

Targeted Anticancer Agent with Original Mode of Action Prepared by Supramolecular Assembly of Antibody Oligonucleotide Conjugates and Cationic Nanoparticles
 , ,
, ,  , and
, and 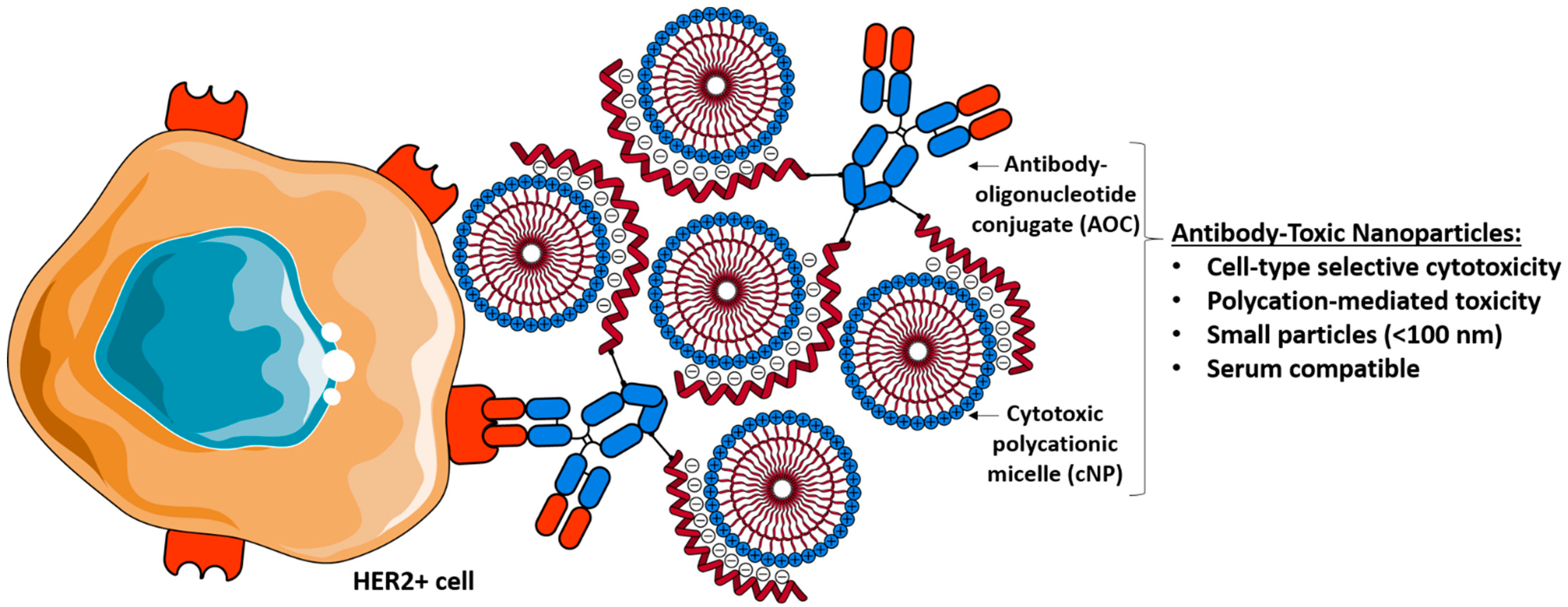{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Micelle Synthesis

2.2.2. Micelle Formulation and Photopolymerisation
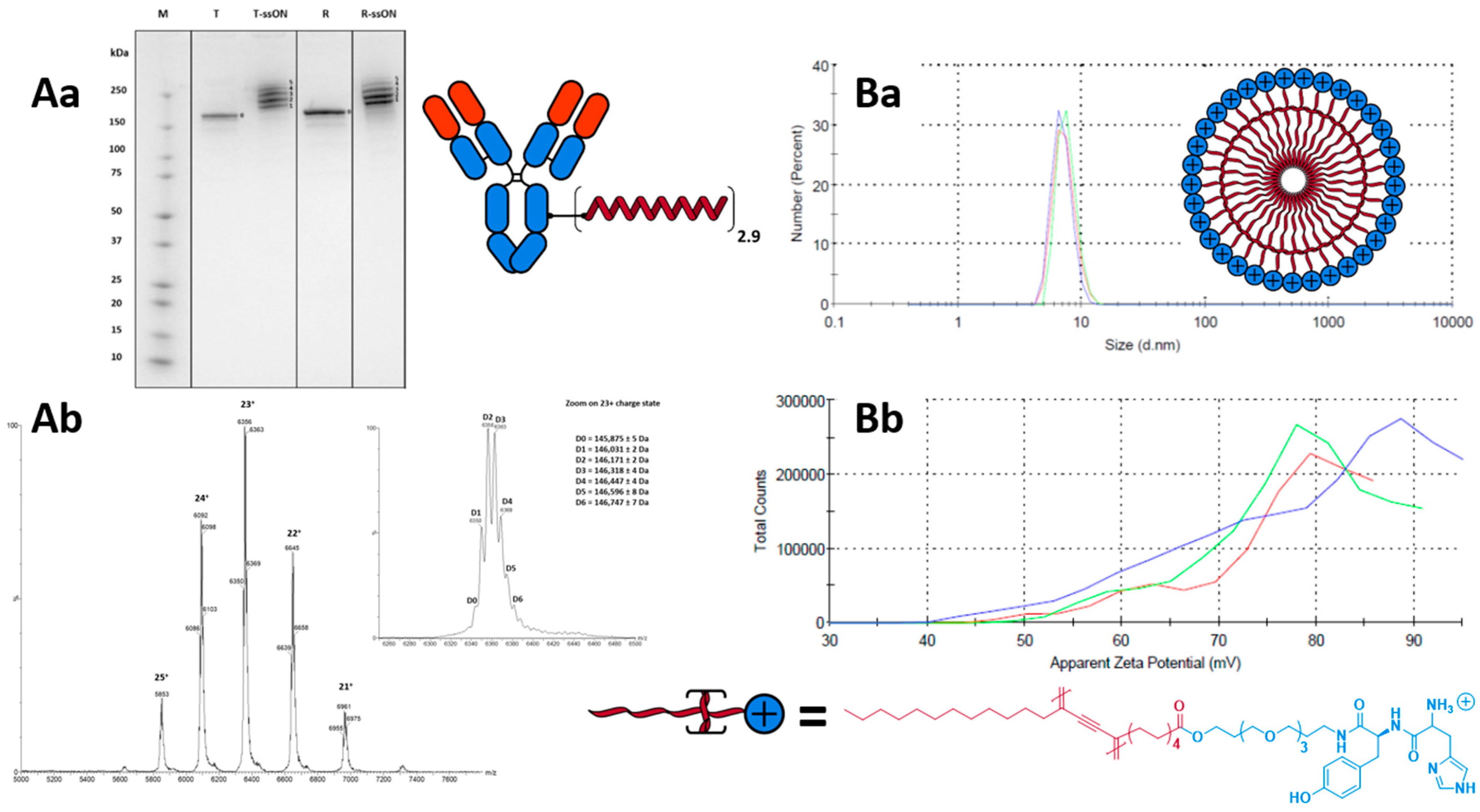
2.2.3. Conjugate Synthesis
2.2.4. Conjugate Characterisation
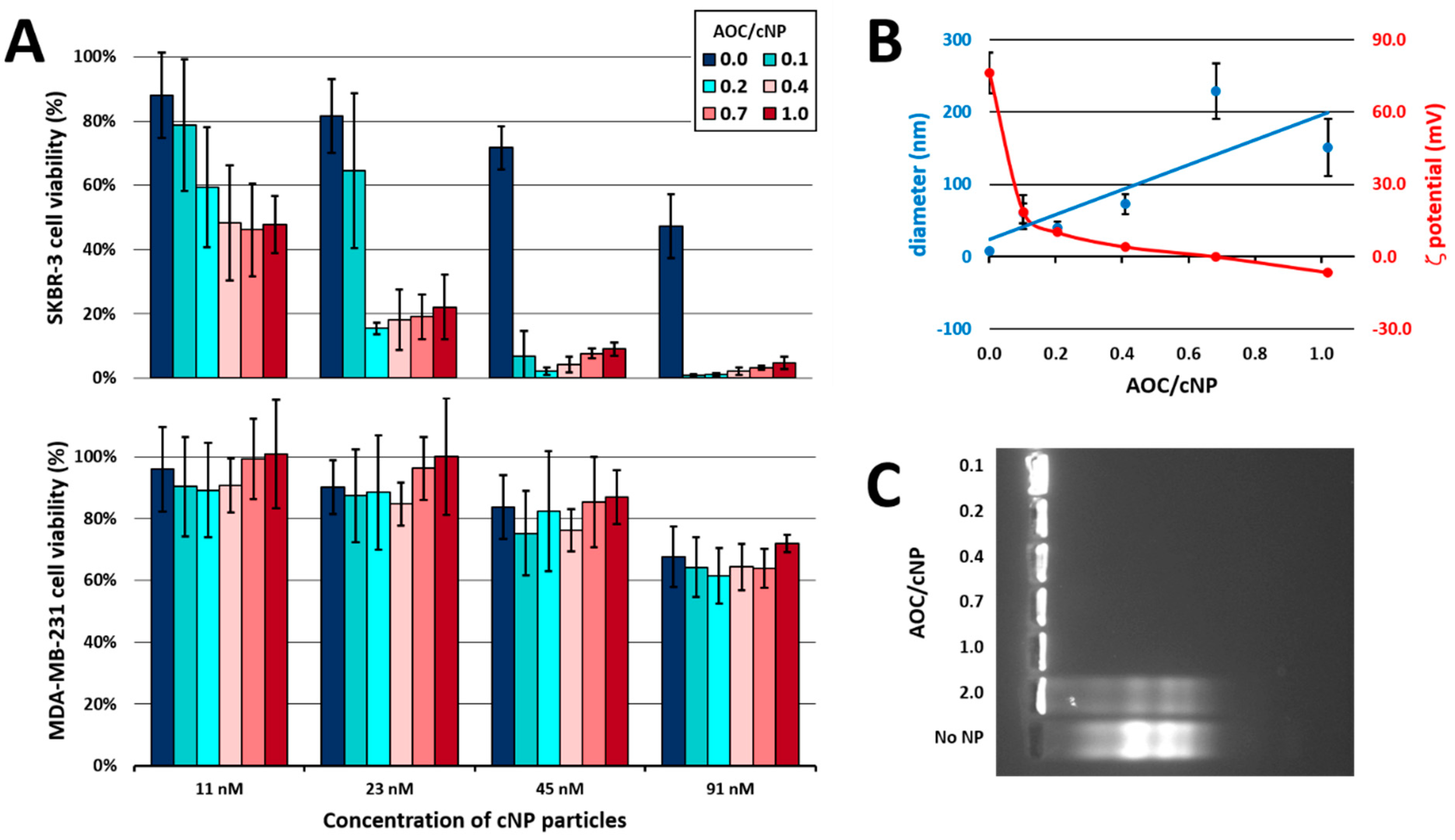
2.2.5. Complex Characterisation
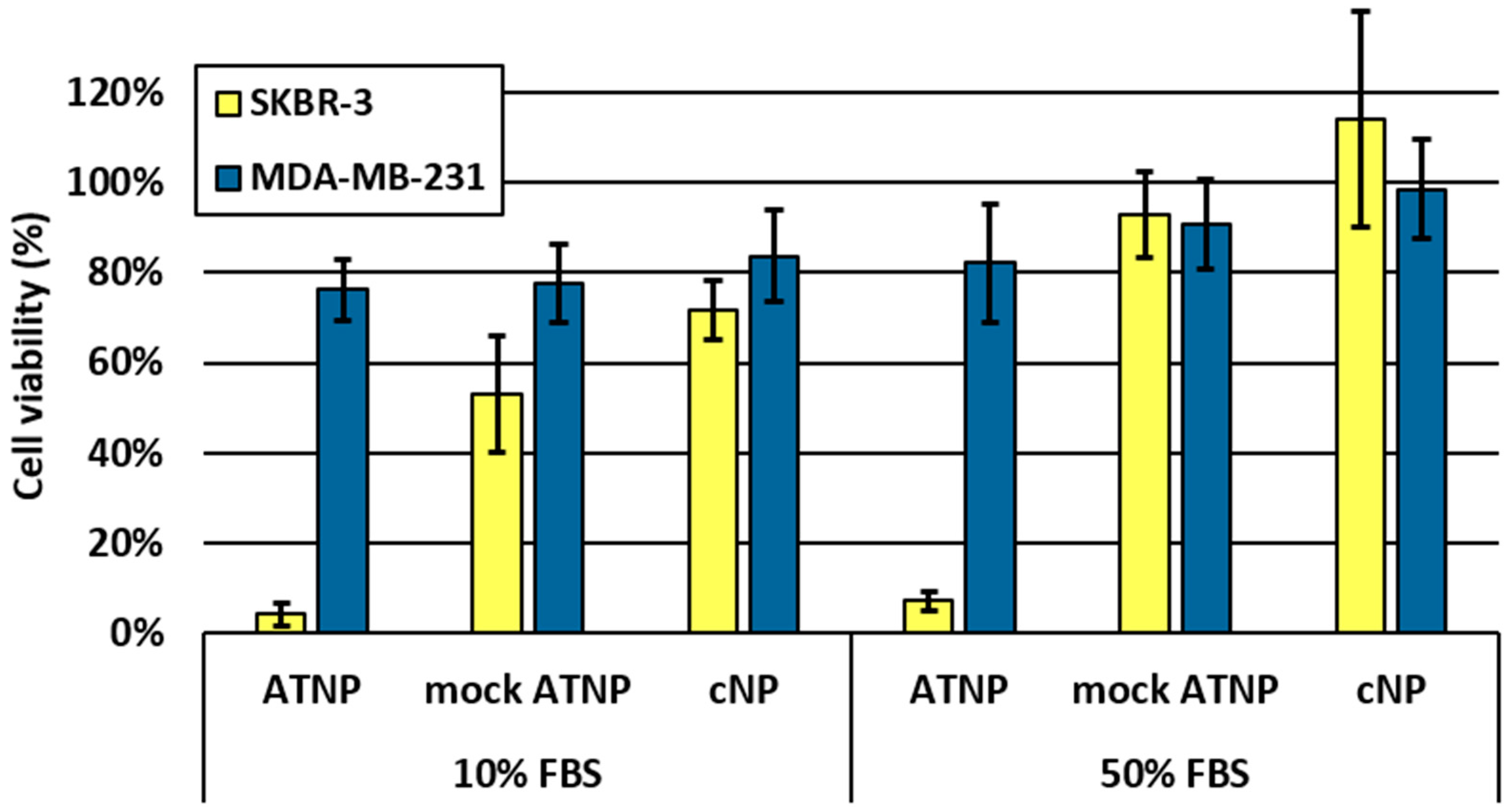
2.3. In Vitro Experiments
2.3.1. Cell Culture
2.3.2. In Vitro Cytotoxicity Assay
2.4. In Vivo Experiments
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaïa, N. Strategies and Challenges for the next Generation of Antibody-Drug Conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef] [PubMed]
- Weldon, J.E.; Pastan, I. A Guide to Taming a Toxin: Recombinant Immunotoxins Constructed from Pseudomonas Exotoxin A for the Treatment of Cancer. FEBS J. 2011, 278, 4683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutz, R.J. CHAPTER 22: The Future of Antibody–Drug Conjugate (ADC) Payloads. In Cytotoxic Payloads for Antibody–Drug Conjugates; Royal Society of Chemistry: London, UK, 2019; pp. 461–471. [Google Scholar] [CrossRef]
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody–Drug Conjugates: A Comprehensive Review. Mol. Cancer Res. 2020, 18, 3–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gingrich, J. How Next Generation ADCs Expand Beyond Cytotoxic Payloads. ADC Review. Available online: https://www.adcreview.com/articles/how-the-next-generation-antibody-drug-conjugates-expands-beyond-cytotoxic-payloads-for-cancer-therapy/ (accessed on 8 July 2021).
- Dovgan, I.; Koniev, O.; Kolodych, S.; Wagner, A. Antibody–Oligonucleotide Conjugates as Therapeutic, Imaging, and Detection Agents. Bioconjug. Chem. 2019, 30, 2483–2501. [Google Scholar] [CrossRef]
- Mi, P.; Cabral, H.; Kataoka, K. Ligand-Installed Nanocarriers toward Precision Therapy. Adv. Mater. 2020, 32, 1902604. [Google Scholar] [CrossRef]
- Kumar, R.; Santa Chalarca, C.F.; Bockman, M.R.; Bruggen, C.V.; Grimme, C.J.; Dalal, R.J.; Hanson, M.G.; Hexum, J.K.; Reineke, T.M. Polymeric Delivery of Therapeutic Nucleic Acids. Chem. Rev. 2021, 121, 11527–11652. [Google Scholar] [CrossRef]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in Oligonucleotide Drug Delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef]
- Chernikov, I.V.; Vlassov, V.V.; Chernolovskaya, E.L. Current Development of SiRNA Bioconjugates: From Research to the Clinic. Front. Pharmacol. 2019, 10, 444. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Liu, X.; Zhu, D.; Wang, Y.; Zhang, Z.; Zhou, X.; Qiu, N.; Chen, X.; Shen, Y. Nonviral Cancer Gene Therapy: Delivery Cascade and Vector Nanoproperty Integration. Adv. Drug Deliv. Rev. 2017, 115, 115–154. [Google Scholar] [CrossRef]
- Gómez-Aguado, I.; Rodríguez-Castejón, J.; Vicente-Pascual, M.; Rodríguez-Gascón, A.; Solinís, M.Á.; del Pozo-Rodríguez, A. Nanomedicines to Deliver MRNA: State of the Art and Future Perspectives. Nanomaterials 2020, 10, 364. [Google Scholar] [CrossRef] [Green Version]
- Ben Djemaa, S.; Munnier, E.; Chourpa, I.; Allard-Vannier, E.; David, S. Versatile Electrostatically Assembled Polymeric SiRNA Nanovectors: Can They Overcome the Limits of SiRNA Tumor Delivery? Int. J. Pharm. 2019, 567, 118432. [Google Scholar] [CrossRef]
- Chen, J.; Wang, K.; Wu, J.; Tian, H.; Chen, X. Polycations for Gene Delivery: Dilemmas and Solutions. Bioconjug. Chem. 2019, 30, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Jeon, T.; Luther, D.C.; Goswami, R.; Bell, C.; Nagaraj, H.; Cicek, Y.A.; Huang, R.; Mas-Rosario, J.A.; Elia, J.L.; Im, J.; et al. Engineered Polymer–SiRNA Polyplexes Provide Effective Treatment of Lung Inflammation. ACS Nano 2023, 17, 4315–4326. [Google Scholar] [CrossRef] [PubMed]
- Moghimi, S.M.; Symonds, P.; Murray, J.C.; Hunter, A.C.; Debska, G.; Szewczyk, A. A Two-Stage Poly(Ethylenimine)-Mediated Cytotoxicity: Implications for Gene Transfer/Therapy. Mol. Ther. 2005, 11, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Kang, S.J.; Kim, Y.J.; Lim, Y.; Chung, H.W. Comparative Studies on the Genotoxicity and Cytotoxicity of Polymeric Gene Carriers Polyethylenimine (PEI) and Polyamidoamine (PAMAM) Dendrimer in Jurkat T-Cells. Drug Chem. Toxicol. 2010, 33, 357–366. [Google Scholar] [CrossRef]
- Hunter, A.C. Molecular Hurdles in Polyfectin Design and Mechanistic Background to Polycation Induced Cytotoxicity. Adv. Drug Deliv. Rev. 2006, 58, 1523–1531. [Google Scholar] [CrossRef]
- Roursgaard, M.; Knudsen, K.B.; Northeved, H.; Persson, M.; Christensen, T.; Kumar, P.E.K.; Permin, A.; Andresen, T.L.; Gjetting, T.; Lykkesfeldt, J.; et al. In Vitro Toxicity of Cationic Micelles and Liposomes in Cultured Human Hepatocyte (HepG2) and Lung Epithelial (A549) Cell Lines. Toxicol. In Vitro 2016, 36, 164–171. [Google Scholar] [CrossRef]
- Monnery, B.D.; Wright, M.; Cavill, R.; Hoogenboom, R.; Shaunak, S.; Steinke, J.H.G.; Thanou, M. Cytotoxicity of Polycations: Relationship of Molecular Weight and the Hydrolytic Theory of the Mechanism of Toxicity. Int. J. Pharm. 2017, 521, 249–258. [Google Scholar] [CrossRef] [Green Version]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of Nanoparticle Design for Overcoming Biological Barriers to Drug Delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef]
- Lochbaum, C.A.; Chew, A.K.; Zhang, X.; Rotello, V.; Van Lehn, R.C.; Pedersen, J.A. Lipophilicity of Cationic Ligands Promotes Irreversible Adsorption of Nanoparticles to Lipid Bilayers. ACS Nano 2021, 15, 6562–6572. [Google Scholar] [CrossRef]
- Fröhlich, E. The Role of Surface Charge in Cellular Uptake and Cytotoxicity of Medical Nanoparticles. Int. J. Nanomed. 2012, 7, 5577–5591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dufès, C.; Keith, W.N.; Bilsland, A.; Proutski, I.; Uchegbu, I.F.; Schätzlein, A.G. Synthetic Anticancer Gene Medicine Exploits Intrinsic Antitumor Activity of Cationic Vector to Cure Established Tumors. Cancer Res. 2005, 65, 8079–8084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.; Agasti, S.S.; Zhu, Z.; Isaacs, L.; Rotello, V.M. Recognition-Mediated Activation of Therapeutic Gold Nanoparticles inside Living Cells. Nat. Chem. 2010, 2, 962–966. [Google Scholar] [CrossRef]
- Dai, Q.; Bertleff-Zieschang, N.; Braunger, J.A.; Björnmalm, M.; Cortez-Jugo, C.; Caruso, F. Particle Targeting in Complex Biological Media. Adv. Healthc. Mater. 2018, 7, 1700575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, T.; Mulholland, P.; Chester, K. Antibody-Targeted Nanoparticles for Cancer Treatment. Immunotherapy 2016, 8, 941–958. [Google Scholar] [CrossRef]
- Wen, Y.; Bai, H.; Zhu, J.; Song, X.; Tang, G.; Li, J. A Supramolecular Platform for Controlling and Optimizing Molecular Architectures of SiRNA Targeted Delivery Vehicles. Sci. Adv. 2020, 6, eabc2148. [Google Scholar] [CrossRef]
- Kim, S.H.; Jeong, J.H.; Chun, K.W.; Park, T.G. Target-Specific Cellular Uptake of PLGA Nanoparticles Coated with Poly(l-Lysine)−Poly(Ethylene Glycol)−Folate Conjugate. Langmuir 2005, 21, 8852–8857. [Google Scholar] [CrossRef]
- Green, J.J.; Chiu, E.; Leshchiner, E.S.; Shi, J.; Langer, R.; Anderson, D.G. Electrostatic Ligand Coatings of Nanoparticles Enable Ligand-Specific Gene Delivery to Human Primary Cells. Nano Lett. 2007, 7, 874–879. [Google Scholar] [CrossRef]
- Eltoukhy, A.A.; Chen, D.; Veiseh, O.; Pelet, J.M.; Yin, H.; Dong, Y.; Anderson, D.G. Nucleic Acid-Mediated Intracellular Protein Delivery by Lipid-like Nanoparticles. Biomaterials 2014, 35, 6454–6461. [Google Scholar] [CrossRef] [Green Version]
- Theodorou, I.; Anilkumar, P.; Lelandais, B.; Clarisse, D.; Doerflinger, A.; Gravel, E.; Ducongé, F.; Doris, E. Stable and Compact Zwitterionic Polydiacetylene Micelles with Tumor-Targeting Properties. Chem. Commun. 2015, 51, 14937–14940. [Google Scholar] [CrossRef]
- Neuberg, P.; Perino, A.; Morin-Picardat, E.; Anton, N.; Darwich, Z.; Weltin, D.; Mely, Y.; Klymchenko, A.S.; Remy, J.-S.; Wagner, A. Photopolymerized Micelles of Diacetylene Amphiphile: Physical Characterization and Cell Delivery Properties. Chem. Commun. 2015, 51, 11595–11598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ripoll, M.; Neuberg, P.; Kichler, A.; Tounsi, N.; Wagner, A.; Remy, J.-S. PH-Responsive Nanometric Polydiacetylenic Micelles Allow for Efficient Intracellular SiRNA Delivery. ACS Appl. Mater. Interfaces 2016, 8, 30665–30670. [Google Scholar] [CrossRef] [PubMed]
- Neuberg, P.; Wagner, A.; Remy, J.-S.; Kichler, A. Design and Evaluation of Ionizable Peptide Amphiphiles for SiRNA Delivery. Int. J. Pharm. 2019, 566, 141–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Midoux, P.; Pichon, C.; Yaouanc, J.-J.; Jaffrès, P.-A. Chemical Vectors for Gene Delivery: A Current Review on Polymers, Peptides and Lipids Containing Histidine or Imidazole as Nucleic Acids Carriers. Br. J. Pharmacol. 2009, 157, 166–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, Y.; Guo, Z.; Du, Z.; Fang, R.; Wu, H.; Zeng, X.; Wang, C.; Feng, M.; Pan, S. Serum Tolerance and Endosomal Escape Capacity of Histidine-Modified PDNA-Loaded Complexes Based on Polyamidoamine Dendrimer Derivatives. Biomaterials 2012, 33, 8111–8121. [Google Scholar] [CrossRef]
- Wang, F.; Hu, K.; Cheng, Y. Structure–Activity Relationship of Dendrimers Engineered with Twenty Common Amino Acids in Gene Delivery. Acta Biomater. 2016, 29, 94–102. [Google Scholar] [CrossRef]
- Ewe, A.; Przybylski, S.; Burkhardt, J.; Janke, A.; Appelhans, D.; Aigner, A. A Novel Tyrosine-Modified Low Molecular Weight Polyethylenimine (P10Y) for Efficient SiRNA Delivery in Vitro and in Vivo. J. Control. Release 2016, 230, 13–25. [Google Scholar] [CrossRef]
- Leyton, J.V. Improving Receptor-Mediated Intracellular Access and Accumulation of Antibody Therapeutics—The Tale of HER2. Antibodies 2020, 9, 32. [Google Scholar] [CrossRef]
- Du, J.; Wang, H.; Zhong, C.; Peng, B.; Zhang, M.; Li, B.; Huo, S.; Guo, Y.; Ding, J. Structural Basis for Recognition of CD20 by Therapeutic Antibody Rituximab. J. Biol. Chem. 2007, 282, 15073–15080. [Google Scholar] [CrossRef] [Green Version]
- Ripoll, M.; Neuberg, P.; Wagner, A.; Remy, J.-S. Amphiphilic Monomers Based Nanovectors and Their Use for Sirna Delivery. WO/2017/167929. 2017. Available online: https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2017167929 (accessed on 9 July 2021).
- Dovgan, I.; Ursuegui, S.; Erb, S.; Michel, C.; Kolodych, S.; Cianférani, S.; Wagner, A. Acyl Fluorides: Fast, Efficient, and Versatile Lysine-Based Protein Conjugation via Plug-and-Play Strategy. Bioconjug. Chem. 2017, 28, 1452–1457. [Google Scholar] [CrossRef]
- Lehot, V.; Kuhn, I.; Nothisen, M.; Erb, S.; Kolodych, S.; Cianférani, S.; Chaubet, G.; Wagner, A. Non-Specific Interactions of Antibody-Oligonucleotide Conjugates with Living Cells. Sci. Rep. 2021, 11, 5881. [Google Scholar] [CrossRef]
- Zhao, Y.H.; Abraham, M.H.; Zissimos, A.M. Fast Calculation of van Der Waals Volume as a Sum of Atomic and Bond Contributions and Its Application to Drug Compounds. J. Org. Chem. 2003, 68, 7368–7373. [Google Scholar] [CrossRef] [PubMed]
- Ripoll, M.; Neuberg, P.; Remy, J.-S.; Kichler, A. Cationic Photopolymerized Polydiacetylenic (PDA) Micelles for SiRNA Delivery. Methods Mol. Biol. 2019, 1943, 101–122. [Google Scholar] [CrossRef] [PubMed]
- Ripoll, M. Synthèse de Nano-Vecteurs Dérivés Des Polydiacétylènes Pour La Co-Délivrance d’un ARN Interférent et d’un Anticancéreux. Ph.D. Thesis, École Doctorale Sciences Chimiques, Strasbourg, France, 2017. Available online: http://www.theses.fr/2017STRAF076 (accessed on 21 July 2021).
- Thielens, N.M.; Belime, A.; Gravel, E.; Ancelet, S.; Caneiro, C.; Doris, E.; Ling, W.L. Impact of the Surface Charge of Polydiacetylene Micelles on Their Interaction with Human Innate Immune Protein C1q and the Complement System. Int. J. Pharm. 2018, 536, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Quan, L.; Zhou, C.; Zhan, Q. Factors Relating to the Biodistribution & Clearance of Nanoparticles & Their Effects on in Vivo Application. Nanomedicine 2018, 13, 1495–1512. [Google Scholar] [CrossRef]
- Mitchell, M.J.; Billingsley, M.M.; Haley, R.M.; Wechsler, M.E.; Peppas, N.A.; Langer, R. Engineering Precision Nanoparticles for Drug Delivery. Nat. Rev. Drug Discov. 2021, 20, 101–124. [Google Scholar] [CrossRef]
- Salvati, A.; Pitek, A.S.; Monopoli, M.P.; Prapainop, K.; Bombelli, F.B.; Hristov, D.R.; Kelly, P.M.; Åberg, C.; Mahon, E.; Dawson, K.A. Transferrin-Functionalized Nanoparticles Lose Their Targeting Capabilities When a Biomolecule Corona Adsorbs on the Surface. Nat. Nanotechnol. 2013, 8, 137–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bros, M.; Nuhn, L.; Simon, J.; Moll, L.; Mailänder, V.; Landfester, K.; Grabbe, S. The Protein Corona as a Confounding Variable of Nanoparticle-Mediated Targeted Vaccine Delivery. Front. Immunol. 2018, 9, 1760. [Google Scholar] [CrossRef]
- Baran, A.; Fırat Baran, M.; Keskin, C.; Hatipoğlu, A.; Yavuz, Ö.; İrtegün Kandemir, S.; Adican, M.T.; Khalilov, R.; Mammadova, A.; Ahmadian, E.; et al. Investigation of Antimicrobial and Cytotoxic Properties and Specification of Silver Nanoparticles (AgNPs) Derived from Cicer Arietinum L. Green Leaf Extract. Front. Bioeng. Biotechnol. 2022, 10, 855136. [Google Scholar] [CrossRef]
- Sellami, H.; Khan, S.A.; Ahmad, I.; Alarfaj, A.A.; Hirad, A.H.; Al-Sabri, A.E. Green Synthesis of Silver Nanoparticles Using Olea Europaea Leaf Extract for Their Enhanced Antibacterial, Antioxidant, Cytotoxic and Biocompatibility Applications. Int. J. Mol. Sci. 2021, 22, 12562. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lehot, V.; Neuberg, P.; Ripoll, M.; Daubeuf, F.; Erb, S.; Dovgan, I.; Ursuegui, S.; Cianférani, S.; Kichler, A.; Chaubet, G.; et al. Targeted Anticancer Agent with Original Mode of Action Prepared by Supramolecular Assembly of Antibody Oligonucleotide Conjugates and Cationic Nanoparticles. Pharmaceutics 2023, 15, 1643. https://doi.org/10.3390/pharmaceutics15061643
Lehot V, Neuberg P, Ripoll M, Daubeuf F, Erb S, Dovgan I, Ursuegui S, Cianférani S, Kichler A, Chaubet G, et al. Targeted Anticancer Agent with Original Mode of Action Prepared by Supramolecular Assembly of Antibody Oligonucleotide Conjugates and Cationic Nanoparticles. Pharmaceutics. 2023; 15(6):1643. https://doi.org/10.3390/pharmaceutics15061643
Chicago/Turabian StyleLehot, Victor, Patrick Neuberg, Manon Ripoll, François Daubeuf, Stéphane Erb, Igor Dovgan, Sylvain Ursuegui, Sarah Cianférani, Antoine Kichler, Guilhem Chaubet, and et al. 2023. "Targeted Anticancer Agent with Original Mode of Action Prepared by Supramolecular Assembly of Antibody Oligonucleotide Conjugates and Cationic Nanoparticles" Pharmaceutics 15, no. 6: 1643. https://doi.org/10.3390/pharmaceutics15061643