

Review: Structure-Activity Relationship of Antimicrobial Peptoids

Abstract
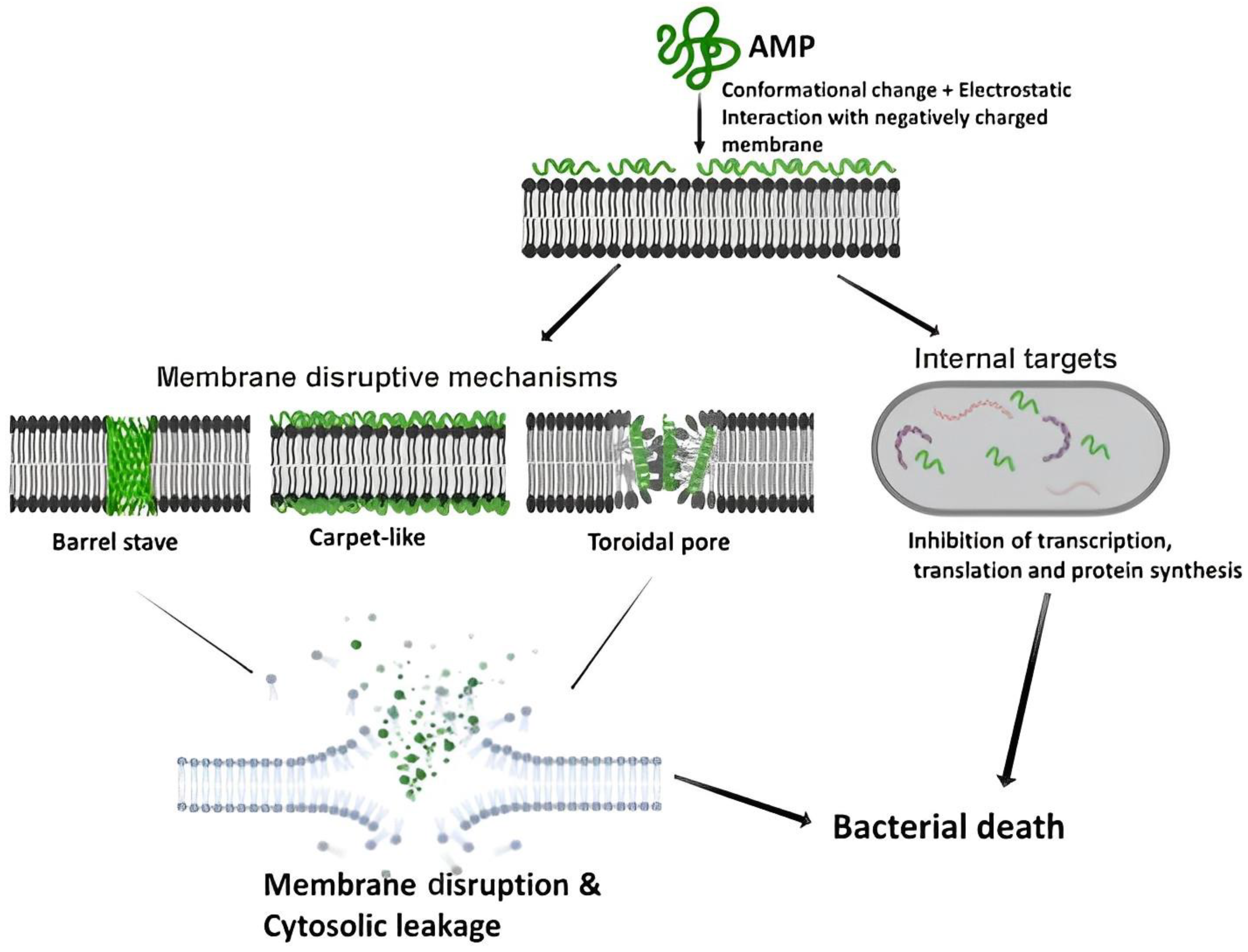
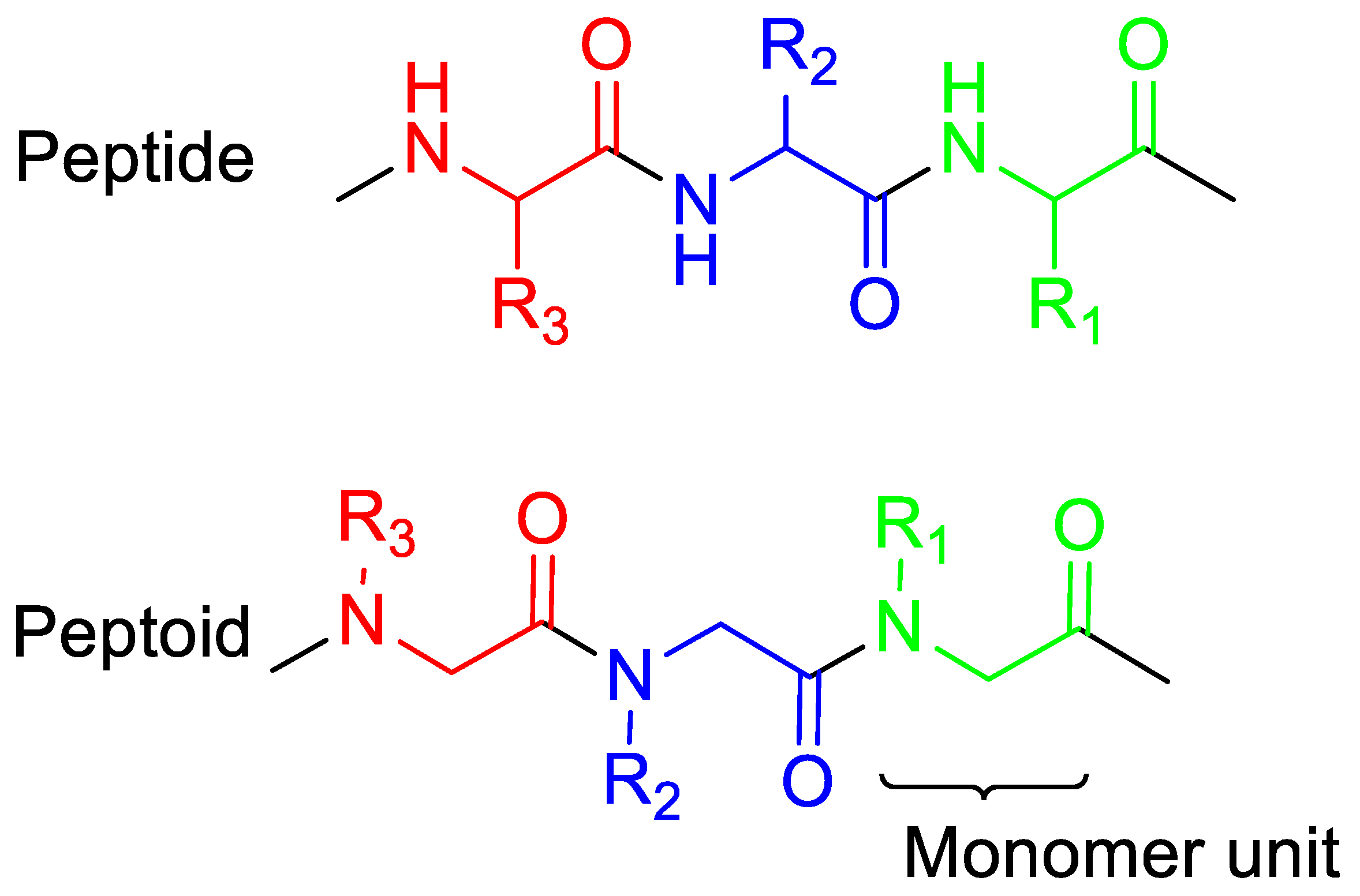
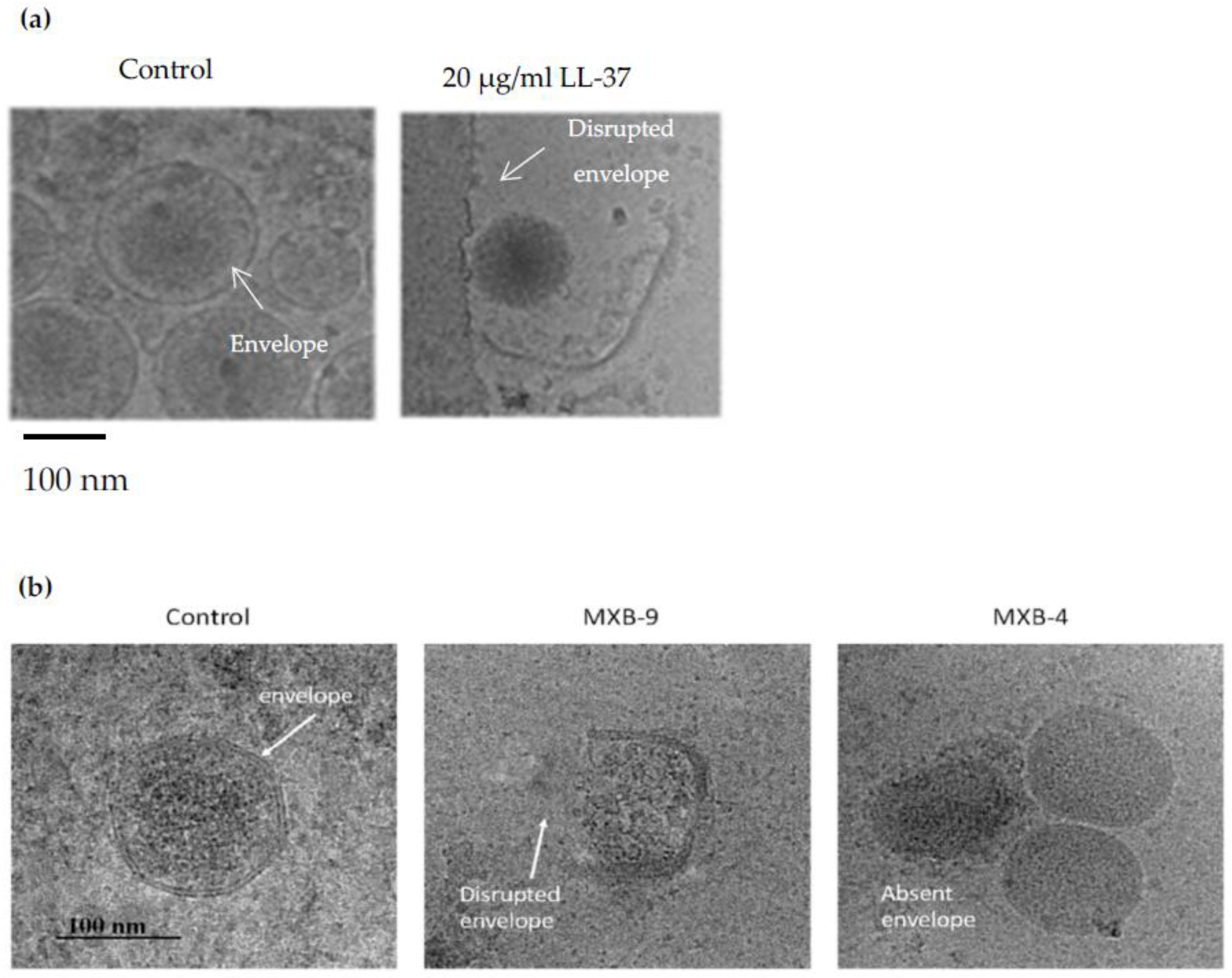
:1. Introduction
2. Main Chain Length
The Effects of Main Chain Length on Antimicrobial Activity
3. Cationic Peptoids
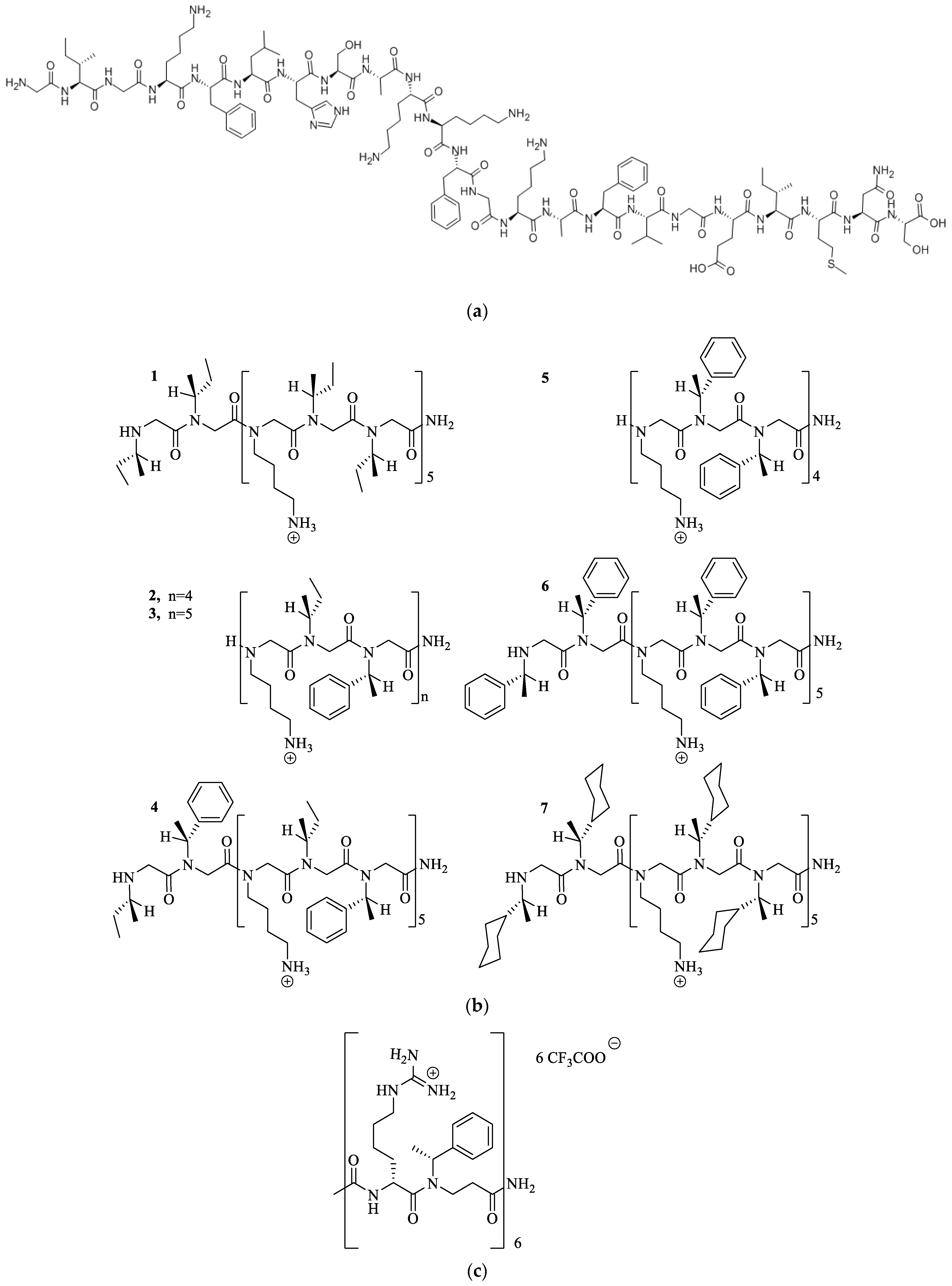
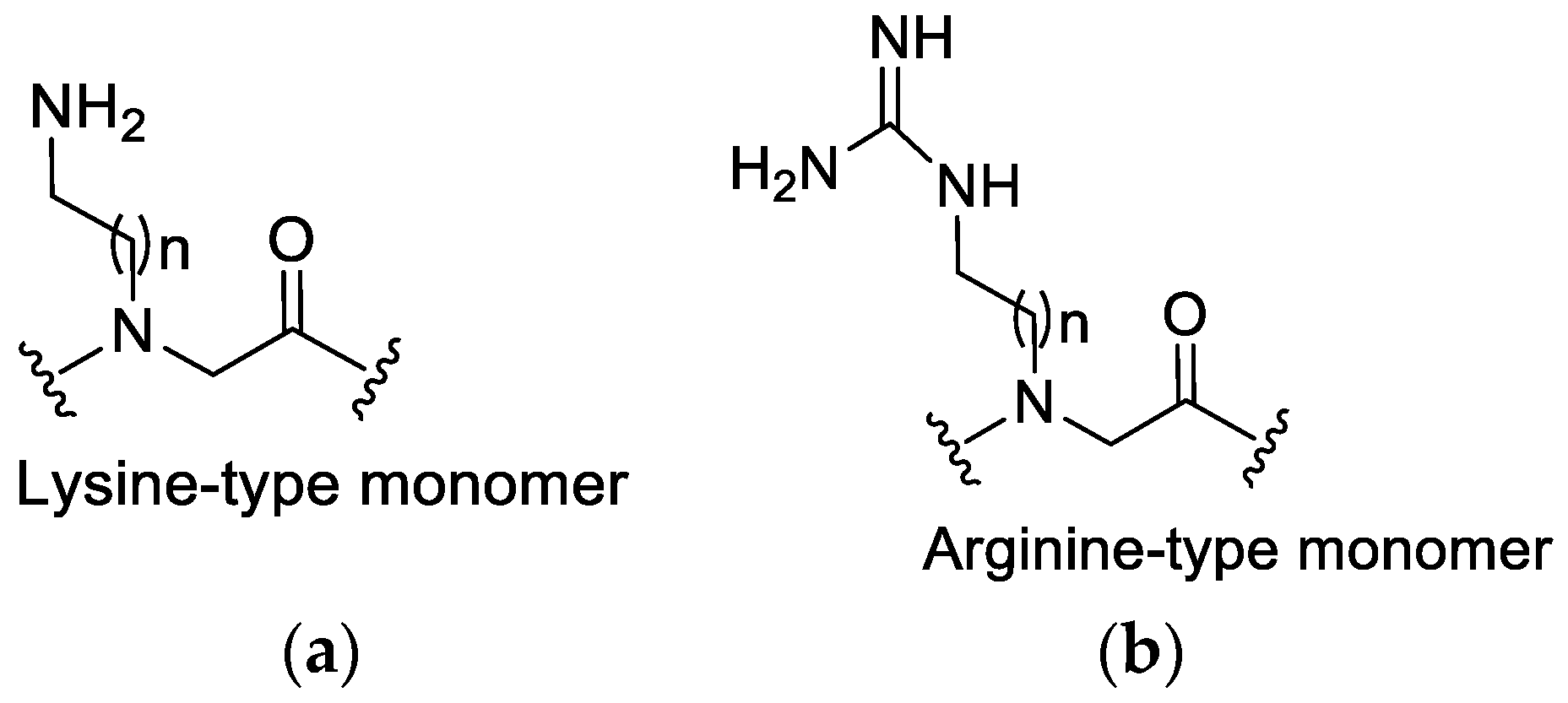
3.1. Lysine or Arginine Type Side Chains
3.2. The Effect of Cationic Side Chains on Antimicrobial Activity
4. Hydrophobicity
4.1. Influence of Hydrophobicity on the Secondary Structure of Peptides
4.2. Effect of Hydrophobic Surface Area (SA) on Antimicrobial Activity
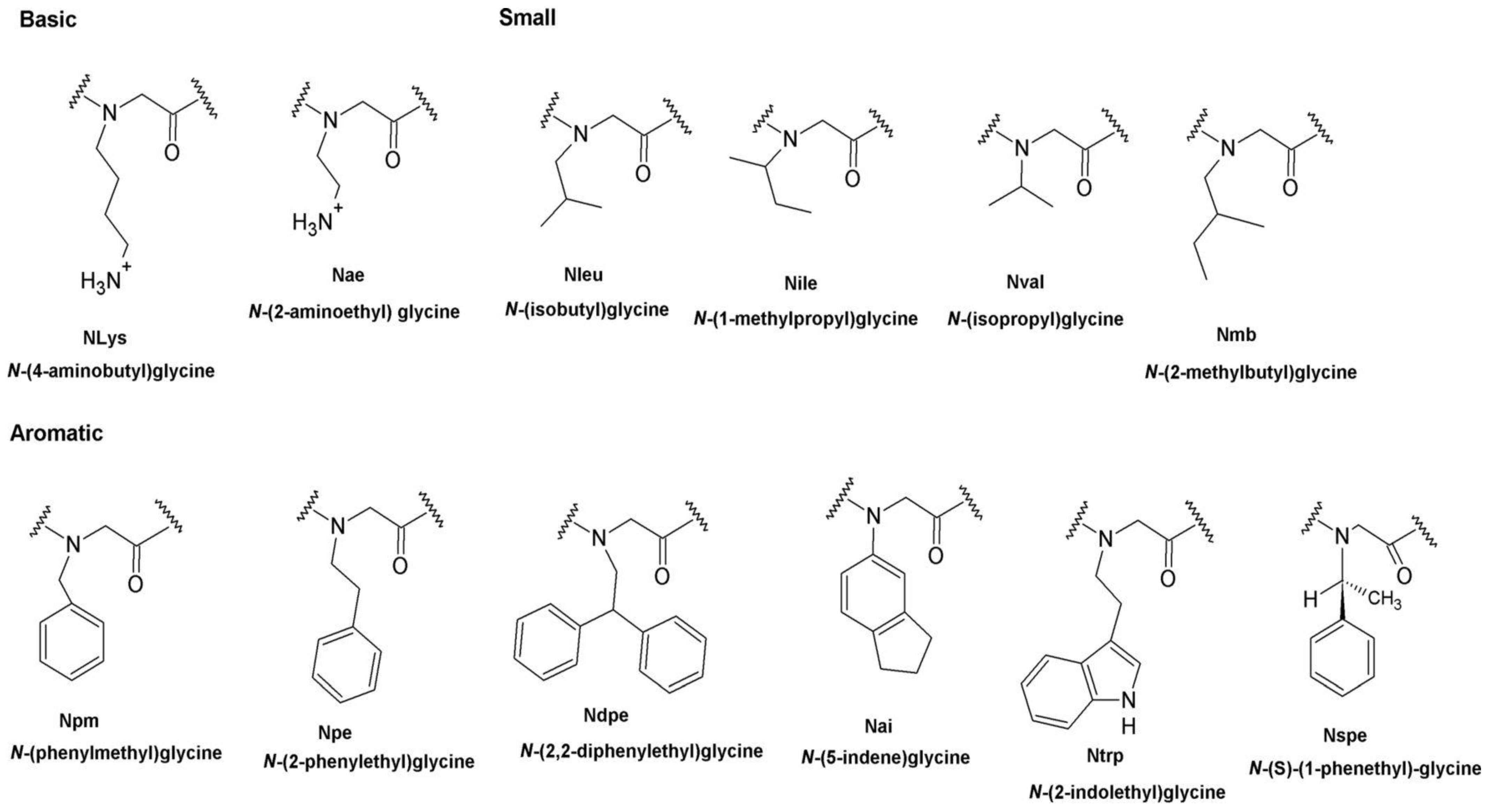
4.2.1. Phenyl Monomers
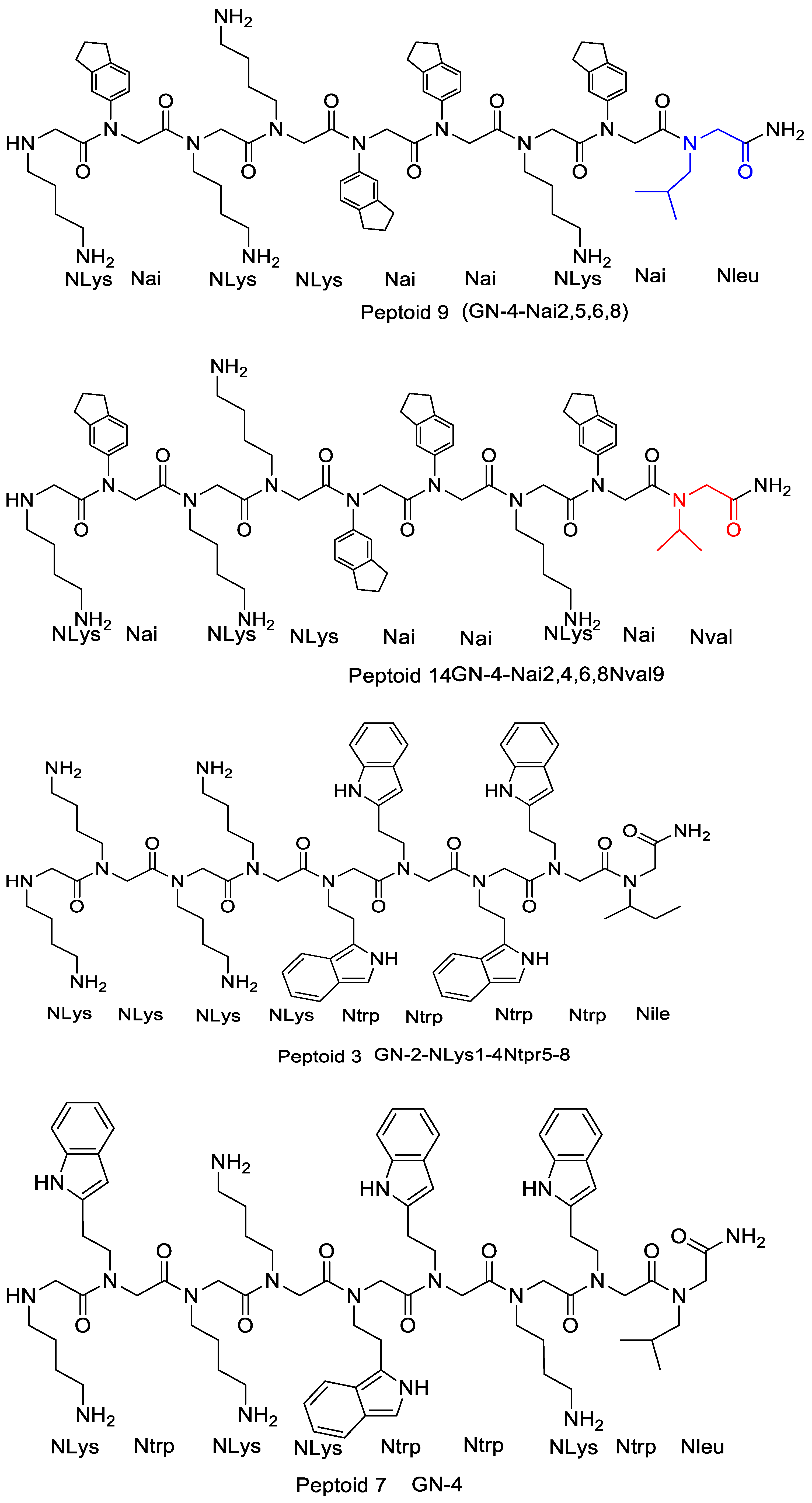
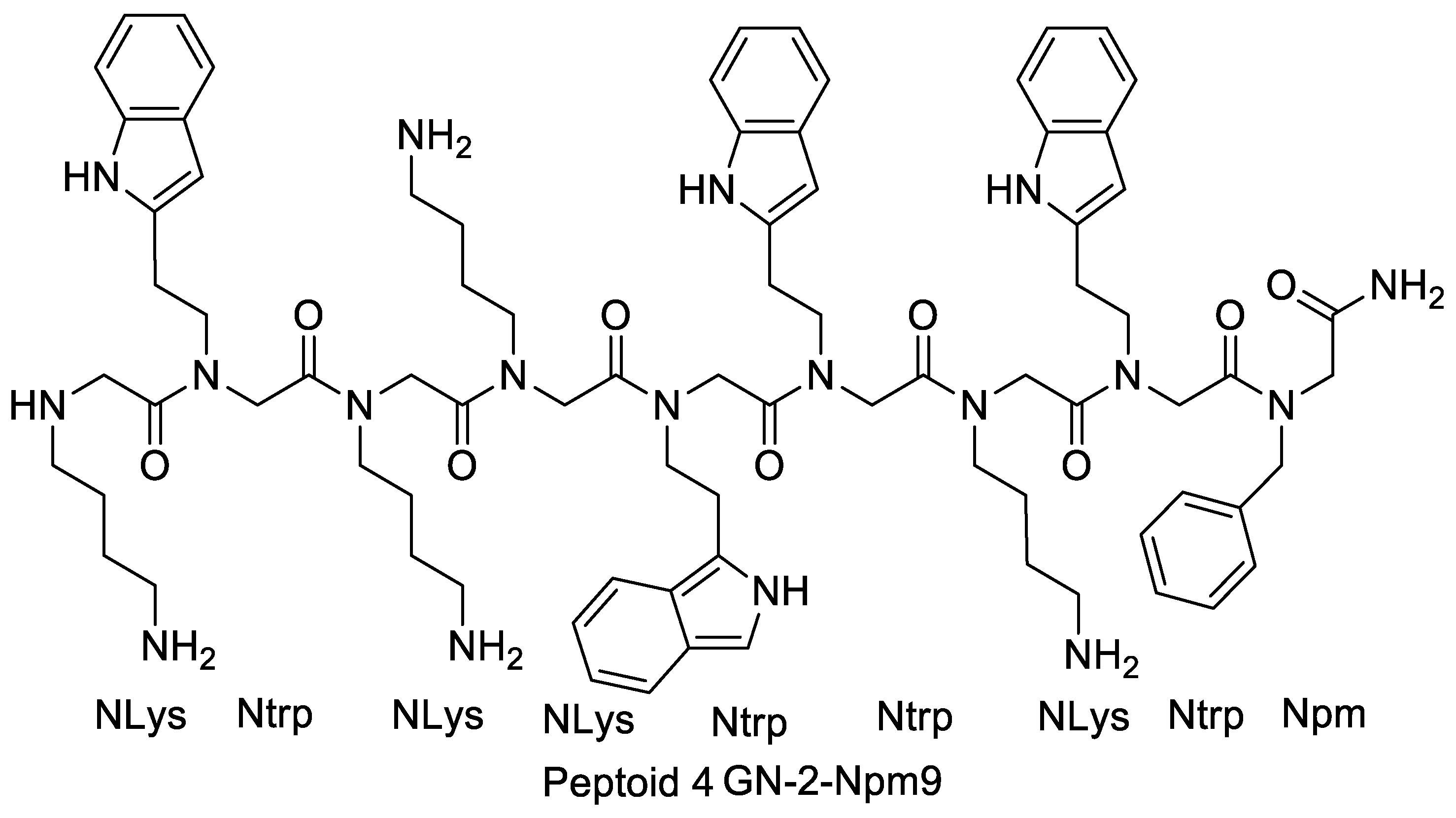
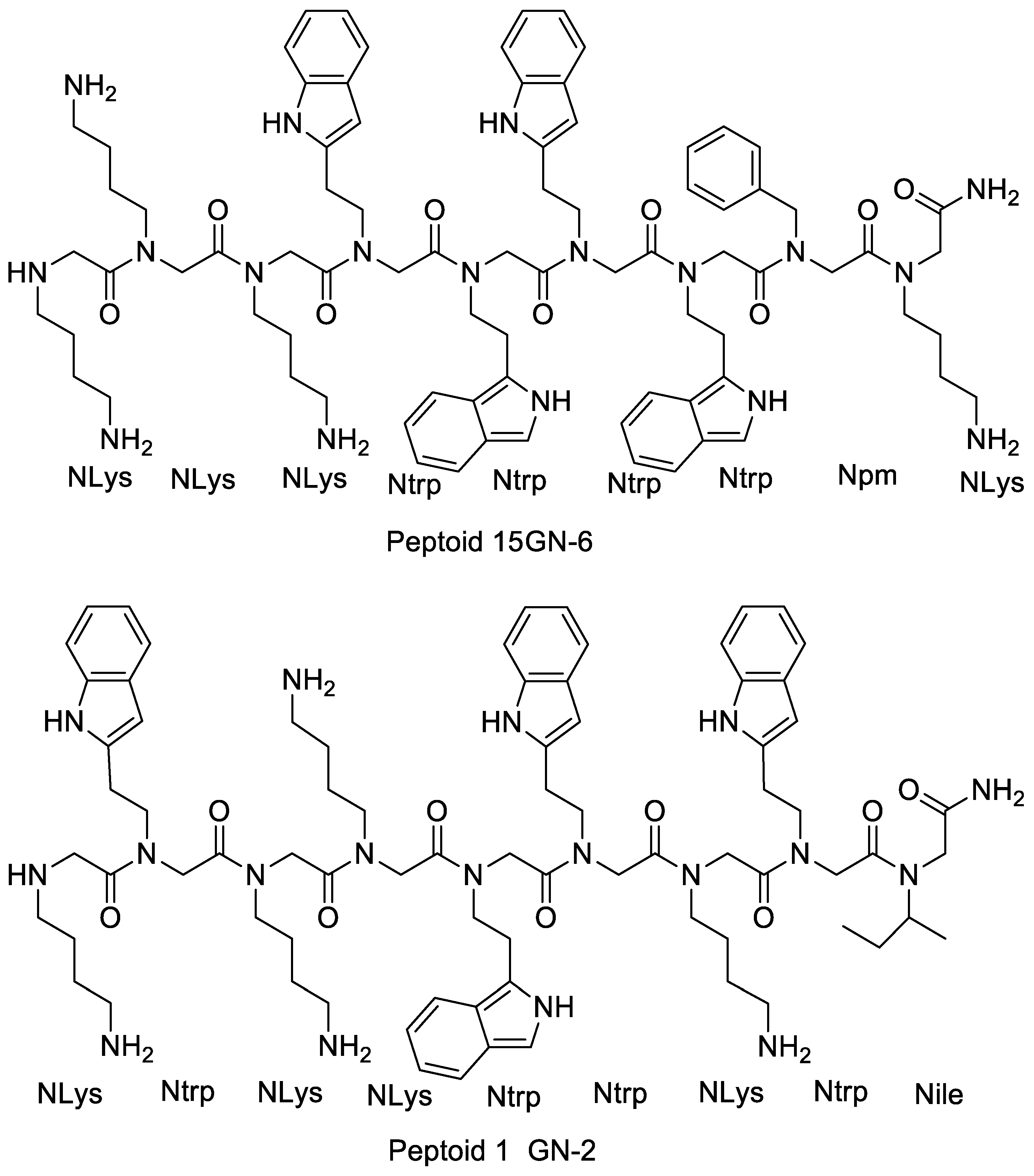
4.2.2. Effects of Increasing Hydrophobicity by Nlys Substitution
4.2.3. Effects of Increasing Hydrophobicity by Ntrp Substitution
4.2.4. The Effect of Increasing Hydrophobicity by Single-Monomer Substitution
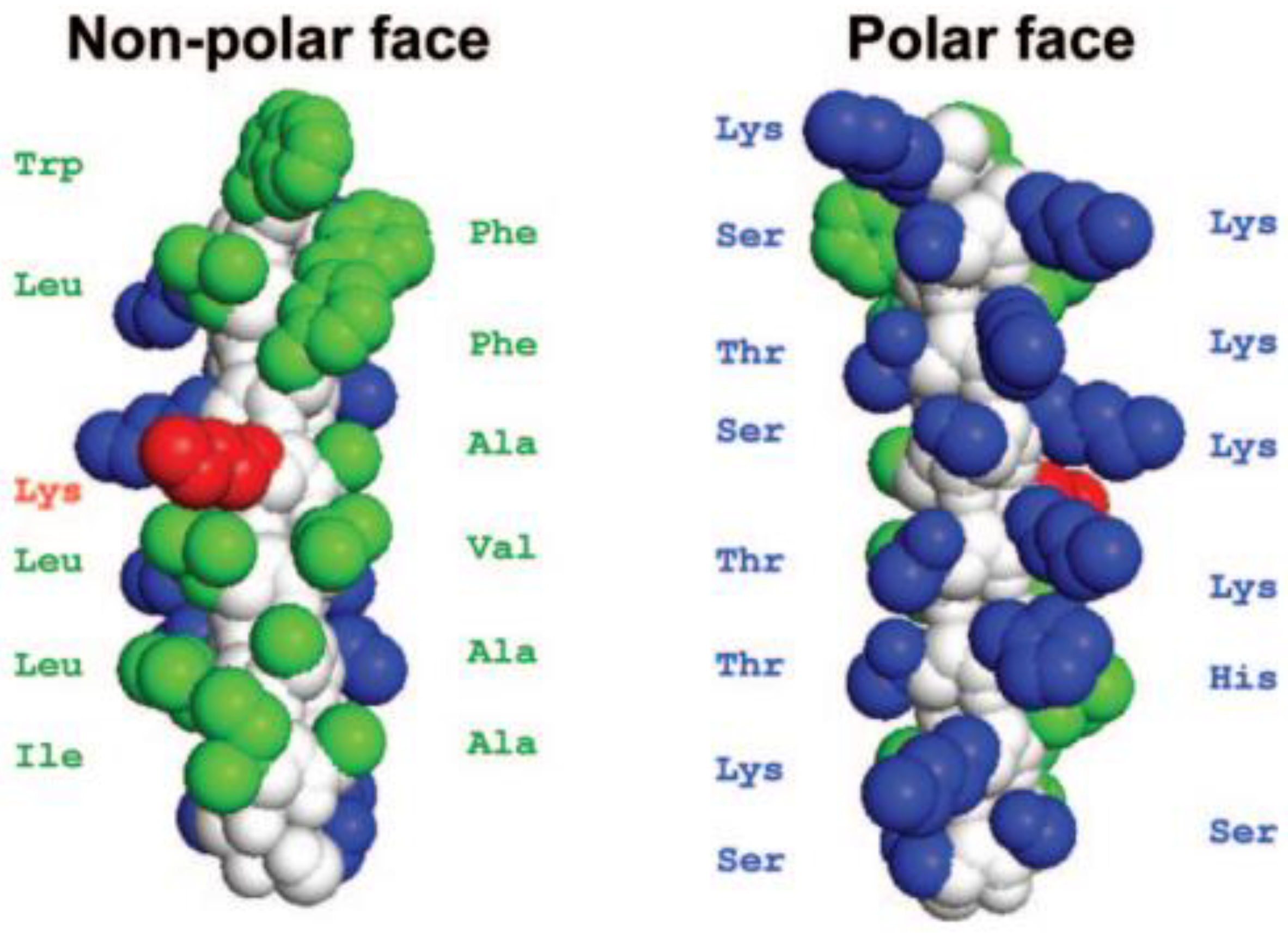
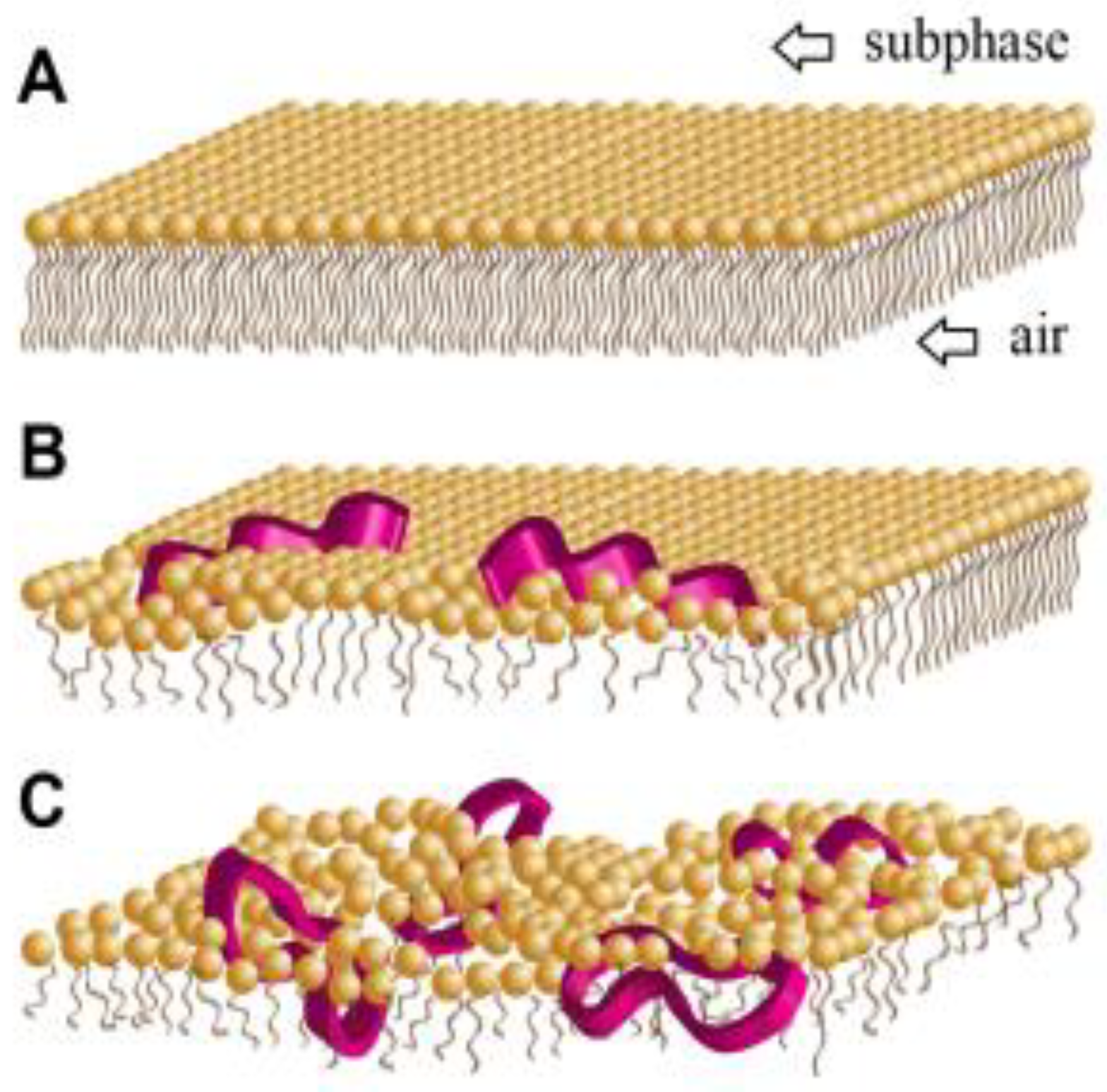
5. Amphiphilicity
Effects of Amphiphilicity by Sequence Rearrangement
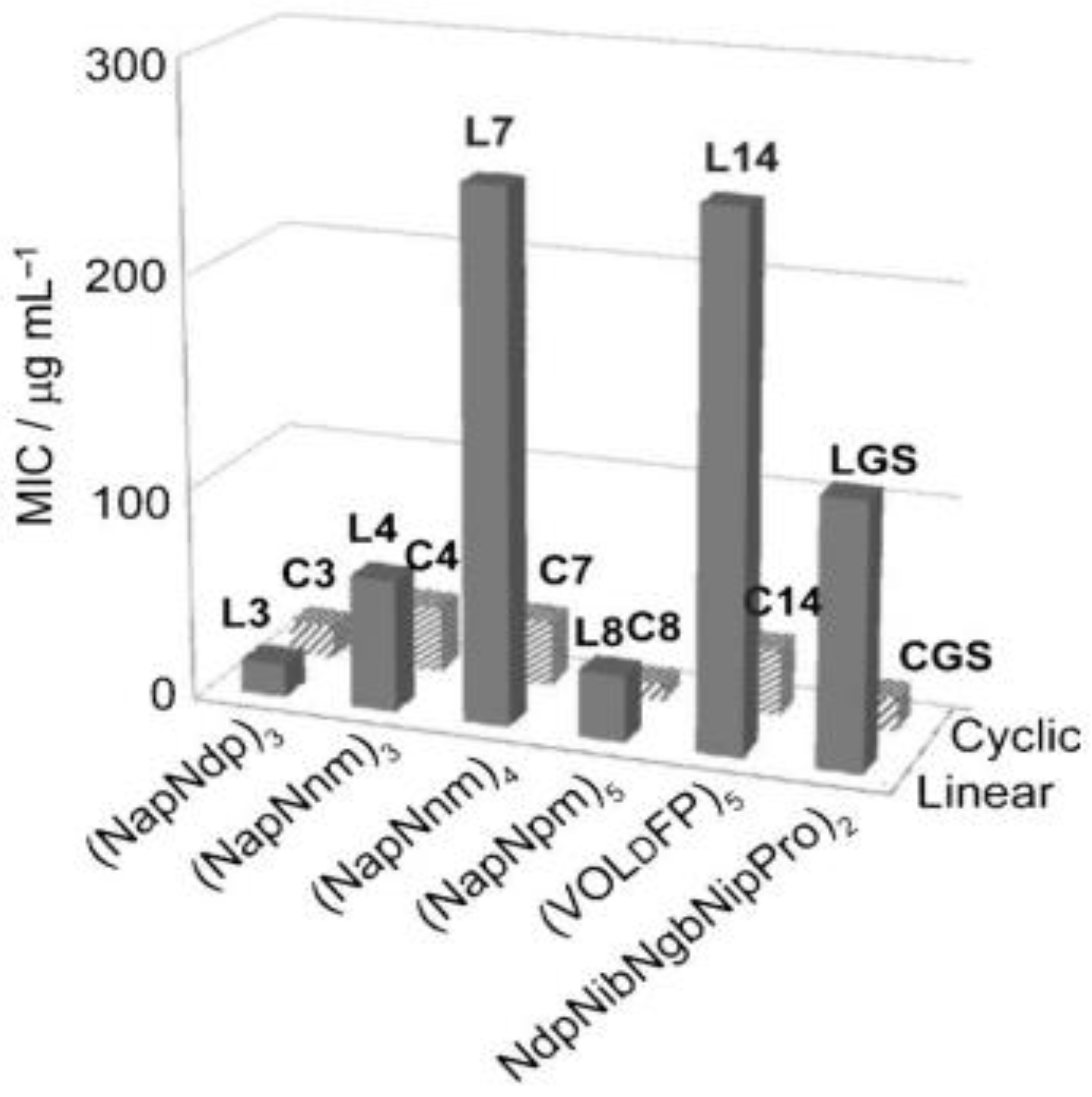
6. Cyclization
Effect of Cyclization on Antimicrobial Activity
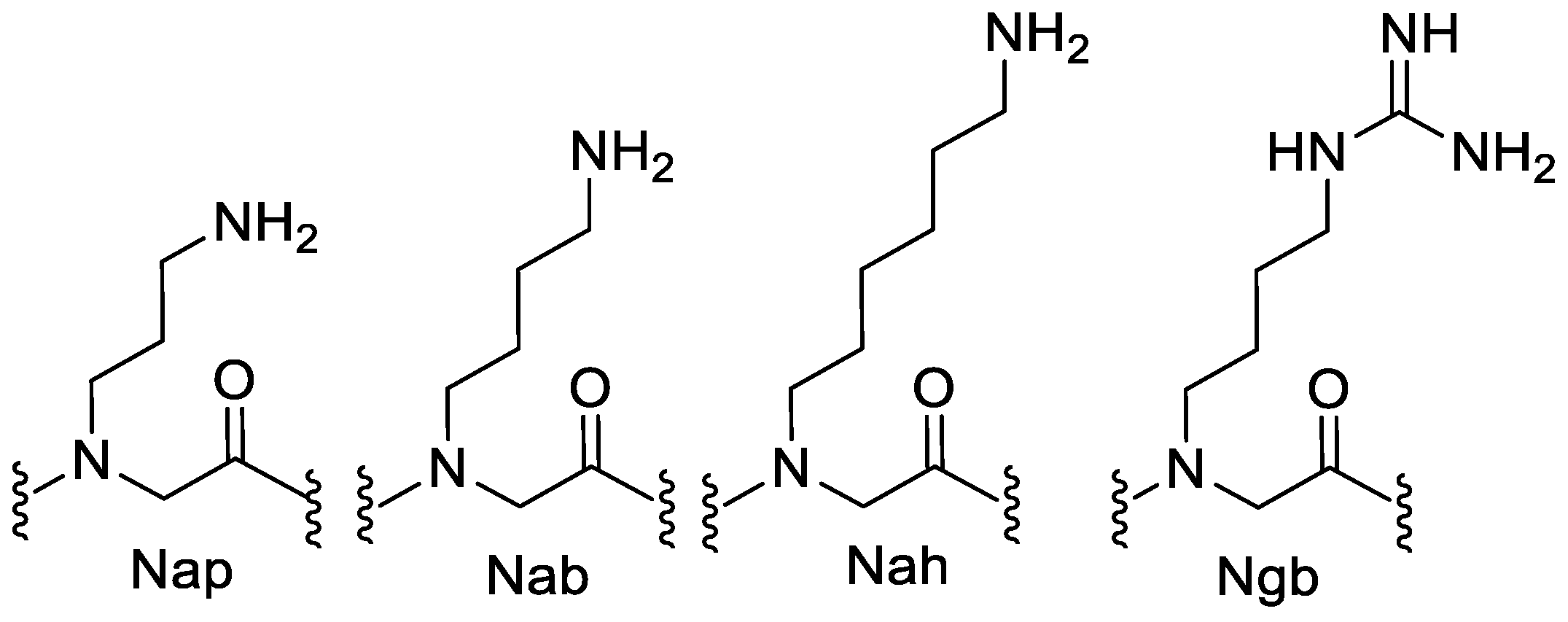
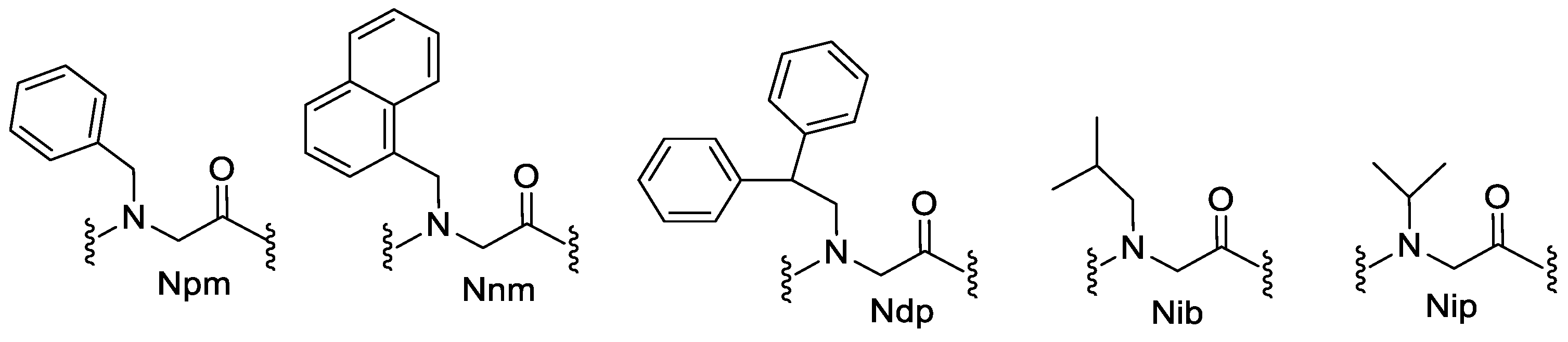
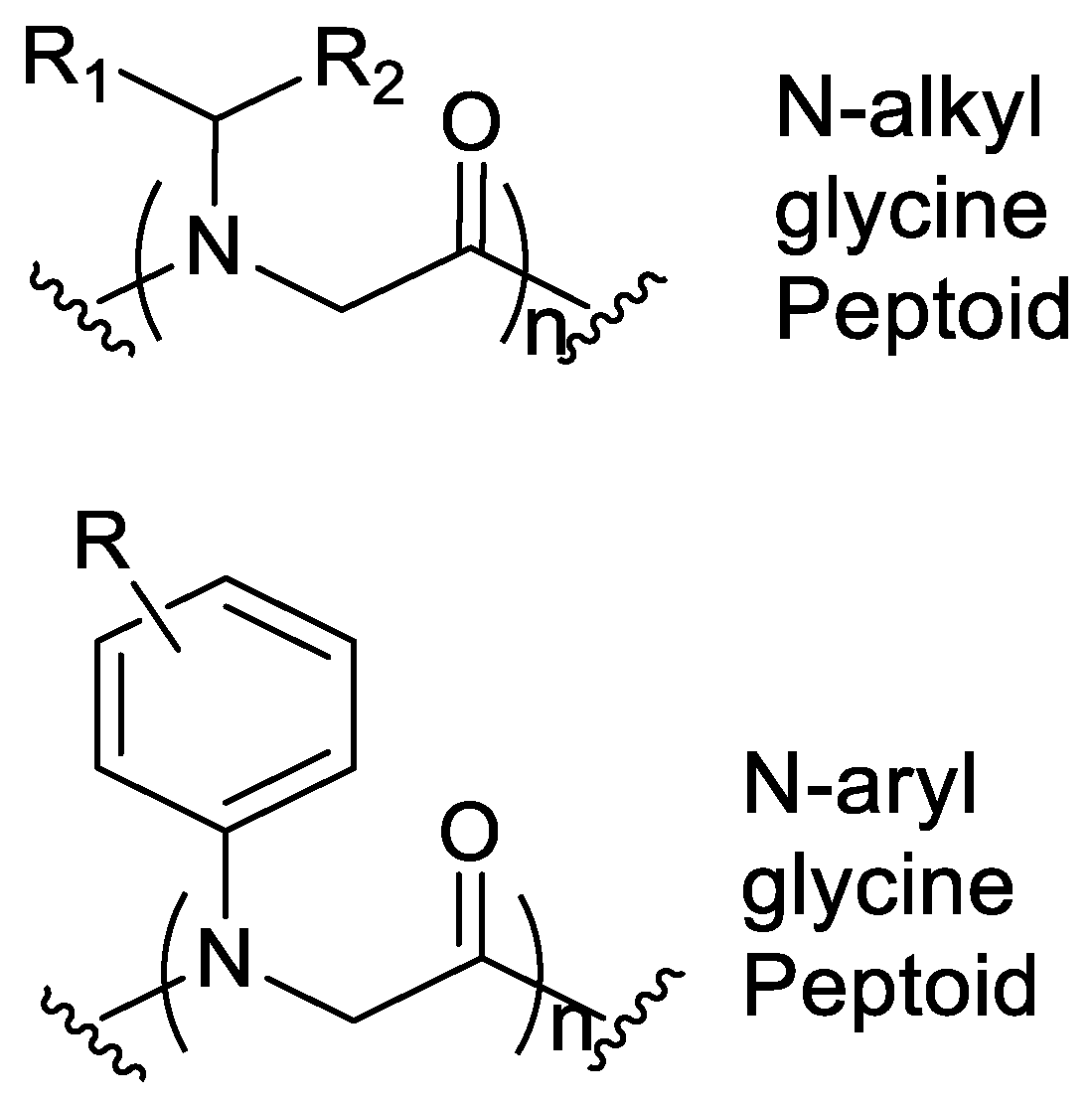
7. Aromatic Side Chains
N-aryl Groups within Peptoid Oligomers
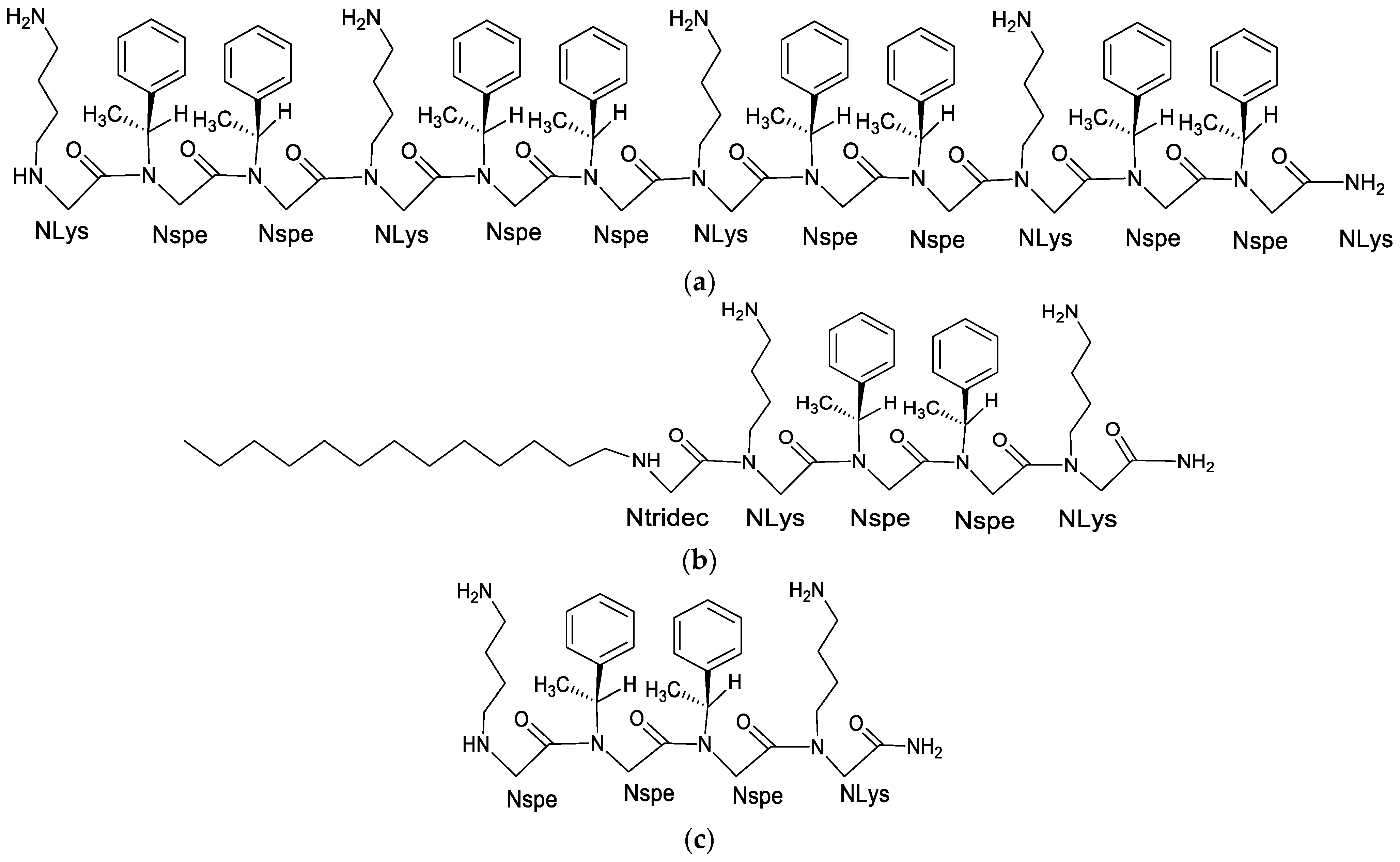
8. Alkylated Peptoids
8.1. Effect of Alkylated Peptoids on Antimicrobial Activity
8.2. Influence of Alkyl Tail on the Formation of Micellar Structures
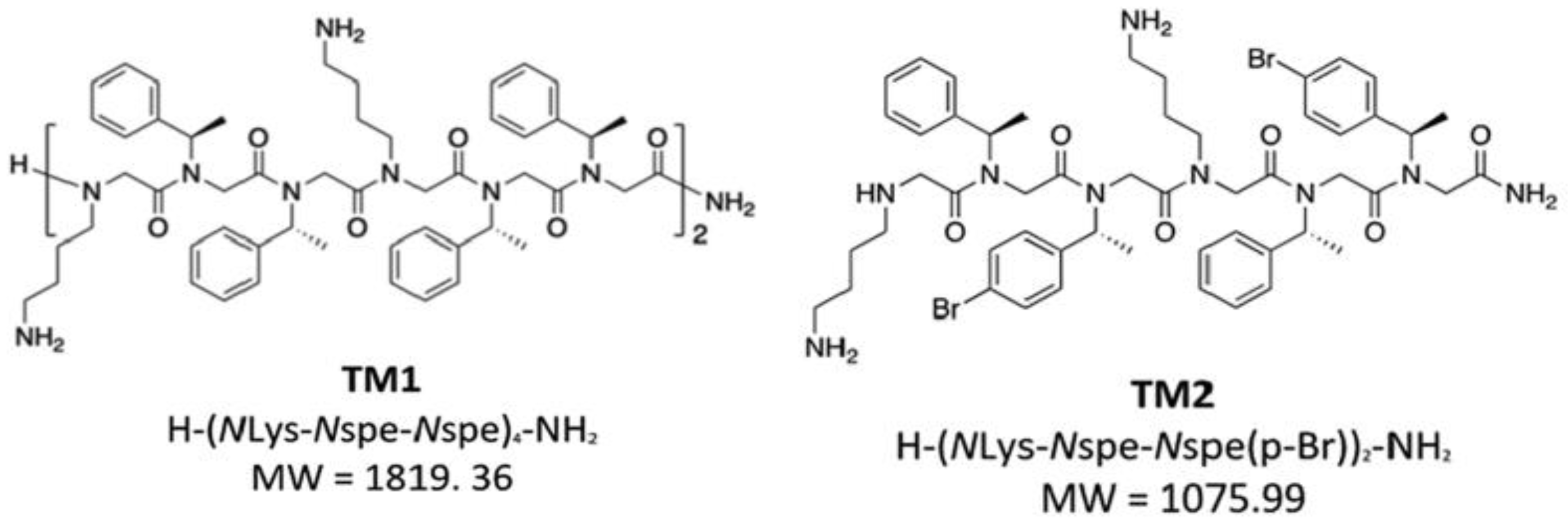
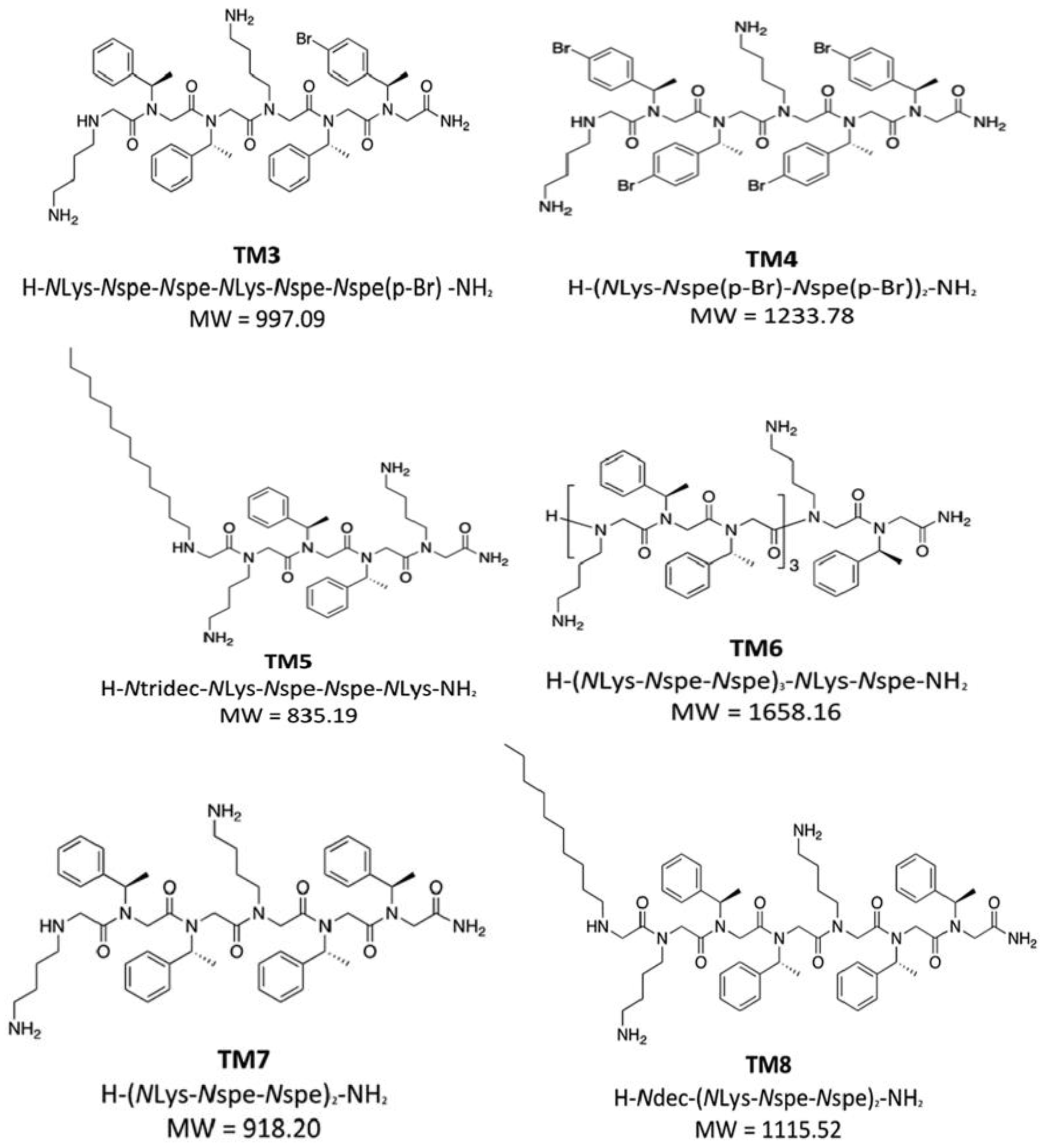
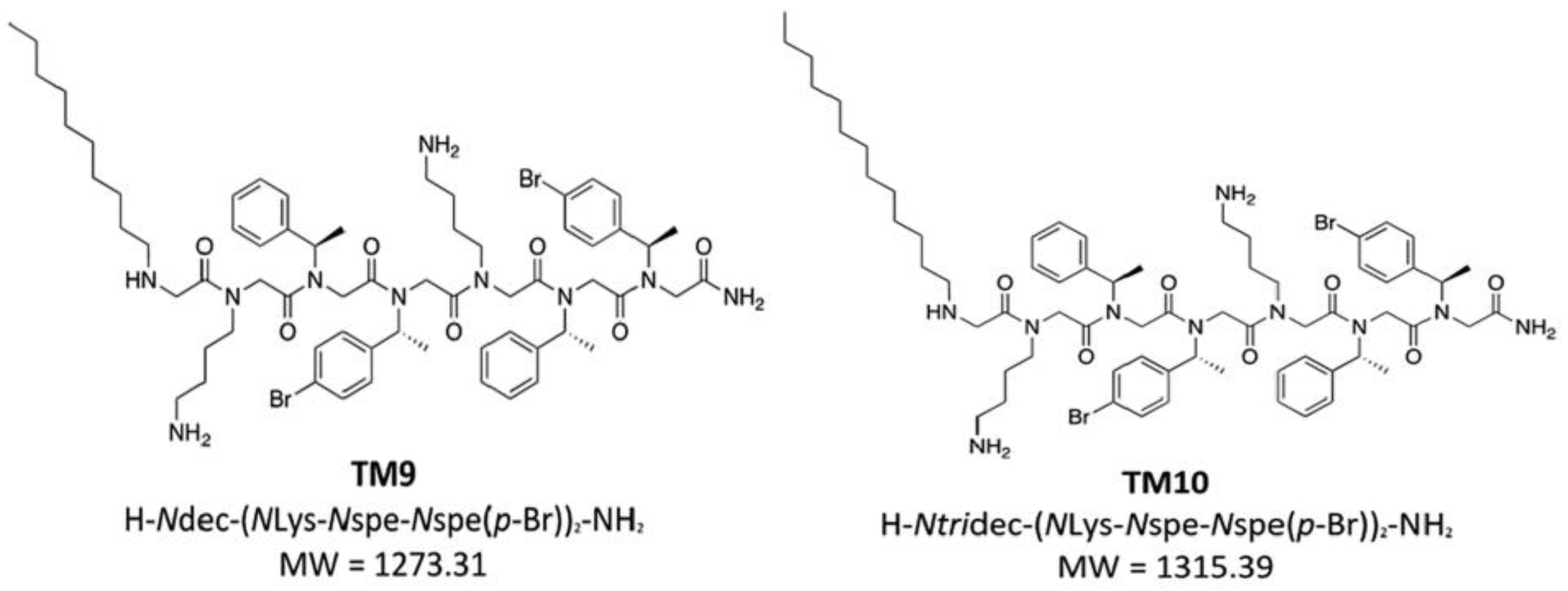
9. Halogen-Substituted Peptoids
10. Future Perspectives
11. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huan, Y.; Kong, Q.; Mou, H.; Yi, H. Antimicrobial Peptides: Classification, Design, Application and Research Progress in Multiple Fields. Front. Microbiol. 2020, 11, 582779. [Google Scholar] [CrossRef] [PubMed]
- Mojsoska, B.; Carretero, G.; Larsen, S.; Mateiu, R.V.; Jenssen, H. Peptoids Successfully Inhibit the Growth of Gram Negative E. coli Causing Substantial Membrane Damage. Sci. Rep. 2017, 7, srep42332. [Google Scholar] [CrossRef] [PubMed]
- Jenssen, H.; Hamill, P.; Hancock, R.E.W. Peptide Antimicrobial Agents. Clin. Microbiol. Rev. 2006, 19, 491–511. [Google Scholar] [CrossRef] [PubMed]
- Mojsoska, B.; Jenssen, H. Peptides and Peptidomimetics for Antimicrobial Drug Design. Pharmaceuticals 2015, 8, 366–415. [Google Scholar] [CrossRef]
- Ageitos, J.M.; Sánchez-Pérez, A.; Calo-Mata, P.; Villa, T.G. Antimicrobial peptides (AMPs): Ancient compounds that represent novel weapons in the fight against bacteria. Biochem. Pharmacol. 2017, 133, 117–138. [Google Scholar] [CrossRef]
- Malkoski, M.; Dashper, S.G.; O’Brien-Simpson, N.M.; Talbo, G.H.; Macris, M.; Cross, K.J.; Reynolds, E.C. Kappacin, a Novel Antibacterial Peptide from Bovine Milk. Antimicrob. Agents Chemother. 2001, 45, 2309–2315. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.L.; Shin, S.B.Y.; Benson, M.A.; Torres, V.J.; Kirshenbaum, K. A Comparison of Linear and Cyclic Peptoid Oligomers as Potent Antimicrobial Agents. ChemMedChem 2012, 7, 114–122. [Google Scholar] [CrossRef]
- Sani, M.A.; Henriques, S.T.; Weber, D.; Separovic, F. Bacteria May Cope Differently from Similar Membrane Damage Caused by the Australian Tree Frog Antimicrobial Peptide Maculatin 1.1. J. Biol. Chem. 2015, 290, 19853–19862. [Google Scholar] [CrossRef]
- Zasloff, M. Antimicrobial Peptides of Multicellular Organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef]
- Mwangi, J.; Hao, X.; Lai, R.; Zhang, Z.Y. Antimicrobial Peptides: New Hope in the War against Multidrug Resistance. Zool. Res. 2019, 40, 488–505. [Google Scholar] [CrossRef]
- Haug, B.E.; Stensen, W.; Kalaaji, M.; Rekdal, Ø.; Svendsen, J.S. Synthetic Antimicrobial Peptidomimetics with Therapeutic Potential. J. Med. Chem. 2008, 51, 4306–4314. [Google Scholar] [CrossRef] [PubMed]
- Mojsoska, B.; Zuckermann, R.N.; Jenssen, H. Structure-Activity Relationship Study of Novel Peptoids That Mimic the Structure of Antimicrobial Peptides. Antimicrob. Agents Chemother. 2015, 59, 4112–4120. [Google Scholar] [CrossRef] [PubMed]
- Turkett, J.A. Design and Screening of Antimicrobial Peptoid Libraries. Ph.D. Thesis, Middle Tennessee State University, Murfreesboro, TN, USA, 2017. [Google Scholar]
- Sun, J.; Chen, M.; Fu, Z.; Yang, J.; Zhou, S.; Yu, G.; Zhou, W.; Ma, Z. A Comparative Study on Low and High Salinity Tolerance of Two Strains of Pinctada Fucata. Front. Mar. Sci. 2021, 8, 704907. [Google Scholar] [CrossRef]
- Khara, J.S.; Mojsoska, B.; Mukherjee, D.; Langford, P.R.; Robertson, B.D.; Jenssen, H.; Ee, P.L.R.; Newton, S.M. Ultra-Short Antimicrobial Peptoids Show Propensity for Membrane Activity Against Multi-Drug Resistant Mycobacterium Tuberculosis. Front. Microbiol. 2020, 11, 417. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.M.; Simon, R.J.; Ng, S.; Zuckermann, R.N.; Kerr, J.M.; Moos, W.H. Proteolytic Studies of Homologous Peptide and N-Substituted Glycine Peptoid Oligomers. Bioorganic Med. Chem. Lett. 1994, 4, 2657–2662. [Google Scholar] [CrossRef]
- Miller, S.M.; Simon, R.J.; Ng, S.; Zuckermann, R.N.; Kerr, J.M.; Moos, W.H. Comparison of the Proteolytic Susceptibilities of Homologous L-Amino Acid, D-Amino Acid, and N-Substituted Clycine Peptide and Peptoid Oligomers. Drug Dev. Res. 1995, 35, 20–32. [Google Scholar] [CrossRef]
- Seo, J.; Lee, B.C.; Zuckermann, R.N. 2.3 Peptoids: Synthesis, Characterization, and Nanostructures. In Comprehensive Biomaterials II; Elsevier: Amsterdam, The Netherlands, 2017; pp. 41–66. [Google Scholar] [CrossRef]
- Kwon, Y.U.; Kodadek, T. Quantitative Evaluation of the Relative Cell Permeability of Peptoids and Peptides. J. Am. Chem. Soc. 2007, 129, 1508–1509. [Google Scholar] [CrossRef]
- Tan, N.C.; Yu, P.; Kwon, Y.-U.; Kodadek, T. High-Throughput Evaluation of Relative Cell Permeability between Peptoids and Peptides. Bioorganic Med. Chem. 2008, 16, 5853–5861. [Google Scholar] [CrossRef]
- Kapoor, R.; Eimerman, P.R.; Hardy, J.W.; Cirillo, J.D.; Contag, C.H.; Barron, A.E. Efficacy of Antimicrobial Peptoids against Mycobacterium Tuberculosis. Antimicrob. Agents Chemother. 2011, 55, 3058–3062. [Google Scholar] [CrossRef]
- Brice, D.C.; Toth, Z.; Diamond, G. LL-37 Disrupts the Kaposi’s Sarcoma-Associated Herpesvirus Envelope and Inhibits Infection in Oral Epithelial Cells; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Diamond, G.; Molchanova, N.; Herlan, C.; Fortkort, J.A.; Lin, J.S.; Figgins, E.; Bopp, N.; Ryan, L.K.; Chung, D.; Adcock, R.S.; et al. Potent Antiviral Activity against HSV-1 and SARS-CoV-2 by Antimicrobial Peptoids. Pharmaceuticals 2021, 14, 304. [Google Scholar] [CrossRef]
- Patch, J.A.; Barron, A.E. Helical Peptoid Mimics of Magainin-2 Amide. J. Am. Chem. Soc. 2003, 125, 12092–12093. [Google Scholar] [CrossRef] [PubMed]
- Chongsiriwatana, N.P.; Patch, J.A.; Czyzewski, A.M.; Dohm, M.T.; Ivankin, A.; Gidalevitz, D.; Zuckermann, R.N.; Barron, A.E. Peptoids That Mimic the Structure, Function, and Mechanism of Helical Antimicrobial Peptides. Proc. Natl. Acad. Sci. USA 2008, 105, 2794–2799. [Google Scholar] [CrossRef] [PubMed]
- Ryge, T.S.; Frimodt-Møller, N.; Hansen, P.R. Antimicrobial Activities of Twenty Lysine-Peptoid Hybrids against Clinically Relevant Bacteria and Fungi. Chemotherapy 2008, 54, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Olsen, C.A.; Bonke, G.; Vedel, L.; Adsersen, A.; Witt, M.; Franzyk, H.; Jaroszewski, J.W. α-Peptide/β-Peptoid Chimeras. Org. Lett. 2007, 9, 1549–1552. [Google Scholar] [CrossRef] [PubMed]
- Chongsiriwatana, N.P.; Miller, T.M.; Wetzler, M.; Vakulenko, S.; Karlsson, A.J.; Palecek, S.P.; Mobashery, S.; Barron, A.E. Short Alkylated Peptoid Mimics of Antimicrobial Lipopeptides. Antimicrob. Agents Chemother. 2011, 55, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Kamysz, W.; Silvestri, C.; Cirioni, O.; Giacometti, A.; Licci, A.; Della Vittoria, A.; Okroj, M.; Scalise, G. In Vitro Activities of the Lipopeptides Palmitoyl (Pal)-Lys-Lys-NH 2 and Pal-Lys-Lys Alone and in Combination with Antimicrobial Agents against Multiresistant Gram-Positive Cocci. Antimicrob. Agents Chemother. 2007, 51, 354–358. [Google Scholar] [CrossRef]
- Laverty, G.; McLaughlin, M.; Shaw, C.; Gorman, S.P.; Gilmore, B.F. Antimicrobial Activity of Short, Synthetic Cationic Lipopeptides. Chem. Biol. Drug. Des. 2010, 75, 563–569. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development q Settings. Adv. Drug. Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Makovitzki, A.; Avrahami, D.; Shai, Y. Ultrashort Antibacterial and Antifungal Lipopeptides. Proc. Natl. Acad. Sci. USA 2006, 103, 15997–16002. [Google Scholar] [CrossRef]
- Makovitzki, A.; Baram, J.; Shai, Y. Antimicrobial Lipopolypeptides Composed of Palmitoyl Di- and Tricationic Peptides: In Vitro and in Vivo Activities, Self-Assembly to Nanostructures, and a Plausible Mode of Action. Biochemistry 2008, 47, 10630–10636. [Google Scholar] [CrossRef]
- Huang, M.L.; Benson, M.A.; Shin, S.B.Y.; Torres, V.J.; Kirshenbaum, K. Amphiphilic Cyclic Peptoids That Exhibit Antimicrobial Activity by Disrupting Staphylococcus Aureus Membranes. Eur. J. Org. Chem. 2013, 2013, 3560–3566. [Google Scholar] [CrossRef]
- Vooturi, S.K.; Firestine, S.M. Synthetic Membrane-Targeted Antibiotics. Curr. Med. Chem. 2010, 17, 2292–2300. [Google Scholar] [CrossRef] [PubMed]
- Blondelle, S.E.; Houghten, R.A. Design of Model Amphipathic Peptides Having Potent Antimicrobial Activities+. Biochemistry 1992, 31, 12688–12694. [Google Scholar] [CrossRef] [PubMed]
- Bolt, H.L.; Cobb, S.L. A Practical Method for the Synthesis of Peptoids Containing Both Lysine-Type and Arginine-Type Monomers. Org. Biomol. Chem. 2016, 14, 1211–1215. [Google Scholar] [CrossRef]
- Wender, P.A.; Mitchell, D.J.; Pattabiraman, K.; Pelkey, E.T.; Steinman, L.; Rothbard, J.B. The Design, Synthesis, and Evaluation of Molecules That Enable or Enhance Cellular Uptake: Peptoid Molecular Transporters. Proc. Natl. Acad. Sci. USA 2000, 97, 13003–13008. [Google Scholar] [CrossRef]
- Nielsen, J.E.; Alford, M.A.; Yung, D.B.Y.; Molchanova, N.; Fortkort, J.A.; Lin, J.S.; Diamond, G.; Hancock, R.E.W.; Jenssen, H.; Pletzer, D.; et al. Self-Assembly of Antimicrobial Peptoids Impacts Their Biological Effects on ESKAPE Bacterial Pathogens. ACS Infect. Dis. 2022, 8, 533–545. [Google Scholar] [CrossRef]
- Amirkhanov, N.V.; Bardasheva, A.V.; Tikunova, N.V.; Pyshnyi, D.V. Synthetic Antimicrobial Peptides: III—Effect of Cationic Groups of Lysine, Arginine, and Histidine on Antimicrobial Activity of Peptides with a Linear Type of Amphipathicity. Russ. J. Bioorganic Chem. 2021, 47, 681–690. [Google Scholar] [CrossRef]
- Andreev, K.; Martynowycz, M.W.; Huang, M.L.; Kuzmenko, I.; Bu, W.; Kirshenbaum, K.; Gidalevitz, D. Hydrophobic Interactions Modulate Antimicrobial Peptoid Selectivity towards Anionic Lipid Membranes. Biochim. Biophys. Acta Biomembr. 2018, 1860, 1414–1423. [Google Scholar] [CrossRef]
- Nielsen, S.L.; Frimodt-Møller, N.; Kragelund, B.B.; Hansen, P.R. Structure-Activity Study of the Antibacterial Peptide Fallaxin. Protein Sci. 2007, 16, 1969–1976. [Google Scholar] [CrossRef]
- Munk, J.K.; Ritz, C.; Fliedner, F.P.; Frimodt-Moller, N.; Hansen, P.R. Novel Method to Identify the Optimal Antimicrobial Peptide in a Combination Matrix, Using Anoplin as an Example. Antimicrob. Agents Chemother. 2014, 58, 1063–1070. [Google Scholar] [CrossRef]
- Chen, Y.; Guarnieri, M.T.; Vasil, A.I.; Vasil, M.L.; Mant, C.T.; Hodges, R.S. Role of Peptide Hydrophobicity in the Mechanism of Action of α-Helical Antimicrobial Peptides. Antimicrob. Agents Chemother. 2007, 51, 1398–1406. [Google Scholar] [CrossRef] [PubMed]
- Maturana, P.; Martinez, M.; Noguera, M.E.; Santos, N.C.; Disalvo, E.A.; Semorile, L.; Maffia, P.C.; Hollmann, A. Lipid Selectivity in Novel Antimicrobial Peptides: Implication on Antimicrobial and Hemolytic Activity. Colloids Surf. B Biointerfaces 2017, 153, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Hollmann, A.; Martínez, M.; Noguera, M.E.; Augusto, M.T.; Disalvo, A.; Santos, N.C.; Semorile, L.; Maffía, P.C. Role of Amphipathicity and Hydrophobicity in the Balance between Hemolysis and Peptide-Membrane Interactions of Three Related Antimicrobial Peptides. Colloids Surf. B Biointerfaces 2016, 141, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Ivankin, A.; Kuzmenko, I.; Gidalevitz, D. Cholesterol Mediates Membrane Curvature during Fusion Events. Phys. Rev. Lett. 2012, 108, 238103. [Google Scholar] [CrossRef]
- Ivankin, A.; Apellániz, B.; Gidalevitz, D.; Nieva, J.L. Mechanism of Membrane Perturbation by the HIV-1 Gp41 Membrane-Proximal External Region and Its Modulation by Cholesterol. Biochim. Biophys. Acta Biomembr. 2012, 1818, 2521–2528. [Google Scholar] [CrossRef]
- Mcphee, J.B.; Scott, M.G.; Hancock, R.E.W. Design of Host Defence Peptides for Antimicrobial and Immunity Enhancing Activities. Comb. Chem. High Throughput Screen. 2005, 8, 257–272. [Google Scholar] [CrossRef]
- Zelezetsky, I.; Tossi, A. Alpha-Helical Antimicrobial Peptides-Using a Sequence Template to Guide Structure-Activity Relationship Studies. Biochim. Biophys. Acta Biomembr. 2006, 1758, 1436–1449. [Google Scholar] [CrossRef]
- Tyagi, A.; Kapoor, P.; Kumar, R.; Chaudhary, K.; Gautam, A.; Raghava, G.P.S. In Silico Models for Designing and Discovering Novel Anticancer Peptides. Sci. Rep. 2013, 3, srep02984. [Google Scholar] [CrossRef] [PubMed]
- Andreev, K.; Martynowycz, M.W.; Ivankin, A.; Huang, M.L.; Kuzmenko, I.; Meron, M.; Lin, B.; Kirshenbaum, K.; Gidalevitz, D. Cyclization Improves Membrane Permeation by Antimicrobial Peptoids. Langmuir 2016, 32, 12905–12913. [Google Scholar] [CrossRef]
- Shah, N.H.; Butterfoss, G.L.; Nguyen, K.; Yoo, B.; Bonneau, R.; Rabenstein, D.L.; Kirshenbaum, K. Oligo(N-Aryl Glycines): A New Twist on Structured Peptoids. J. Am. Chem. Soc. 2008, 130, 16622–16632. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, R.; Wadman, M.W.; Dohm, M.T.; Czyzewski, A.M.; Spormann, A.M.; Barron, A.E. Antimicrobial Peptoids Are Effective against Pseudomonas Aeruginosa Biofilms. Antimicrob. Agents Chemother. 2011, 55, 3054–3057. [Google Scholar] [CrossRef] [PubMed]
- Avrahami, D.; Shai, Y. Bestowing Antifungal and Antibacterial Activities by Lipophilic Acid Conjugation to D,L-Amino Acid-Containing Antimicrobial Peptides: A Plausible Mode of Action. Biochemistry 2003, 42, 14946–14956. [Google Scholar] [CrossRef] [PubMed]
- Lockwood, N.A.; Haseman, J.R.; Tirrell, M.V.; Mayo, K.H. Acylation of SC4 Dodecapeptide Increases Bactericidal Potency against Gram-Positive Bacteria, Including Drug-Resistant Strains. Biochem. J. 2004, 378, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, Y.; Yokomine, M.; Kuroda, D.; Tsumoto, K.; Morimoto, J.; Sando, S. Peptoid-Based Reprogrammable Template for Cell-Permeable Inhibitors of Protein-Protein Interactions. Chem. Sci. 2021, 12, 13292–13300. [Google Scholar] [CrossRef] [PubMed]




















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptoid No. | Peptoid Nomenclature | Sequence f (N-C) | Rt (min) a | MIC (µg/mL) for Strain b: | Hemolytic Concn (µg/mL) c | SR d | Cytotoxicity (µg/mL) e | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S. aureus | E. coli | P. aeruginosa | ||||||||||
| ATCC 21213 | C623 MRSA | ATCC 25922 | 63103 ESBL | PAO1 | H1027 MDR | |||||||
| 1 | GN-2 | H-Nlys-Ntrp-Nlys-Nlys-Ntrp-Ntrp-Nlys-Ntrp-Nile-NH2 | 10.54 | 64 | 32 | 32 | 32 | 32 | 4 | >128 | >2–32 | 168 |
| 3 | GN-2-Nlys1–4Ntrp5–8 | H-Nlys-Nlys-Nlys-Nlys-Ntrp-Ntrp-Ntrp-Ntrp-Nile-NH2 | 11.48 | 8 | 8 | 32 | 8 | 32 | 4 | 128 | 4–32 | 110 |
| 7 | GN-4 | H-Nlys-Ntrp-Nlys-Nlys-Ntrp-Ntrp-Nlys-Ntrp-Nleu-NH2 | 10.51 | 32 | ND | 64 | ND | 64 | ND | >128 | >2–4 | 166 |
| 9 | GN-4-Nai2,5,6,8 | H-Nlys-Nai-Nlys-Nlys-Nai-Nai-Nlys-Nai-Nleu-NH2 | 12.31 | 4 | 4 | 16–32 | 32 | 16–32 | 4 | 32 | 1–8 | 172 |
| 21 | Nlys1–4Ntrp5–8 | H-Nlys-Nlys-Nlys-Nlys-Ntrp-Ntrp-Ntrp-Ntrp-NH2 | 11.06 | 4 | ND | 16 | ND | ND | ND | 64 | 4–16 | ND |
| Sequence a | MIC [µg·mL−1] b | HC [µg·mL−1] c | SR d | ||||
|---|---|---|---|---|---|---|---|
| B. sub. | E. coli | S. aur. | HC50 | HC10 | |||
| L3 | Ac(NapNdp)3 | 1 | 15.6 | 15.6 | >250 | >62.5 | >4 |
| L4 | Ac(NapNnm)3 | 2 | 62.5 | 125 | >250 | >250 | >4 |
| L6 | Ac(NapNpm)3 | 125 | 125 | 250 | >250 | >250 | >2 |
| C3 | C(NapNnm)3 | 1 | 15.6 | 7.8 | >250 | 31.3 | >2 |
| C4 | C(NapNnm)3 | 0.5 | 31.3 | 31.3 | >250 | >250 | >8 |
| C6 | C(NapNpm)3 | 500 | >500 | 500 | >250 | >250 | NA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nyembe, P.L.; Ntombela, T.; Makatini, M.M. Review: Structure-Activity Relationship of Antimicrobial Peptoids. Pharmaceutics 2023, 15, 1506. https://doi.org/10.3390/pharmaceutics15051506
Nyembe PL, Ntombela T, Makatini MM. Review: Structure-Activity Relationship of Antimicrobial Peptoids. Pharmaceutics. 2023; 15(5):1506. https://doi.org/10.3390/pharmaceutics15051506
Chicago/Turabian StyleNyembe, Priscilla L., Thandokuhle Ntombela, and Maya M. Makatini. 2023. "Review: Structure-Activity Relationship of Antimicrobial Peptoids" Pharmaceutics 15, no. 5: 1506. https://doi.org/10.3390/pharmaceutics15051506