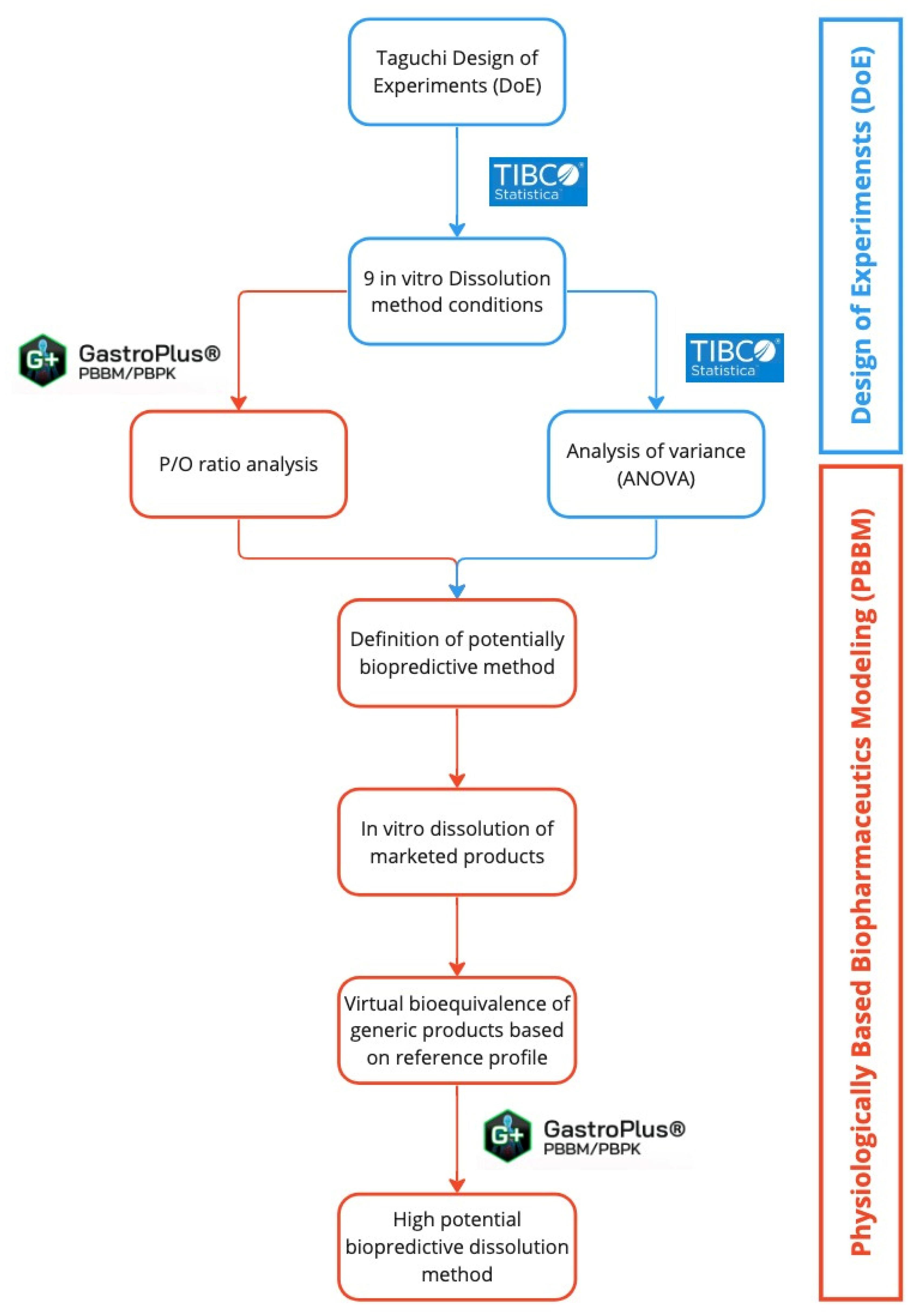
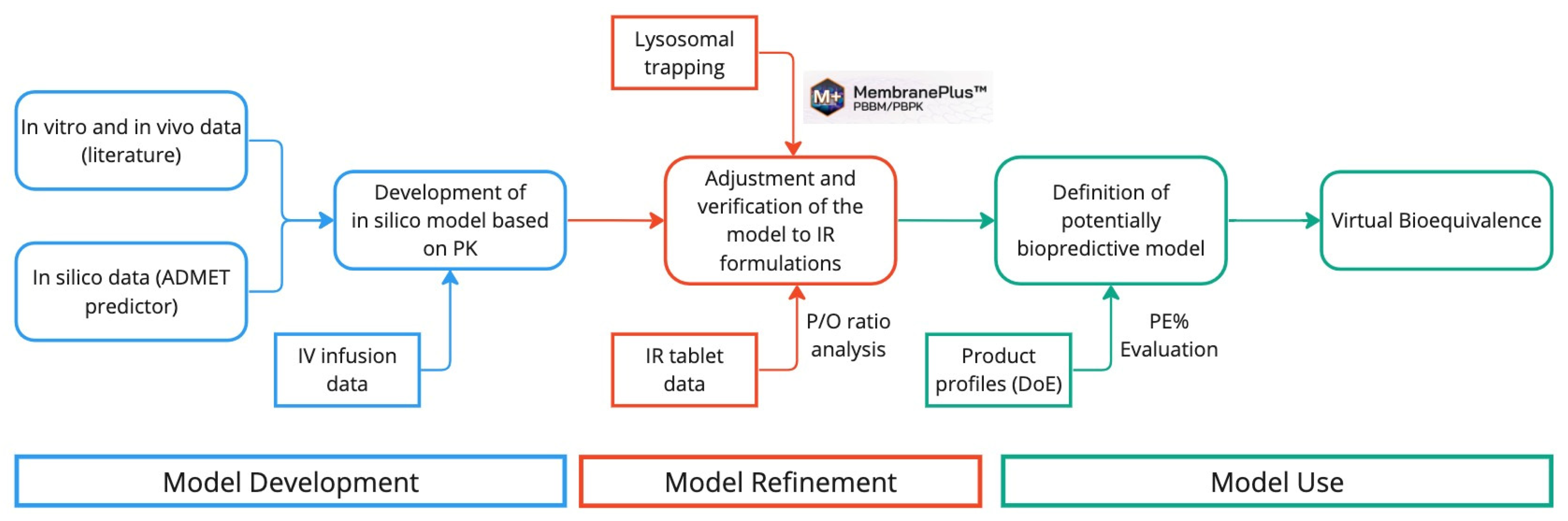
3.1. PBBM Development and Refinement
A pH-dependent permeability was observed and described for desvenlafaxine IR tablets [
4]. Experimental values were incorporated by these authors in their model to explain a delay in the T
max of desvenlafaxine IR tablets.
However, in our model in GastroPlus®, this pH-dependent permeability is automatically considered by the software due to the absorption scale factors (ASF) in the Advanced Compartmental Absorption and Transit (ACAT™) model. The ASF uses the permeability (Peff) and the logD values of the drug, considering the differences in permeability that occur in the gastrointestinal tract due to the pH. Since it is already considered in the software, and the drug presents characteristics of a lysosomal tropic agent, we decided to evaluate it using the MembranePlus™ software.
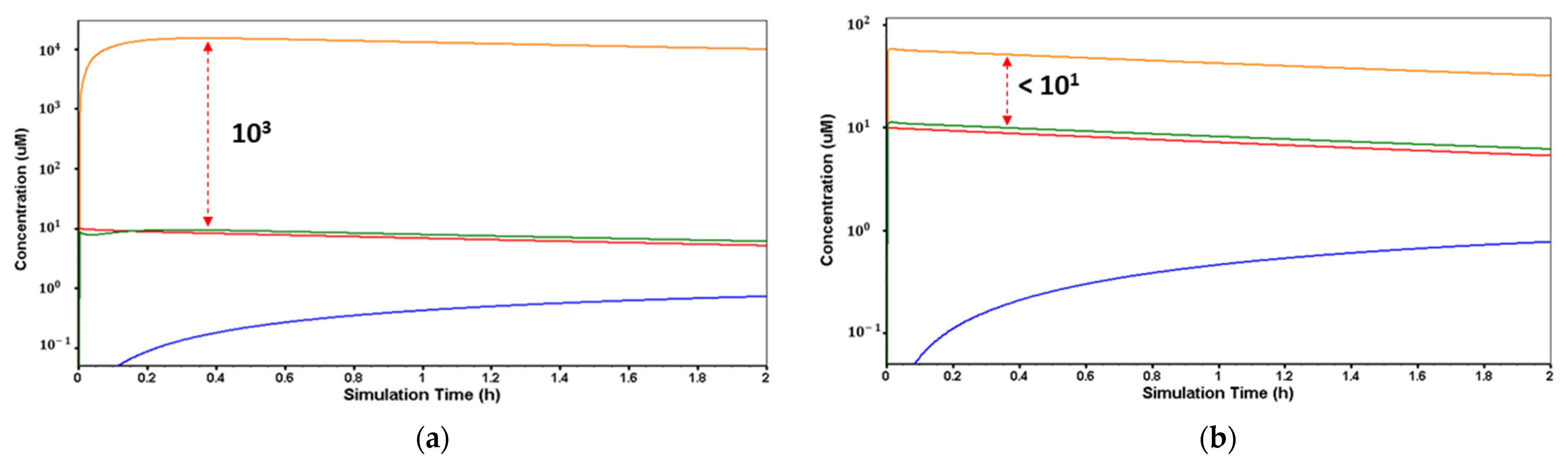
As it was expected, due to the pka and logP values of desvenlafaxine, the lysosomal trapping evaluation of the drug using the MembranePlus™ software showed that the drug concentration in the lysosomes at pH = 4.0 (
Figure 3a) is 10
3 times higher compared to the cytosol concentration, and at pH = 6.5 (
Figure 3b), this concentration is less than 10
1 times higher compared to cytosol. This reveals that the lysosomal trapping is significantly lower at pH 6.5.
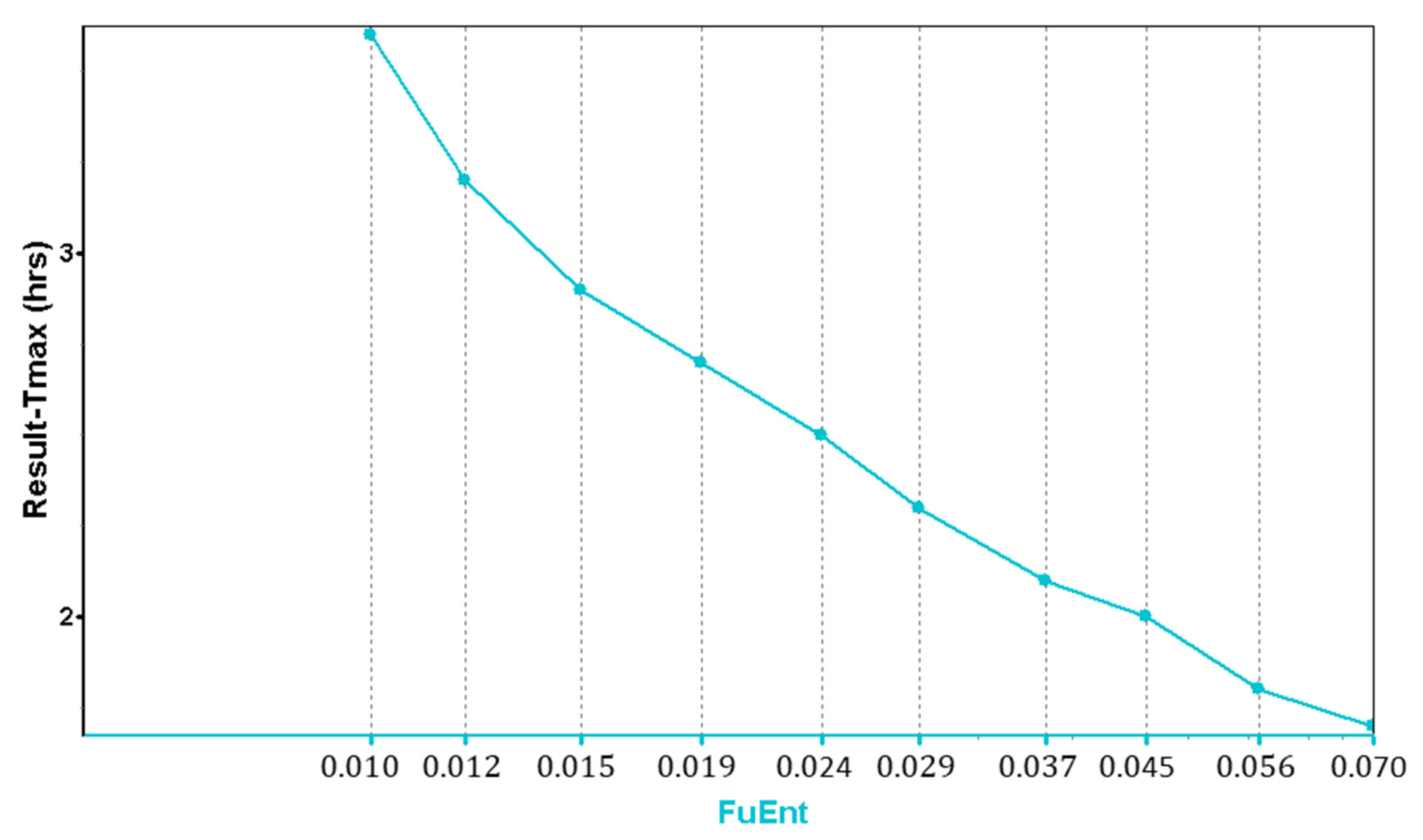
In addition to the lysosomal trapping evaluation in MembranePlus™, a parameter sensitivity analysis (PSA) was run in GastroPlus®, showing the influence of the fraction unbound in the enterocytes (Fuent) in the Tmax values.
For IR tablets, the observed T
max values range from 0.5 to 6 h for the 50 mg dose [
14], and from 2–4 h for the 100 mg dose [
14]. Based on these values, and according to the PSA graph (
Figure 4), a value of F
uent = 2% was used in the model for the simulations.
A two-compartment pharmacokinetic model was established using the IV and IR data as a top-down approach, in which values of the PK parameters are: T1/2 = 9.7 h, Cl = 0.2031 L/h/kg, Vss = 2.2343 L/kg, K12 = 0.15764 1/h, and K21 = 0.19244 1/h. These values are within the values described in the literature [
13,
14]. The values of the predicted versus observed PK parameters are shown in
Table 4.
3.2. Statistical and In Silico Evaluation to Define Biopredictive Method
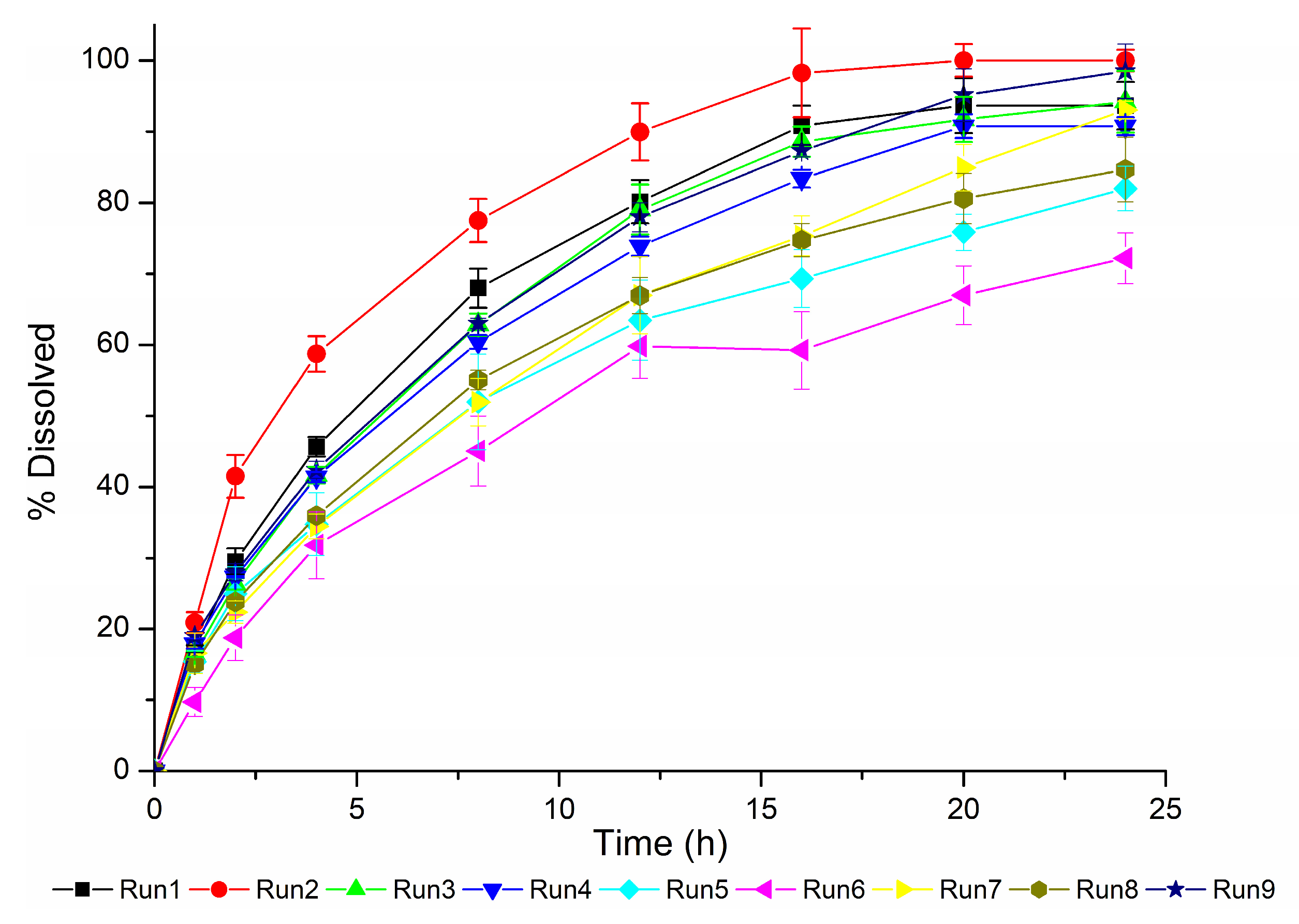
The dissolution profiles obtained based on the DoE are presented in
Figure 5.
The DoE statistical analyses are presented in
Tables S1–S3 (Supplementary Materials). These results revealed that the apparatus and formulation employed had a significant influence on the percentage of drug dissolved at all time points studied, as indicated by the
p-value < 0.05. This finding suggests that the apparatus and formulation (drug product) are crucial factors affecting the dissolution of desvenlafaxine ER tablets. Conversely, the analysis did not demonstrate that the rotation speed had a significant impact on dissolution, as evidenced by the
p-value > 0.05 for all studied responses.
The influence of the dissolution medium warrants further investigation. The Q%2 h and Q%24 h results indicate that the dissolution medium has an influence on drug dissolution (p-value < 0.05), despite the high solubility of desvenlafaxine in all studied dissolution media. However, this information conflicts with the finding that the dissolution medium had no effect on Q%12 h (p-value > 0.05). The discrepancy may be attributed to the indirect influence of other dissolution parameters on the result, particularly given the ultra-fractionated Taguchi experimental design employed.
This kind of design is defined by an orthogonal array, which is generally used for variable screening, allowing the evaluation of different factors performing a limited number of experiments [
18]. Considering the adopted strategy (DoE + in silico model), it was decided to carry out the minimum amount of dissolution tests. As desvenlafaxine is a class I drug [
4], we may infer that the dissolution medium does not significantly have influence on desvenlafaxine dissolution from the ER tablets analyzed.
Additionally, it is also worth mentioning that the Taguchi array is an experimental design that is already used in pharmaceutical technology to support the development of controlled release formulations, for example, in the evaluation of process and formulation variables in the development of a gastroretentive rivastigmine system [
19], the development and optimization of solid lipid nanoparticles of isradipine [
20], and in the production of carbamazepine [
21] and cilostazol [
22] osmotic pumps.
The average weight, thickness, and diameter/length of each drug product tablets are shown in
Table 5. These results reveal that the SA/V ratio and weight of Generic #1 are, respectively, higher and lower than the other products analyzed. The higher SA/V ratio should favor the drug release since all tablets analyzed relies on a hypromellose (HPMC) hydrophilic matrix to extend the drug release, and a higher SA/V ratio, in these cases, is directly related to a faster dissolution profile [
15]. The lower weight may also be indirectly influencing the drug release since it means that the HPMC is at a lower concentration in the final formulation, resulting in a less consistent gel after matrix hydration, and therefore favoring the drug release [
23].
The dissolution profiles obtained (
Figure 5) confirm this hypothesis as the Generic #1 profiles have the higher dissolution rates. The information that this product has a facilitated release is crucial to properly analyze the in silico results of the predictive errors obtained throughout convolution of the in vitro dissolution results for the DoE runs.
Table 6 contains the results of the C
max, AUC
0-t and their respective prediction errors (PE%) obtained using Gastroplus
® to convolute the in vitro dissolution results (
Figure 5) for each DoE run condition. A method was considered biopredictive for the evaluated drug product when both C
max and AUC
0-t predictive errors showed values within ±10% [
24].
For the Generic #1 product, runs 1 and 3 are more biopredictive, based on the results obtained. These methods provide conditions that do not contribute to drug release from the matrix, since the method used in Run 1 consists of the use of basket apparatus at 50 rpm (
Table 3), which promotes low hydrodynamics, and Run 3 does not use a sinker to hold the tablet during the assay, which implies in the adherence of the tablet in the bottom of the dissolution vessel. The Run 2 method, which is not biopredictive for Generic #1, is based on the use of paddle apparatus and sinkers to hold the tablets (
Table 3). This condition promotes high hydrodynamics and prevents tablet adherence, fully hydrating the tablets by the end of the assay; therefore, it is a condition that facilitates drug release [
25].
A different behavior was observed for Generic #2 and Reference products, compared to Generic #1, most likely due to the higher tablet weight and lower SA/V ratio (
Table 5). In this case, the methods that promoted drug release (Run 4 and Run 9) were more biopredictive. At Run 4, the Reference product was evaluated using paddle at 100 rpm and sinkers (
Table 3). This condition promotes high hydrodynamics and prevents tablet adherence, being a condition that facilitated drug release. Generic #2 was evaluated in Run 9, using the paddle apparatus at 50 rpm and sinkers (
Table 3), conditions that prevent tablet adherence and promotes gentler agitation, compared to Run 4. The other methods studied would either use the basket apparatus that promotes low hydrodynamics or promotes tablet adherence by using the paddle apparatus with no sinker holding the tablets, thus not considered to be biopredictive [
25].
Figure 6a,b show the Reference product tablets after a dissolution assay with and without the use of sinkers to hold the tablets.
Figure 6a represents the table adhered to the bottom of the dissolution vessel.
Figure 6b represents the difference in tablet gel formation and hydration after the assays, with the use of a sinker, in the left, and without this use, in the right. The same behavior was observed for all products studied.
These results show that, since Generic #1 have lower tablet weight and SA/V ratio, than Generic #2 and Reference products, for a method to be biopredictive for all drug products, it would have to promote drug release in Generic #2 and Reference, but also cannot promote such high hydrodynamics that would intensify drug release in Generic #1. In this case, the use of the basket apparatus may be discarded, since even with the higher rotation speed applied (Run 8), this condition is not biopredictive for Generic #2. In addition, the use of the paddle apparatus with no sinker holding the tablets is not a biopredictive condition, since the adherence of the tablets in the vessel bottom has a high impact on the matrix hydration, preventing the desvenlafaxine from being fully released from the Generic #2 and Reference tablets. Therefore, the most potential condition of being biopredictive is the paddle apparatus at 50 rpm and the use of sinkers. This condition promoted a relatively lower hydrodynamic in the vessel that would favor Generic #1 and would not inhibit drug release from Generic #2 and Reference tablets.
According to the previous discussion, the dissolution media do not have influence on the drug release. Thus, it was maintained as 0.9% NaCl, as suggested by the FDA for desvenlafaxine ER tablets [
9]. In a recent study, Da Silva et al. (2020) [
16] suggested the use of the basket apparatus at 75 rpm and a reduced volume of dissolution medium (500 mL) as a biorelevant method. However, according to the low hydrodynamics of the basket, as previously discussed, we decided to consider, as biopredictive, the following dissolution method: 900 mL of 0.9% NaCl and paddle at 50 rpm with sinker, which does not interfere with the performance of the matrix system.
3.3. In Vitro Dissolution Using Biopredictive Method and Virtual Bioequivalence Studies
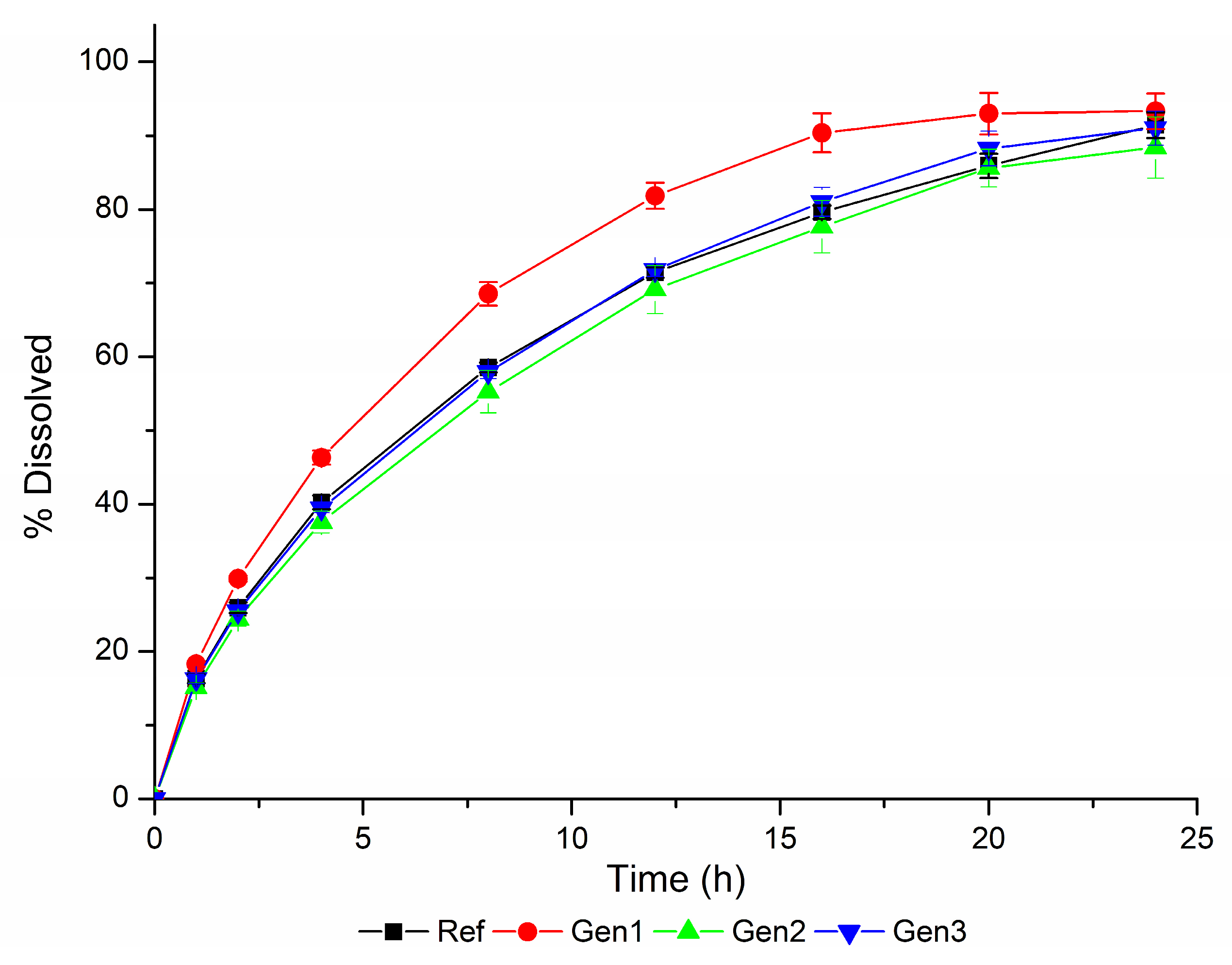
The in vitro dissolution profiles obtained using the biopredictive dissolution method are shown in
Figure 7.
The VBE studies of the drug products Generic #1, Generic #2, and Generic #3 versus Reference, using the dissolution profiles generated using the biopredictive dissolution method (
Figure 7) led to the results presented in
Tables S4–S6. Ten trials were performed for each VBE, and, in summary, the number of pass/fails are presented in
Table 7.
Based on these results, we confirm that the dissolution method developed is biopredictive, since it is capable of detecting the potential of all Generic products studied to pass bioequivalence. The Generic #3, used as external validation, passed in all 10 VBE. The Generic #2 passed in 9 out of 10 VBE, which would be considered a good candidate for a Generic product. Lastly, Generic #1 passed in 7 out of 10 VBE, which is the majority of the tests and expresses the capability of the method to identify it as a good candidate; still, it shows that this product may be reproved on bioequivalence study depending on the individual characteristics of the participants on the bioequivalence study.
Although any datum from a non-bioequivalent ER desvenlafaxine formulation was used, and a dissolution/bioequivalence safe space was not defined, it was possible to obtain a biopredictive dissolution method. The ability of this method to detect non-bioequivalent products can be observed in
Figure 7 and
Table 7, in which, as the dissolution profile deviates from that of the reference drug product (Generic #1), the chance of failing bioequivalence increases.
Considering this behavior, we decided to set dissolution specifications based on the best results in virtual bioequivalence (Generic #2 and Generic #3) in addition to the reference drug product profile. Thus, a possible specification for desvenlafaxine ER tablets to ensure bioequivalence would be: 1 h (<20%), 4 h (38–48%), 8 h (50–72%), 16 h (72–93%), and 24 h (not less than 80%).
When comparing this specification with the one presented by Da Silva et al. (2020) [
16], a difference can be observed, mainly at 4 h and 8 h, since the authors proposed a slightly lower range (4 h: 28–48%; 8 h: 48–68%), which may be attributed to the use of basket apparatus. It is worth mentioning that each specification is related to the dissolution method employed.

 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}