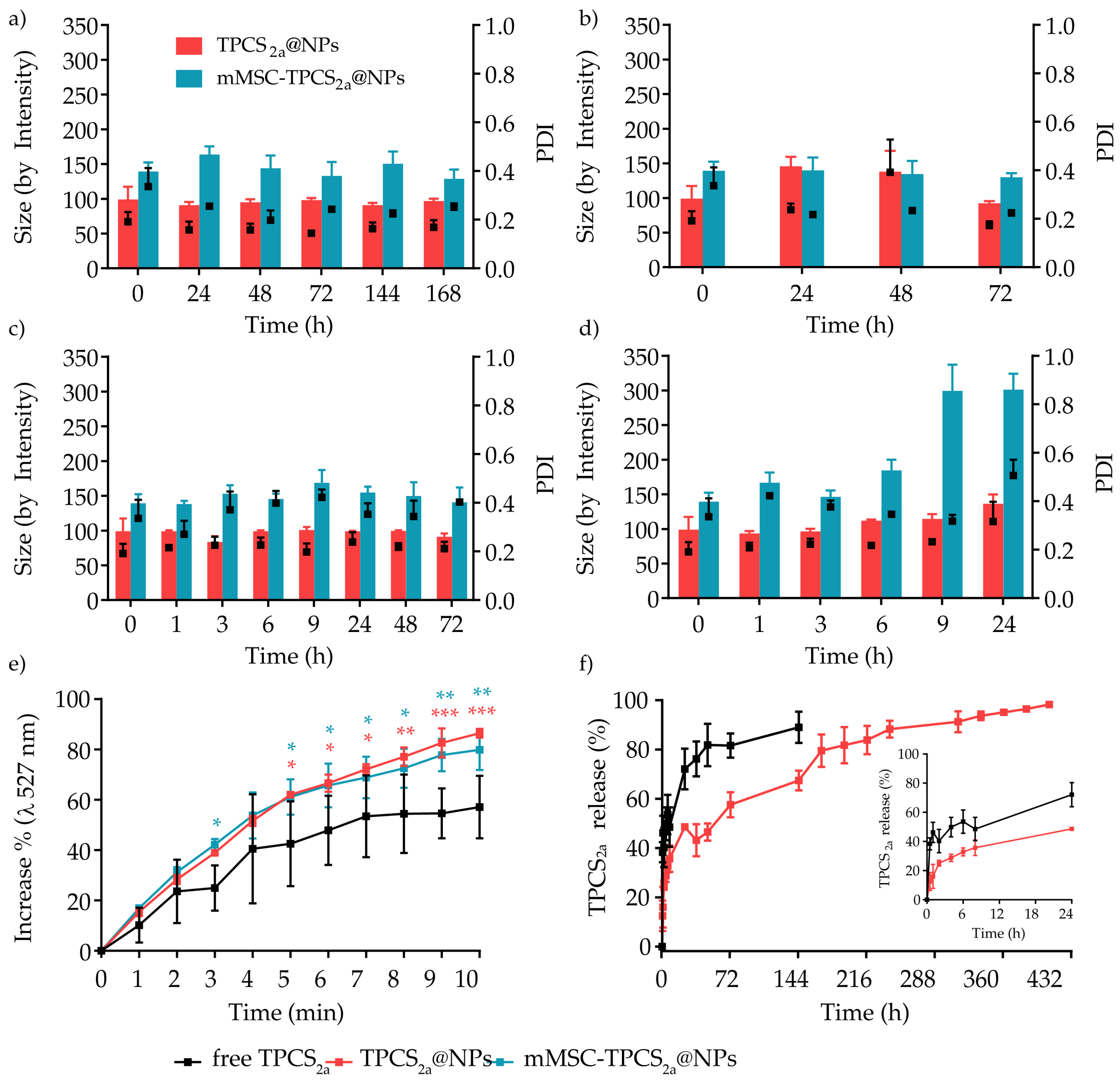
2.4.3. Release Kinetic of TPCS2a from TPCS2a@NPs
2.4.4. SDS-PAGE
2.4.5. Transmission Electron Microscopy (TEM)
2.4.6. Stability Studies
2.4.7. Spectroscopic Studies
2.4.8. Singlet Oxygen (1O2) Generation
2.5. NP Capture by Human Macrophages
2.5.1. Isolation and Differentiation of Monocytes from Buffy Coat
2.5.2. NP Internalization by Human Macrophages
2.6. In Vitro Studies on BC Cell Monolayers
2.6.1. Uptake Studies
2.6.2. Confocal Microscopy Uptake and Localization Studies
2.6.3. Dark and Phototoxicity Studies
2.7. In Vitro Studies on Spheroids
2.7.1. Generation of Multicellular Tumor Spheroids
2.7.2. Dark and Phototoxicity Studies
2.7.3. Uptake Studies
2.8. Statistical Analysis
3. Results and Discussion
3.1. Preparation and Characterization of mMSC-TPCS2a@NPs
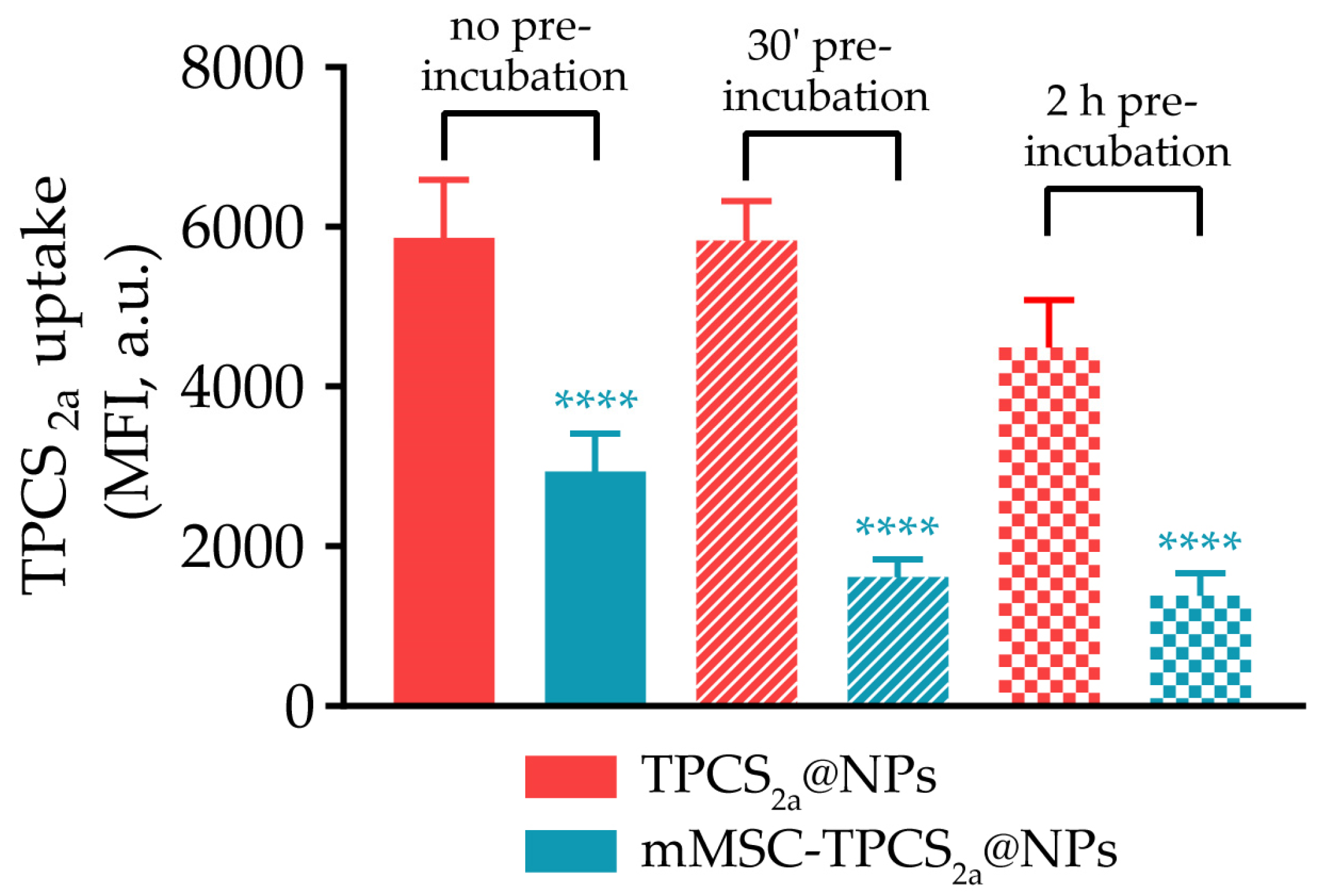
3.2. In Vitro NP Capture by Human Macrophages
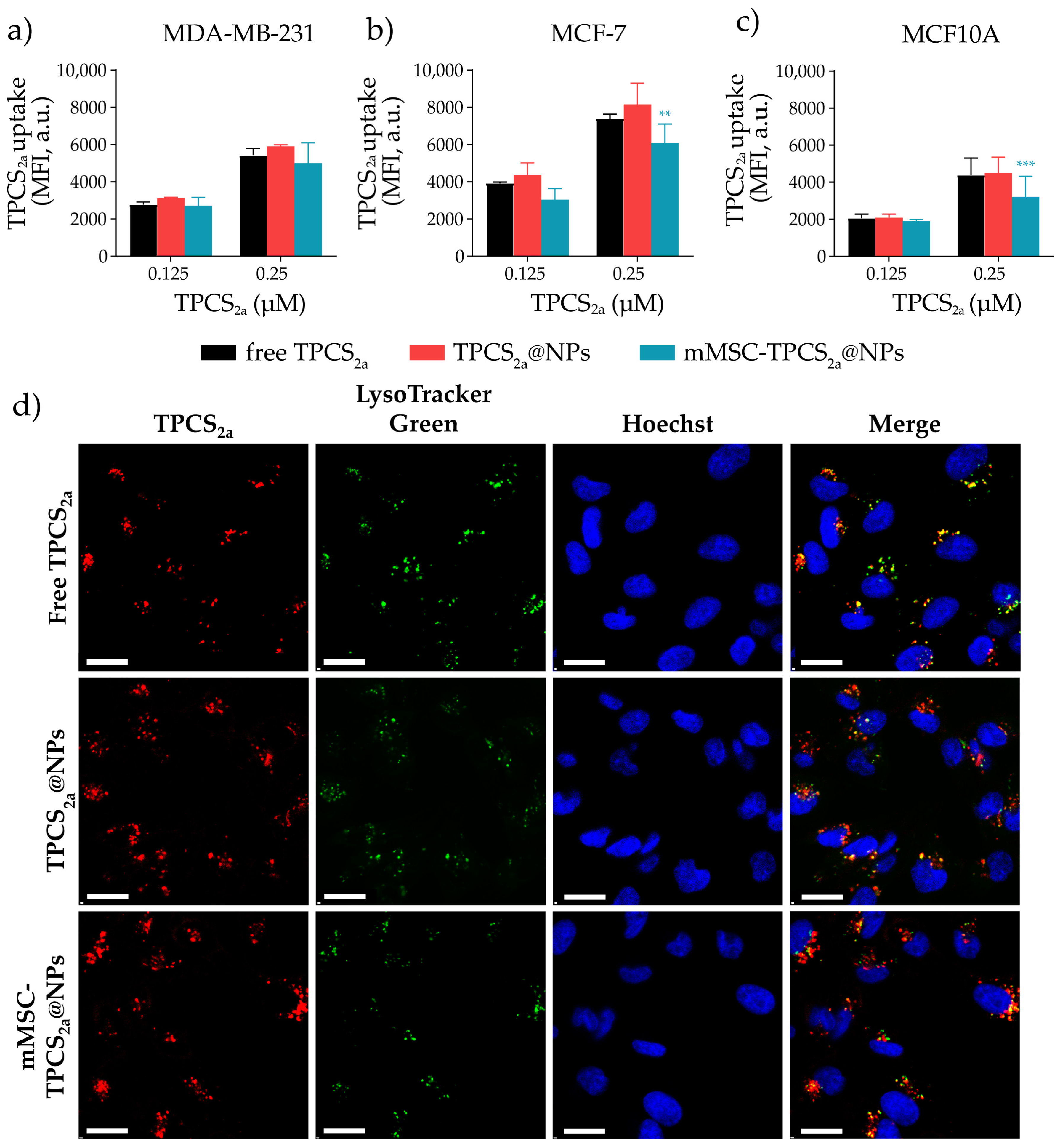
3.3. In Vitro Uptake and Localization Studies in Monolayered Breast Cancer Cells
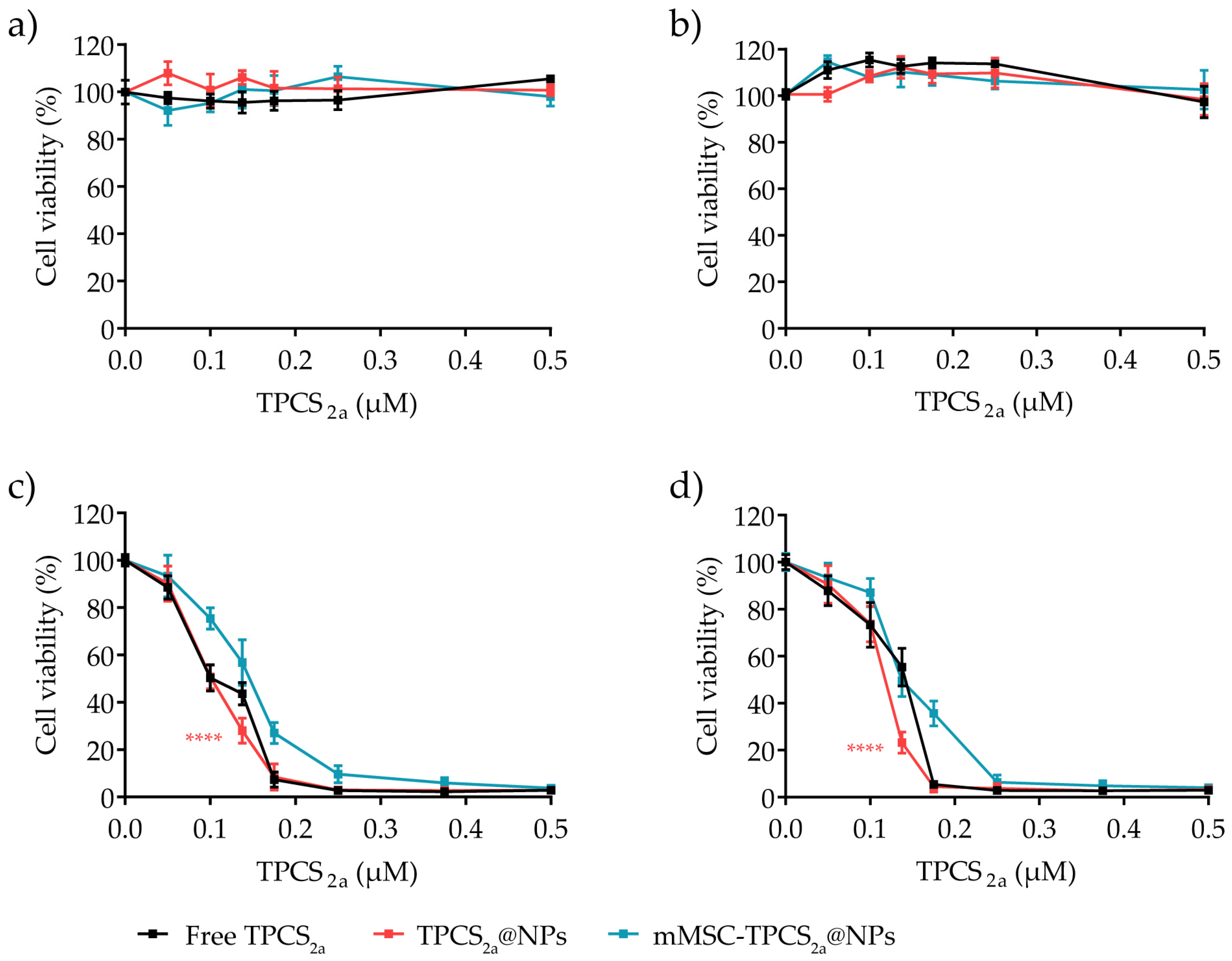
3.4. In Vitro Cytotoxicity in Monolayered Breast Cancer Cells
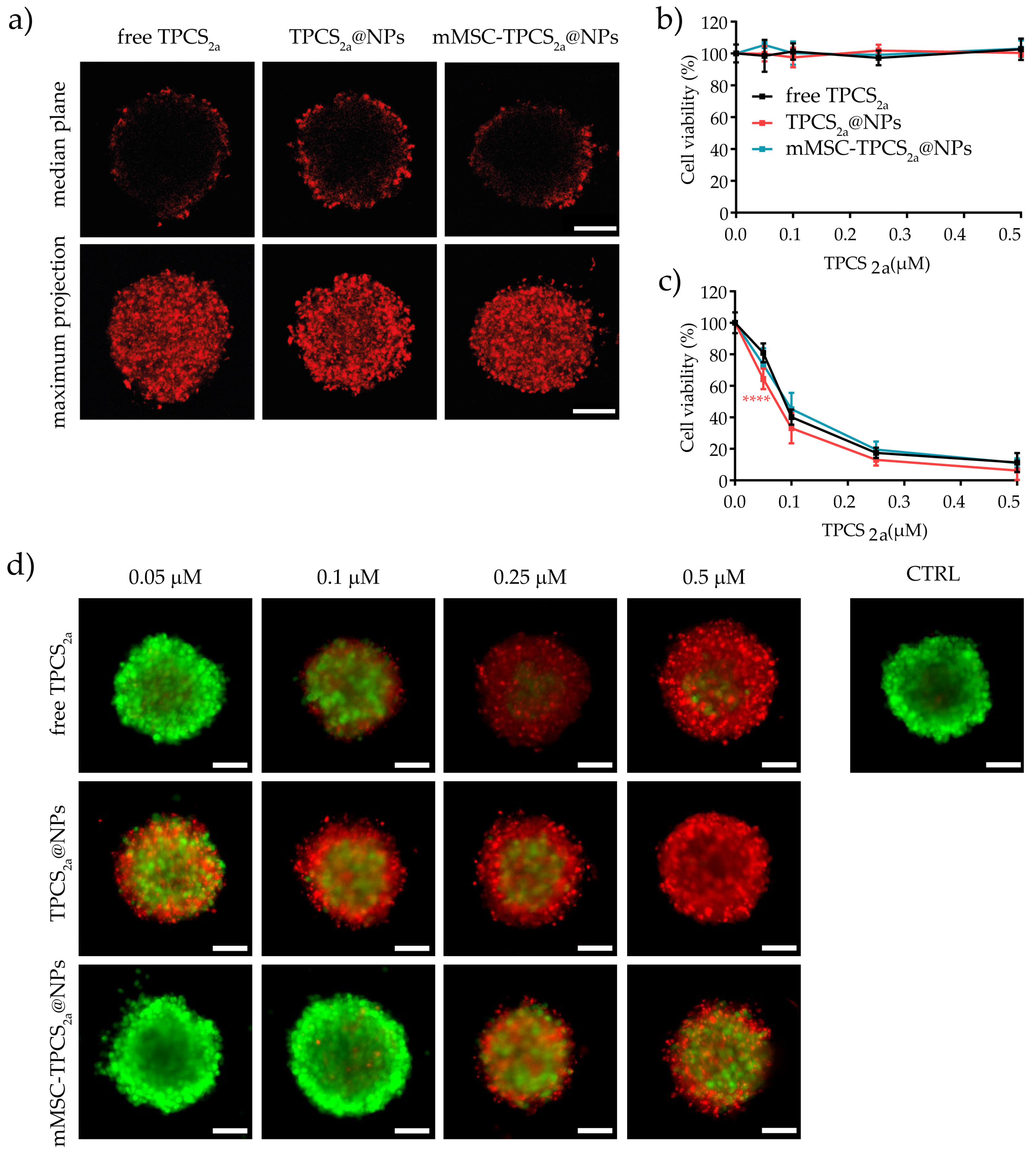
3.5. In Vitro Uptake and Cytotoxicity in 3D Spheroids
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Kassam, F.; Enright, K.; Dent, R.; Dranitsaris, G.; Myers, J.; Flynn, C.; Fralick, M.; Kumar, R.; Clemons, M. Survival Outcomes for Patients with Metastatic Triple-Negative Breast Cancer: Implications for Clinical Practice and Trial Design. Clin. Breast Cancer 2009, 9, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D.; et al. Photodynamic Therapy of Cancer: An Update. CA Cancer J. Clin. 2011, 61, 250–281. [Google Scholar] [CrossRef] [PubMed]
- Alsaab, H.O.; Alghamdi, M.S.; Alotaibi, A.S.; Alzhrani, R.; Alwuthaynani, F.; Althobaiti, Y.S.; Almalki, A.H.; Sau, S.; Iyer, A.K. Progress in Clinical Trials of Photodynamic Therapy for Solid Tumors and the Role of Nanomedicine. Cancers 2020, 12, 2793. [Google Scholar] [CrossRef]
- Ostańska, E.; Aebisher, D.; Bartusik-Aebisher, D. The Potential of Photodynamic Therapy in Current Breast Cancer Treatment Methodologies. Biomed. Pharmacother. 2021, 137, 111302. [Google Scholar] [CrossRef]
- Cuenca, R.E.; Allison, R.R.; Sibata, C.; Downie, G.H. Breast Cancer with Chest Wall Progression: Treatment with Photodynamic Therapy. Ann. Surg. Oncol. 2004, 11, 322–327. [Google Scholar] [CrossRef]
- Khan, S.A.; Dougherty, T.J.; Mang, T.S. An Evaluation of Photodynamic Therapy in the Management of Cutaneous Metastases of Breast Cancer. Eur. J. Cancer 1993, 29, 1686–1690. [Google Scholar] [CrossRef]
- Wyss, P.; Schwarz, V.; Dobler-Girdziunaite, D.; Hornung, R.; Walt, H.; Degen, A.; Fehr, M. Photodynamic Therapy of Locoregional Breast Cancer Recurrences Using a Chlorin-Type Photosensitizer. Int. J. Cancer 2001, 93, 720–724. [Google Scholar] [CrossRef]
- Banerjee, S.M.; El-Sheikh, S.; Malhotra, A.; Mosse, C.A.; Parker, S.; Williams, N.R.; MacRobert, A.J.; Hamoudi, R.; Bown, S.G.; Keshtgar, M.R.S. Photodynamic Therapy in Primary Breast Cancer. J. Clin. Med. 2020, 9, 483. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.M.; MacRobert, A.J.; Mosse, C.A.; Periera, B.; Bown, S.G.; Keshtgar, M.R.S. Photodynamic Therapy: Inception to Application in Breast Cancer. Breast 2017, 31, 105–113. [Google Scholar] [CrossRef] [Green Version]
- Brodin, N.P.; Guha, C.; Tomé, W.A. Photodynamic Therapy and Its Role in Combined Modality Anticancer Treatment. Technol. Cancer Res. Treat. 2015, 14, 355–368. [Google Scholar] [CrossRef] [PubMed]
- van Straten, D.; Mashayekhi, V.; de Bruijn, H.S.; Oliveira, S.; Robinson, D.J. Oncologic Photodynamic Therapy: Basic Principles, Current Clinical Status and Future Directions. Cancers 2017, 9, 19. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, D.K.; Fong, L.S.; Zhang, Y. Nanoparticles in Photodynamic Therapy: An Emerging Paradigm. Adv. Drug. Deliv. Rev. 2008, 60, 1627–1637. [Google Scholar] [CrossRef] [PubMed]
- Moret, F.; Reddi, E. Strategies for Optimizing the Delivery to Tumors of Macrocyclic Photosensitizers Used in Photodynamic Therapy (PDT). J. Porphyr. Phthalocyanines 2017, 21, 239–256. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Lee, Y.-K.; Park, I.-K.; Hwang, S.R. Current Limitations and Recent Progress in Nanomedicine for Clinically Available Photodynamic Therapy. Biomedicines 2021, 9, 85. [Google Scholar] [CrossRef] [PubMed]
- Menilli, L.; Milani, C.; Reddi, E.; Moret, F. Overview of Nanoparticle-Based Approaches for the Combination of Photodynamic Therapy (PDT) and Chemotherapy at the Preclinical Stage. Cancers 2022, 14, 4462. [Google Scholar] [CrossRef] [PubMed]
- Wu, J. The Enhanced Permeability and Retention (EPR) Effect: The Significance of the Concept and Methods to Enhance Its Application. J. Pers. Med. 2021, 11, 771. [Google Scholar] [CrossRef]
- Bottini, M.; Foldvari, M. Targeted Nanosystems as Therapeutic and Diagnostic Tools: The Beautiful Voyage of Nanomedicine. Nanomedicine 2016, 12, 253–254. [Google Scholar] [CrossRef]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C.W. Analysis of Nanoparticle Delivery to Tumours. Nat. Rev. Mater. 2016, 1, 16014. [Google Scholar] [CrossRef]
- Meyer, R.A.; Sunshine, J.C.; Green, J.J. Biomimetic Particles as Therapeutics. Trends Biotechnol. 2015, 33, 514–524. [Google Scholar] [CrossRef] [Green Version]
- Sushnitha, M.; Evangelopoulos, M.; Tasciotti, E.; Taraballi, F. Cell Membrane-Based Biomimetic Nanoparticles and the Immune System: Immunomodulatory Interactions to Therapeutic Applications. Front. Bioeng. Biotechnol. 2020, 8, 627. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Yan, C. Research Progress of Cell Membrane Biomimetic Nanoparticles for Tumor Therapy. Nanoscale Res. Lett. 2022, 17, 36. [Google Scholar] [CrossRef] [PubMed]
- Prajapati, S.; Hinchliffe, T.; Roy, V.; Shah, N.; Jones, C.N.; Obaid, G. Biomimetic Nanotechnology: A Natural Path Forward for Tumor-Selective and Tumor-Specific NIR Activable Photonanomedicines. Pharmaceutics 2021, 13, 786. [Google Scholar] [CrossRef]
- Min, H.; Wang, J.; Qi, Y.; Zhang, Y.; Han, X.; Xu, Y.; Xu, J.; Li, Y.; Chen, L.; Cheng, K.; et al. Biomimetic Metal–Organic Framework Nanoparticles for Cooperative Combination of Antiangiogenesis and Photodynamic Therapy for Enhanced Efficacy. Adv. Mater. 2019, 31, 1808200. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Wang, Y.; Wang, B.; Chen, Q.; Wan, G.; Yang, X.; Zhang, J.; Zhang, L.; Li, C.; Wang, Y. Homologous-Targeting Biomimetic Nanoparticles for Photothermal Therapy and Nrf2-SiRNA Amplified Photodynamic Therapy against Oral Tongue Squamous Cell Carcinoma. Chem. Eng. J. 2020, 388, 124268. [Google Scholar] [CrossRef]
- Wu, M.; Ling, W.; Wei, J.; Liao, R.; Sun, H.; Li, D.; Zhao, Y.; Zhao, L. Biomimetic Photosensitizer Nanocrystals Trigger Enhanced Ferroptosis for Improving Cancer Treatment. J. Control. Release 2022, 352, 1116–1133. [Google Scholar] [CrossRef]
- Zhang, Y.; Ma, N.; Luo, C.; Zhu, J.; Bao, C. Photosensitizer-Loaded Cell Membrane Biomimetic Nanoparticles for Enhanced Tumor Synergetic Targeted Therapy. RSC Adv. 2020, 10, 9378–9386. [Google Scholar] [CrossRef] [Green Version]
- Jerjes, W.; Theodossiou, T.A.; Hirschberg, H.; Høgset, A.; Weyergang, A.; Selbo, P.K.; Hamdoon, Z.; Hopper, C.; Berg, K. Photochemical Internalization for Intracellular Drug Delivery. From Basic Mechanisms to Clinical Research. J. Clin. Med. 2020, 9, 528. [Google Scholar] [CrossRef] [Green Version]
- Rezvantalab, S.; Keshavarz Moraveji, M. Microfluidic Assisted Synthesis of PLGA Drug Delivery Systems. RSC Adv. 2019, 9, 2055–2072. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.-M.; Cheng, T.-L.; Roffler, S.R. Polyethylene Glycol Immunogenicity: Theoretical, Clinical, and Practical Aspects of Anti-Polyethylene Glycol Antibodies. ACS Nano 2021, 15, 14022–14048. [Google Scholar] [CrossRef]
- Fang, R.H.; Gao, W.; Zhang, L. Targeting Drugs to Tumours Using Cell Membrane-Coated Nanoparticles. Nat. Rev. Clin. Oncol. 2023, 20, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Cho, S.-G. Clinical Applications of Mesenchymal Stem Cells. Korean J. Intern. Med. 2013, 28, 387. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Lin, Z.; Jurado-Sánchez, B.; Lin, X.; Wu, Z.; He, Q. Stem Cell Membrane-Coated Nanogels for Highly Efficient In Vivo Tumor Targeted Drug Delivery. Small 2016, 12, 4056–4062. [Google Scholar] [CrossRef]
- Walker, J.M. The Bicinchoninic Acid (BCA) Assay for Protein Quantitation. In Basic. Protein and Peptide Protocols; Humana Press: Totowa, NJ, USA, 1994; pp. 5–8. [Google Scholar]
- Menck, K.; Behme, D.; Pantke, M.; Reiling, N.; Binder, C.; Pukrop, T.; Klemm, F. Isolation of Human Monocytes by Double Gradient Centrifugation and Their Differentiation to Macrophages in Teflon-Coated Cell Culture Bags. J. Vis. Exp. 2014, 91, e51554. [Google Scholar] [CrossRef] [Green Version]
- Rapozzi, V.; Moret, F.; Menilli, L.; Guerrini, A.; Tedesco, D.; Naldi, M.; Bartolini, M.; Gani, M.; Zorzet, S.; Columbaro, M.; et al. HSA-Binding Prodrugs-Based Nanoparticles Endowed with Chemo and Photo-Toxicity against Breast Cancer. Cancers 2022, 14, 877. [Google Scholar] [CrossRef]
- Gaio, E.; Conte, C.; Esposito, D.; Miotto, G.; Quaglia, F.; Moret, F.; Reddi, E. Co-Delivery of Docetaxel and Disulfonate Tetraphenyl Chlorin in One Nanoparticle Produces Strong Synergism between Chemo- and Photodynamic Therapy in Drug-Sensitive and -Resistant Cancer Cells. Mol. Pharm. 2018, 15, 4599–4611. [Google Scholar] [CrossRef]
- Gaio, E.; Conte, C.; Esposito, D.; Reddi, E.; Quaglia, F.; Moret, F. CD44 Targeting Mediated by Polymeric Nanoparticles and Combination of Chlorine TPCS2a-PDT and Docetaxel-Chemotherapy for Efficient Killing of Breast Differentiated and Stem Cancer Cells In Vitro. Cancers 2020, 12, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, T.L.; Rodriguez-Lorenzo, L.; Hirsch, V.; Balog, S.; Urban, D.; Jud, C.; Rothen-Rutishauser, B.; Lattuada, M.; Petri-Fink, A. Nanoparticle Colloidal Stability in Cell Culture Media and Impact on Cellular Interactions. Chem. Soc. Rev. 2015, 44, 6287–6305. [Google Scholar] [CrossRef] [Green Version]
- Inam, W.; Bhadane, R.; Akpolat, R.N.; Taiseer, R.A.; Filippov, S.K.; Salo-Ahen, O.M.H.; Rosenholm, J.M.; Zhang, H. Interactions between Polymeric Nanoparticles and Different Buffers as Investigated by Zeta Potential Measurements and Molecular Dynamics Simulations. VIEW 2022, 3, 20210009. [Google Scholar] [CrossRef]
- Lilletvedt, M.; Tønnesen, H.H.; Høgset, A.; Sande, S.A.; Kristensen, S. Evaluation of Physicochemical Properties and Aggregation of the Photosensitizers TPCS2a and TPPS2a in Aqueous Media. Pharmazie 2011, 66, 325–333. [Google Scholar]
- Lilletvedt, M.; Tønnesen, H.H.; Høgset, A.; Nardo, L.; Kristensen, S. Physicochemical Characterization of the Photosensitizers TPCS2a and TPPS2a 1. Spectroscopic Evaluation of Drug--Solvent Interactions. Pharmazie 2010, 65, 588–595. [Google Scholar] [PubMed]
- Bruschi, M.L. Mathematical Models of Drug Release. In Strategies to Modify the Drug. Release from Pharmaceutical Systems; Elsevier: Amsterdam, The Netherlands, 2015; pp. 63–86. [Google Scholar]
- Costa, P.; Sousa Lobo, J.M. Modeling and Comparison of Dissolution Profiles. Eur. J. Pharm. Sci. 2001, 13, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Boraschi, D.; Italiani, P.; Palomba, R.; Decuzzi, P.; Duschl, A.; Fadeel, B.; Moghimi, S.M. Nanoparticles and Innate Immunity: New Perspectives on Host Defence. Semin. Immunol. 2017, 34, 33–51. [Google Scholar] [CrossRef] [PubMed]
- Marshall, S.K.; Angsantikul, P.; Pang, Z.; Nasongkla, N.; Hussen, R.S.D.; Thamphiwatana, S.D. Biomimetic Targeted Theranostic Nanoparticles for Breast Cancer Treatment. Molecules 2022, 27, 6473. [Google Scholar] [CrossRef]
- Li, R.; He, Y.; Zhang, S.; Qin, J.; Wang, J. Cell Membrane-Based Nanoparticles: A New Biomimetic Platform for Tumor Diagnosis and Treatment. Acta Pharm. Sin. B 2018, 8, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Fontana, F.; Lindstedt, H.; Correia, A.; Chiaro, J.; Kari, O.K.; Ndika, J.; Alenius, H.; Buck, J.; Sieber, S.; Mäkilä, E.; et al. Influence of Cell Membrane Wrapping on the Cell–Porous Silicon Nanoparticle Interactions. Adv. Healthc. Mater. 2020, 9, 2000529. [Google Scholar] [CrossRef] [PubMed]
- Moan, J.; Berg, K.; Kvam, E.; Western, A.; Malik, Z.; Rück, A.; Schneckenburger, H. Intracellular Localization of Photosensitizers. In Ciba Foundation Symposium 146-Photosensitizing Compounds: Their Chemistry, Biology and Clinical Use: Photosensitizing Compounds: Their Chemistry, Biology and Clinical Use: Ciba Foundation Symposium 146; John Wiley & Sons, Ltd.: Chichester, UK, 2007; pp. 95–111. [Google Scholar]
- Oliveira, C.S.; Turchiello, R.; Kowaltowski, A.J.; Indig, G.L.; Baptista, M.S. Major Determinants of Photoinduced Cell Death: Subcellular Localization versus Photosensitization Efficiency. Free. Radic. Biol. Med. 2011, 51, 824–833. [Google Scholar] [CrossRef]
- Berg, K.; Nordstrand, S.; Selbo, P.K.; Tran, D.T.T.; Angell-Petersen, E.; Høgset, A. Disulfonated Tetraphenyl Chlorin (TPCS2a), a Novel Photosensitizer Developed for Clinical Utilization of Photochemical Internalization. Photochem. Photobiol. Sci. 2011, 10, 1637–1651. [Google Scholar] [CrossRef]
- Cuccato, N.; Nardo, L.; Kristensen, S.; Hjorth Tønnesen, H.; Lilletvedt Tovsen, M. Solubilization of the Chlorin TPCS 2a in the Presence of Pluronic® F127/Tween 80 Mixtures. Pharm. Dev. Technol. 2019, 24, 513–520. [Google Scholar] [CrossRef]
- Abrahamse, H.; Hamblin, M.R. New Photosensitizers for Photodynamic Therapy. Biochem. J. 2016, 473, 347–364. [Google Scholar] [CrossRef] [Green Version]
- Kwiatkowska, E.; Wojtala, M.; Gajewska, A.; Soszyński, M.; Bartosz, G.; Sadowska-Bartosz, I. Effect of 3-Bromopyruvate Acid on the Redox Equilibrium in Non-Invasive MCF-7 and Invasive MDA-MB-231 Breast Cancer Cells. J. Bioenerg. Biomembr. 2016, 48, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Theodossiou, T.A.; Olsen, C.E.; Jonsson, M.; Kubin, A.; Hothersall, J.S.; Berg, K. The Diverse Roles of Glutathione-Associated Cell Resistance against Hypericin Photodynamic Therapy. Redox Biol. 2017, 12, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Aguilar Cosme, J.R.; Gagui, D.C.; Green, N.H.; Bryant, H.E.; Claeyssens, F. In Vitro Low-Fluence Photodynamic Therapy Parameter Screening Using 3D Tumor Spheroids Shows That Fractionated Light Treatments Enhance Phototoxicity. ACS Biomater. Sci. Eng. 2021, 7, 5078–5089. [Google Scholar] [CrossRef] [PubMed]
- Gaio, E.; Scheglmann, D.; Reddi, E.; Moret, F. Uptake and Photo-Toxicity of Foscan®, Foslip® and Fospeg® in Multicellular Tumor Spheroids. J. Photochem. Photobiol. B. 2016, 161, 244–252. [Google Scholar] [CrossRef]
- Gaio, E.; Guerrini, A.; Ballestri, M.; Varchi, G.; Ferroni, C.; Martella, E.; Columbaro, M.; Moret, F.; Reddi, E. Keratin Nanoparticles Co-Delivering Docetaxel and Chlorin E6 Promote Synergic Interaction between Chemo- and Photo-Dynamic Therapies. J. Photochem. Photobiol. B 2019, 199, 111598. [Google Scholar] [CrossRef]
- Wang, Y.; Xie, Y.; Li, J.; Peng, Z.-H.; Sheinin, Y.; Zhou, J.; Oupický, D. Tumor-Penetrating Nanoparticles for Enhanced Anticancer Activity of Combined Photodynamic and Hypoxia-Activated Therapy. ACS Nano 2017, 11, 2227–2238. [Google Scholar] [CrossRef] [Green Version]
- Dahle, J. The Bystander Effect in Photodynamic Inactivation of Cells. Biochim. et Biophys. Acta (BBA)-Gen. Subj. 2000, 1475, 273–280. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | MDA-MB-231 IC50 (μM) | MCF-7 IC50 (μM) |
|---|---|---|
| Free TPCS2a | 0.1055 | 0.1315 |
| TPCS2a@NPs | 0.1009 | 0.1160 |
| mMSC-TPCS2a@NPs | 0.1392 | 0.1446 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Avancini, G.; Menilli, L.; Visentin, A.; Milani, C.; Mastrotto, F.; Moret, F. Mesenchymal Stem Cell Membrane-Coated TPCS2a-Loaded Nanoparticles for Breast Cancer Photodynamic Therapy. Pharmaceutics 2023, 15, 1654. https://doi.org/10.3390/pharmaceutics15061654
Avancini G, Menilli L, Visentin A, Milani C, Mastrotto F, Moret F. Mesenchymal Stem Cell Membrane-Coated TPCS2a-Loaded Nanoparticles for Breast Cancer Photodynamic Therapy. Pharmaceutics. 2023; 15(6):1654. https://doi.org/10.3390/pharmaceutics15061654
Chicago/Turabian StyleAvancini, Greta, Luca Menilli, Adele Visentin, Celeste Milani, Francesca Mastrotto, and Francesca Moret. 2023. "Mesenchymal Stem Cell Membrane-Coated TPCS2a-Loaded Nanoparticles for Breast Cancer Photodynamic Therapy" Pharmaceutics 15, no. 6: 1654. https://doi.org/10.3390/pharmaceutics15061654