
DA7R: A 7-Letter Zip Code to Target PDAC
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemistry
2.1.1. Materials and Instruments
2.1.2. Peptide Synthesis
2.1.3. Synthesis of FITC-DA7R
2.1.4. Synthesis of PAPTPL-I-DA7R
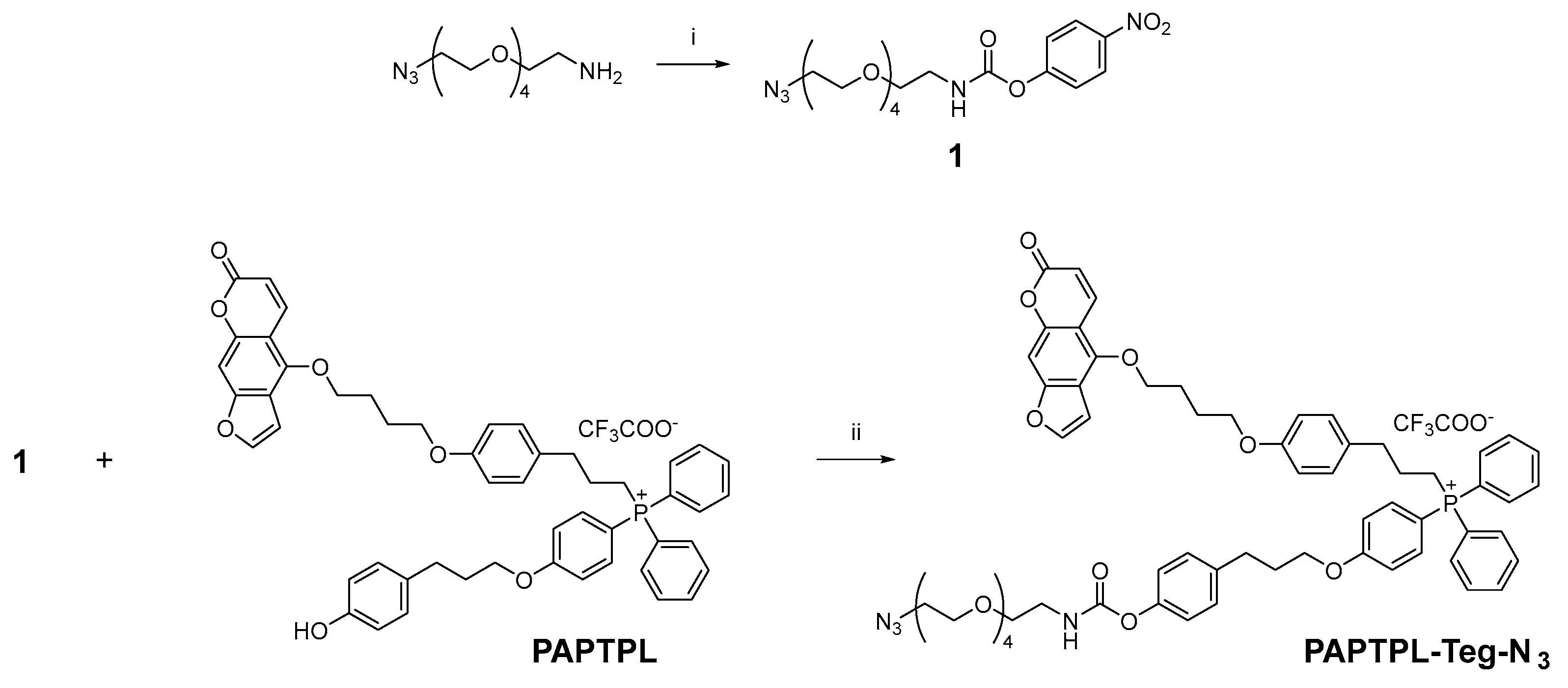
2.1.5. Synthesis of PAPTPL-Teg-DA7R and PAPTPL-Teg-cA7R
PAPTPL-Teg-N3 Synthesis
Synthesis of PAPTPL-Teg-DA7R
Synthesis of PAPTPL-Teg-cA7R
2.2. Cell Cultures
2.3. Protein Extraction
2.4. Western Blot
2.5. Blood Stability
2.6. Hemolysis Assay
2.7. Flow Cytometry
2.8. Cell Uptake of PAPTPL-I-DA7R
2.9. Statistics
3. Results
3.1. Chemistry
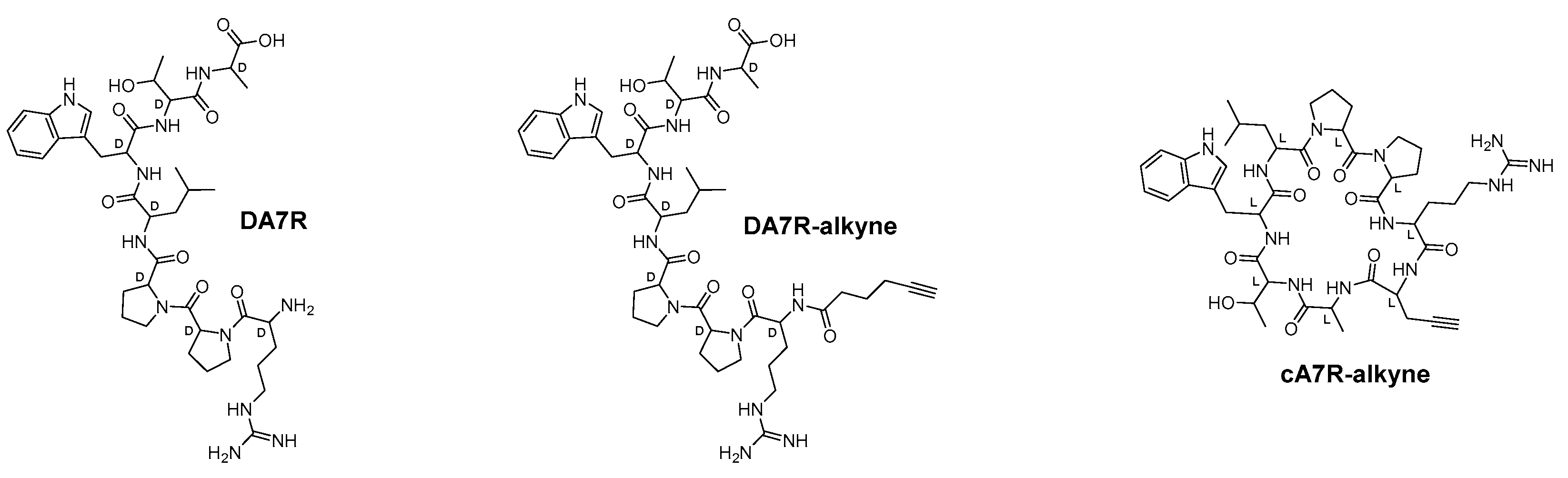
3.1.1. Peptide Design and Synthesis
3.1.2. Synthesis of PAPTPL-I-DA7R
3.1.3. Synthesis of PAPTPL-Teg-DA7R and PAPTPL-Teg-cA7R
Synthesis of PAPTPL-Teg-N3
Synthesis of PAPTPL-Teg-DA7R
Synthesis of PAPTPL-Teg-cA7R
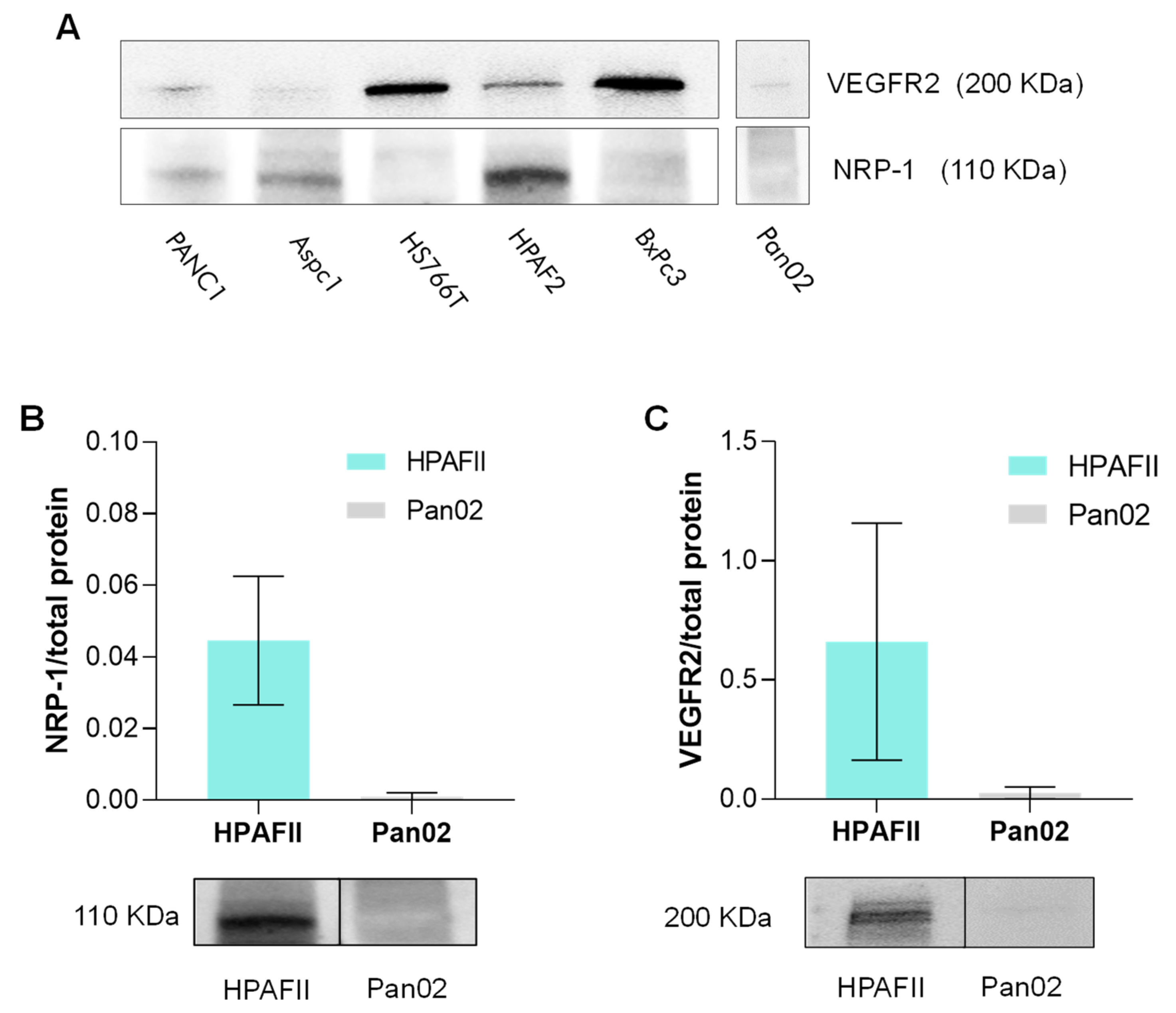
3.2. Neuropilin-1 and VEGFR2 Expression in PDAC Cell Lines
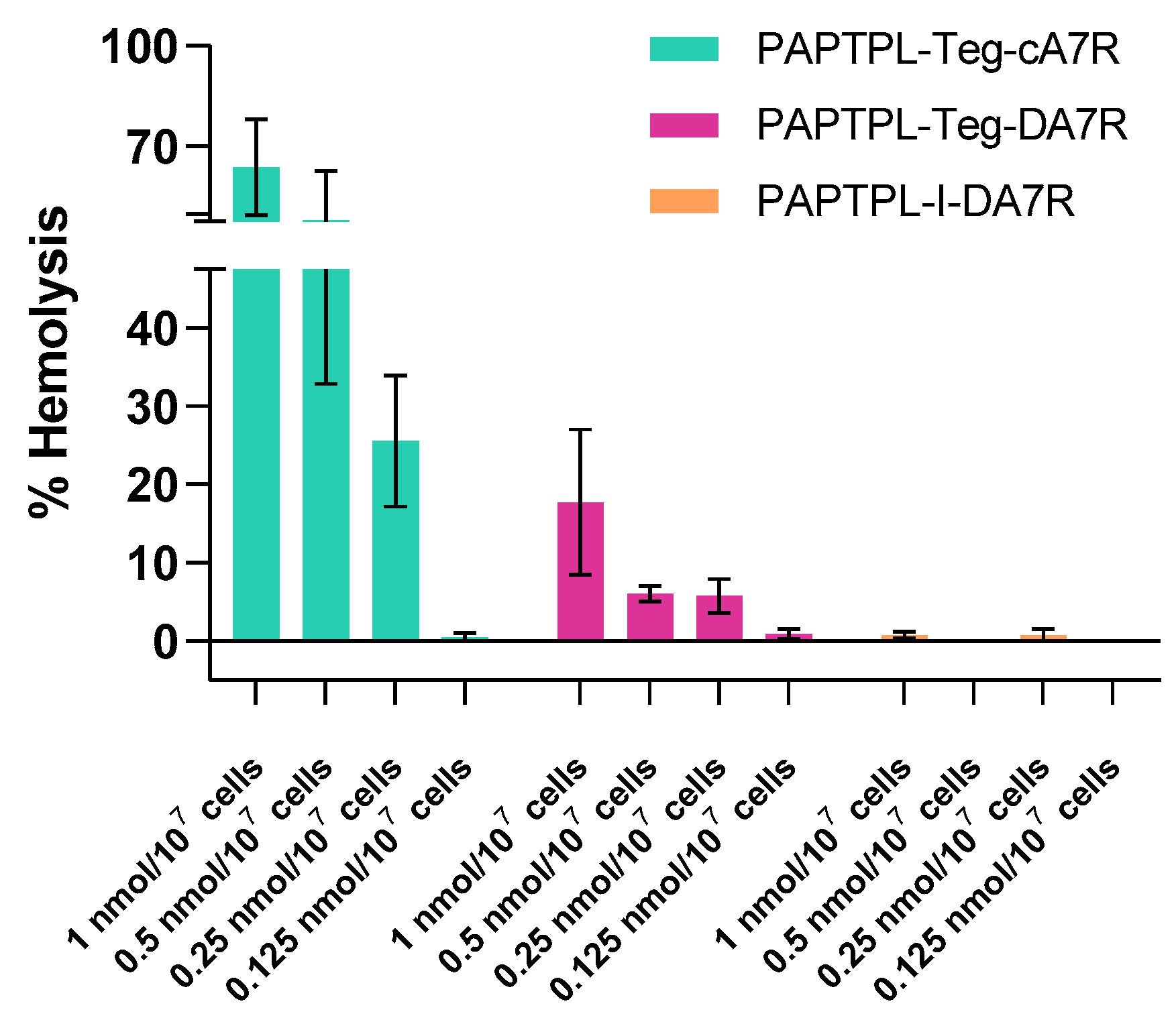
3.3. Blood Stability and Hemolysis
3.4. DA7R-FITC Cell Uptake
3.5. Cellular Uptake of PAPTPL-I-DA7R
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BBB | Blood–brain barrier |
| DCM | Dichloromethane |
| DIPEA | N,N-diisopropylethylamine |
| DMAP | 4-dimethylaminopyridine |
| DMEM | Dulbecco’s modified Eagle medium |
| DMF | N,N-dimethylformamide |
| DMSO | Dimethylsulfoxide |
| ESI-MS | Electrospray ionization-mass spectrometry |
| FBS | Fetal bovine serum |
| FITC | Fluorescein-5-isothiocyanate |
| Fmoc | Fluorenylmethoxycarbonyl |
| HATU | Hexafluorophosphate azabenzotriazole tetramethyl uronium |
| HBTU | Hexafluorophosphate benzotriazole tetramethyl uronium |
| HOBt | Hydroxybenzotriazole |
| NRP-1 | Neuropilin 1 |
| PBS | Phosphate buffer saline |
| PDAC | Pancreatic ductal adenocarcinoma |
| TFA | Trifluoroacetic acid |
| VEGFR2 | Vascular endothelial growth factor receptor 2 |
References
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- Halbrook, C.J.; Lyssiotis, C.A.; Pasca di Magliano, M.; Maitra, A. Pancreatic cancer: Advances and challenges. Cell 2023, 186, 1729–1754. [Google Scholar] [CrossRef] [PubMed]
- Parrasia, S.; Zoratti, M.; Szabò, I.; Biasutto, L. Targeting Pancreatic Ductal Adenocarcinoma (PDAC). Cell. Physiol. Biochem. 2021, 55, 61–90. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Fagman, J.B.; Ma, Y.; Liu, J.; Vihav, C.; Engstrom, C.; Liu, B.; Chen, C. A comprehensive review of pancreatic cancer and its therapeutic challenges. Aging 2022, 14, 7635–7649. [Google Scholar] [CrossRef]
- Anderson, E.M.; Thomassian, S.; Gong, J.; Hendifar, A.; Osipov, A. Advances in Pancreatic Ductal Adenocarcinoma Treatment. Cancers 2021, 13, 5510. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Gong, Y.; Fan, Z.; Luo, G.; Huang, Q.; Deng, S.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; et al. Molecular alterations and targeted therapy in pancreatic ductal adenocarcinoma. J. Hematol. Oncol. 2020, 13, 130. [Google Scholar] [CrossRef]
- Hu, H.F.; Ye, Z.; Qin, Y.; Xu, X.W.; Yu, X.J.; Zhuo, Q.F.; Ji, S.R. Mutations in key driver genes of pancreatic cancer: Molecularly targeted therapies and other clinical implications. Acta Pharmacol. Sin. 2021, 42, 1725–1741. [Google Scholar] [CrossRef]
- Hsu, S.K.; Jadhao, M.; Liao, W.T.; Chang, W.T.; Hung, C.T.; Chiu, C.C. Culprits of PDAC resistance to gemcitabine and immune checkpoint inhibitor: Tumour microenvironment components. Front. Mol. Biosci. 2022, 9, 1020888. [Google Scholar] [CrossRef] [PubMed]
- Truong, L.H.; Pauklin, S. Pancreatic Cancer Microenvironment and Cellular Composition: Current Understandings and Therapeutic Approaches. Cancers 2021, 13, 5028. [Google Scholar] [CrossRef]
- Gautam, S.K.; Basu, S.; Aithal, A.; Dwivedi, N.V.; Gulati, M.; Jain, M. Regulation of pancreatic cancer therapy resistance by chemokines. Semin. Cancer Biol. 2022, 86, 69–80. [Google Scholar] [CrossRef]
- Ayasun, R.; Saridogan, T.; Gaber, O.; Sahin, I.H. Systemic Therapy for Patients with Pancreatic Cancer: Current Approaches and Opportunities for Novel Avenues toward Precision Medicine. Clin. Color. Cancer 2022, 22, 2–11. [Google Scholar] [CrossRef]
- Timur, S.S.; Gürsoy, R.N. The role of peptide-based therapeutics in oncotherapy. J. Drug Target. 2021, 29, 1048–1062. [Google Scholar] [CrossRef] [PubMed]
- Bose, D.; Roy, L.; Chatterjee, S. Peptide therapeutics in the management of metastatic cancers. RSC Adv. 2022, 12, 21353–21373. [Google Scholar] [CrossRef]
- Karami Fath, M.; Babakhaniyan, K.; Zokaei, M.; Yaghoubian, A.; Akbari, S.; Khorsandi, M.; Soofi, A.; Nabi-Afjadi, M.; Zalpoor, H.; Jalalifar, F.; et al. Anti-cancer peptide-based therapeutic strategies in solid tumors. Cell. Mol. Biol. Lett. 2022, 27, 33. [Google Scholar] [CrossRef] [PubMed]
- Binétruy-Tournaire, R.; Demangel, C.; Malavaud, B.; Vassy, R.; Rouyre, S.; Kraemer, M.; Plouët, J.; Derbin, C.; Perret, G.; Mazié, J.C. Identification of a peptide blocking vascular endothelial growth factor (VEGF)-mediated angiogenesis. EMBO J. 2000, 19, 1525–1533. [Google Scholar] [CrossRef]
- Lu, L.; Chen, H.; Hao, D.; Zhang, X.; Wang, F. The functions and applications of A7R in anti-angiogenic therapy, imaging and drug delivery systems. Asian J. Pharm. Sci. 2019, 14, 595–608. [Google Scholar] [CrossRef]
- Thomas, N.; Tirand, L.; Chatelut, E.; Plénat, F.; Frochot, C.; Dodeller, M.; Guillemin, F.; Barberi-Heyob, M. Tissue distribution and pharmacokinetics of an ATWLPPR-conjugated chlorin-type photosensitizer targeting neuropilin-1 in glioma-bearing nude mice. Photochem. Photobiol. Sci. 2008, 7, 433–441. [Google Scholar] [CrossRef]
- Tirand, L.; Thomas, N.; Dodeller, M.; Dumas, D.; Frochot, C.; Maunit, B.; Guillemin, F.; Barberi-Heyob, M. Metabolic profile of a peptide-conjugated chlorin-type photosensitizer targeting neuropilin-1: An in vivo and in vitro study. Drug Metab. Dispos. 2007, 35, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Ying, M.; Shen, Q.; Liu, Y.; Yan, Z.; Wei, X.; Zhan, C.; Gao, J.; Xie, C.; Yao, B.; Lu, W. Stabilized Heptapeptide A7R for Enhanced Multifunctional Liposome-Based Tumor-Targeted Drug Delivery. ACS Appl. Mater. Interfaces 2016, 8, 13232–13241. [Google Scholar] [CrossRef] [PubMed]
- Ying, M.; Shen, Q.; Zhan, C.; Wei, X.; Gao, J.; Xie, C.; Yao, B.; Lu, W. A stabilized peptide ligand for multifunctional glioma targeted drug delivery. J. Control. Release 2016, 243, 86–98. [Google Scholar] [CrossRef]
- Lucana, M.C.; Arruga, Y.; Petrachi, E.; Roig, A.; Lucchi, R.; Oller-Salvia, B. Protease-Resistant Peptides for Targeting and Intracellular Delivery of Therapeutics. Pharmaceutics 2021, 13, 2065. [Google Scholar] [CrossRef]
- Rai, J. Peptide and protein mimetics by retro and retroinverso analogs. Chem. Biol. Drug Des. 2019, 93, 724–736. [Google Scholar] [CrossRef] [PubMed]
- Ellis, L.M. The role of neuropilins in cancer. Mol. Cancer Ther. 2006, 5, 1099–1107. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.T.; Radin, D.P.; Tsirka, S.E. From protein-protein interactions to immune modulation: Therapeutic prospects of targeting Neuropilin-1 in high-grade glioma. Front. Immunol. 2022, 13, 958620. [Google Scholar] [CrossRef]
- Zhao, L.; Chen, H.; Lu, L.; Wang, L.; Zhang, X.; Guo, X. New insights into the role of co-receptor neuropilins in tumour angiogenesis and lymphangiogenesis and targeted therapy strategies. J. Drug Target. 2021, 29, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Starzec, A.; Ladam, P.; Vassy, R.; Badache, S.; Bouchemal, N.; Navaza, A.; du Penhoat, C.H.; Perret, G.Y. Structure-function analysis of the antiangiogenic ATWLPPR peptide inhibiting VEGF(165) binding to neuropilin-1 and molecular dynamics simulations of the ATWLPPR/neuropilin-1 complex. Peptides 2007, 28, 2397–2402. [Google Scholar] [CrossRef]
- Pang, H.B.; Braun, G.B.; Friman, T.; Aza-Blanc, P.; Ruidiaz, M.E.; Sugahara, K.N.; Teesalu, T.; Ruoslahti, E. An endocytosis pathway initiated through neuropilin-1 and regulated by nutrient availability. Nat. Commun. 2014, 5, 4904. [Google Scholar] [CrossRef]
- Pang, H.B.; Braun, G.B.; Ruoslahti, E. Neuropilin-1 and heparan sulfate proteoglycans cooperate in cellular uptake of nanoparticles functionalized by cationic cell-penetrating peptides. Sci. Adv. 2015, 1, e1500821. [Google Scholar] [CrossRef]
- Parrasia, S.; Szabò, I.; Zoratti, M.; Biasutto, L. Peptides as Pharmacological Carriers to the Brain: Promises, Shortcomings and Challenges. Mol. Pharm. 2022, 19, 3700–3729. [Google Scholar] [CrossRef]
- von Marschall, Z.; Cramer, T.; Höcker, M.; Burde, R.; Plath, T.; Schirner, M.; Heidenreich, R.; Breier, G.; Riecken, E.O.; Wiedenmann, B.; et al. De novo expression of vascular endothelial growth factor in human pancreatic cancer: Evidence for an autocrine mitogenic loop. Gastroenterology 2000, 119, 1358–1372. [Google Scholar] [CrossRef]
- Itakura, J.; Ishiwata, T.; Shen, B.; Kornmann, M.; Korc, M. Concomitant over-expression of vascular endothelial growth factor and its receptors in pancreatic cancer. Int. J. Cancer 2000, 85, 27–34. [Google Scholar] [CrossRef]
- Doi, Y.; Yashiro, M.; Yamada, N.; Amano, R.; Ohira, G.; Komoto, M.; Noda, S.; Kashiwagi, S.; Kato, Y.; Fuyuhiro, Y.; et al. Significance of phospho-vascular endothelial growth factor receptor-2 expression in pancreatic cancer. Cancer Sci. 2010, 101, 1529–1535. [Google Scholar] [CrossRef] [PubMed]
- Fukahi, K.; Fukasawa, M.; Neufeld, G.; Itakura, J.; Korc, M. Aberrant expression of neuropilin-1 and -2 in human pancreatic cancer cells. Clin. Cancer Res. 2004, 10, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Ben, Q.; Zheng, J.; Fei, J.; An, W.; Li, P.; Li, Z.; Yuan, Y. High neuropilin 1 expression was associated with angiogenesis and poor overall survival in resected pancreatic ductal adenocarcinoma. Pancreas 2014, 43, 744–749. [Google Scholar] [CrossRef]
- Parikh, A.A.; Liu, W.B.; Fan, F.; Stoeltzing, O.; Reinmuth, N.; Bruns, C.J.; Bucana, C.D.; Evans, D.B.; Ellis, L.M. Expression and regulation of the novel vascular endothelial growth factor receptor neuropilin-1 by epidermal growth factor in human pancreatic carcinoma. Cancer 2003, 98, 720–729. [Google Scholar] [CrossRef]
- Chung, G.G.; Yoon, H.H.; Zerkowski, M.P.; Ghosh, S.; Thomas, L.; Harigopal, M.; Charette, L.A.; Salem, R.R.; Camp, R.L.; Rimm, D.L.; et al. Vascular endothelial growth factor, FLT-1, and FLK-1 analysis in a pancreatic cancer tissue microarray. Cancer 2006, 106, 1677–1684. [Google Scholar] [CrossRef]
- Matkar, P.N.; Jong, E.D.; Ariyagunarajah, R.; Prud’homme, G.J.; Singh, K.K.; Leong-Poi, H. Jack of many trades: Multifaceted role of neuropilins in pancreatic cancer. Cancer Med. 2018, 7, 5036–5046. [Google Scholar] [CrossRef]
- Zeng, Z.; Fang, C.; Zhang, Y.; Chen, C.X.; Zhang, Y.F.; Zhang, K. Mitochondria-Targeted Nanocarriers Promote Highly Efficient Cancer Therapy: A Review. Front. Bioeng. Biotechnol. 2021, 9, 784602. [Google Scholar] [CrossRef]
- Guo, X.; Yang, N.; Ji, W.; Zhang, H.; Dong, X.; Zhou, Z.; Li, L.; Shen, H.M.; Yao, S.Q.; Huang, W. Mito-Bomb: Targeting Mitochondria for Cancer Therapy. Adv. Mater. 2021, 33, e2007778. [Google Scholar] [CrossRef]
- Fialova, J.L.; Raudenska, M.; Jakubek, M.; Kejik, Z.; Martasek, P.; Babula, P.; Matkowski, A.; Filipensky, P.; Masarik, M. Novel Mitochondria-targeted Drugs for Cancer Therapy. Mini Rev. Med. Chem. 2021, 21, 816–832. [Google Scholar] [CrossRef]
- Dong, L.; Gopalan, V.; Holland, O.; Neuzil, J. Mitocans Revisited: Mitochondrial Targeting as Efficient Anti-Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 7941. [Google Scholar] [CrossRef]
- Schmitz, A.; Sankaranarayanan, A.; Azam, P.; Schmidt-Lassen, K.; Homerick, D.; Hänsel, W.; Wulff, H. Design of PAP-1, a selective small molecule Kv1.3 blocker, for the suppression of effector memory T cells in autoimmune diseases. Mol. Pharmacol. 2005, 68, 1254–1270. [Google Scholar] [CrossRef] [PubMed]
- Zielonka, J.; Joseph, J.; Sikora, A.; Hardy, M.; Ouari, O.; Vasquez-Vivar, J.; Cheng, G.; Lopez, M.; Kalyanaraman, B. Mitochondria-Targeted Triphenylphosphonium-Based Compounds: Syntheses, Mechanisms of Action, and Therapeutic and Diagnostic Applications. Chem. Rev. 2017, 117, 10043–10120. [Google Scholar] [CrossRef]
- Leanza, L.; Romio, M.; Becker, K.A.; Azzolini, M.; Trentin, L.; Managò, A.; Venturini, E.; Zaccagnino, A.; Mattarei, A.; Carraretto, L.; et al. Direct Pharmacological Targeting of a Mitochondrial Ion Channel Selectively Kills Tumor Cells In Vivo. Cancer Cell 2017, 31, 516–531.e10. [Google Scholar] [CrossRef] [PubMed]
- Venturini, E.; Leanza, L.; Azzolini, M.; Kadow, S.; Mattarei, A.; Weller, M.; Tabatabai, G.; Edwards, M.J.; Zoratti, M.; Paradisi, C.; et al. Targeting the Potassium Channel Kv1.3 Kills Glioblastoma Cells. Neuro-Signals 2017, 25, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wilson, G.C.; Bachmann, M.; Wang, J.; Mattarei, A.; Paradisi, C.; Edwards, M.J.; Szabo, I.; Gulbins, E.; Ahmad, S.A.; et al. Inhibition of a Mitochondrial Potassium Channel in Combination with Gemcitabine and Abraxane Drastically Reduces Pancreatic Ductal Adenocarcinoma in an Immunocompetent Orthotopic Murine Model. Cancers 2022, 14, 2618. [Google Scholar] [CrossRef]
- Parrasia, S.; Rossa, A.; Varanita, T.; Checchetto, V.; De Lorenzi, R.; Zoratti, M.; Paradisi, C.; Ruzza, P.; Mattarei, A.; Szabò, I.; et al. An Angiopep2-PAPTP Construct Overcomes the Blood-Brain Barrier. New Perspectives against Brain Tumors. Pharmaceuticals 2021, 14, 129. [Google Scholar] [CrossRef]
- Lakkadwala, S.; Singh, J. Co-delivery of doxorubicin and erlotinib through liposomal nanoparticles for glioblastoma tumor regression using an in vitro brain tumor model. Colloids Surfaces. B Biointerfaces 2019, 173, 27–35. [Google Scholar] [CrossRef]
- Greco, I.; Molchanova, N.; Holmedal, E.; Jenssen, H.; Hummel, B.D.; Watts, J.L.; Håkansson, J.; Hansen, P.R.; Svenson, J. Correlation between hemolytic activity, cytotoxicity and systemic in vivo toxicity of synthetic antimicrobial peptides. Sci. Rep. 2020, 10, 13206. [Google Scholar] [CrossRef]
- Severin, F.; Urbani, A.; Varanita, T.; Bachmann, M.; Azzolini, M.; Martini, V.; Pizzi, M.; Tos, A.P.D.; Frezzato, F.; Mattarei, A.; et al. Pharmacological modulation of Kv1.3 potassium channel selectively triggers pathological B lymphocyte apoptosis in vivo in a genetic CLL model. J. Exp. Clin. Cancer Res. CR 2022, 41, 64. [Google Scholar] [CrossRef]
- Kadow, S.; Schumacher, F.; Kramer, M.; Hessler, G.; Scholtysik, R.; Oubari, S.; Johansson, P.; Hüttmann, A.; Reinhardt, H.C.; Kleuser, B.; et al. Mitochondrial Kv1.3 Channels as Target for Treatment of Multiple Myeloma. Cancers 2022, 14, 1955. [Google Scholar] [CrossRef] [PubMed]
- Peruzzo, R.; Mattarei, A.; Azzolini, M.; Becker-Flegler, K.A.; Romio, M.; Rigoni, G.; Carrer, A.; Biasutto, L.; Parrasia, S.; Kadow, S.; et al. Insight into the mechanism of cytotoxicity of membrane-permeant psoralenic Kv1.3 channel inhibitors by chemical dissection of a novel member of the family. Redox Biol. 2020, 37, 101705. [Google Scholar] [CrossRef] [PubMed]
- Ying, M.; Wang, S.; Zhang, M.; Wang, R.; Zhu, H.; Ruan, H.; Ran, D.; Chai, Z.; Wang, X.; Lu, W. Myristic Acid-Modified (D)A7R Peptide for Whole-Process Glioma-Targeted Drug Delivery. ACS Appl. Mater. Interfaces 2018, 10, 19473–19482. [Google Scholar] [CrossRef]
- Douyère, M.; Chastagner, P.; Boura, C. Neuropilin-1: A Key Protein to Consider in the Progression of Pediatric Brain Tumors. Front. Oncol. 2021, 11, 665634. [Google Scholar] [CrossRef] [PubMed]
- Park, W.; Chawla, A.; O’Reilly, E.M. Pancreatic Cancer: A Review. JAMA 2021, 326, 851–862. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parrasia, S.; Rossa, A.; Roncaglia, N.; Mattarei, A.; Honisch, C.; Szabò, I.; Ruzza, P.; Biasutto, L. DA7R: A 7-Letter Zip Code to Target PDAC. Pharmaceutics 2023, 15, 1508. https://doi.org/10.3390/pharmaceutics15051508
Parrasia S, Rossa A, Roncaglia N, Mattarei A, Honisch C, Szabò I, Ruzza P, Biasutto L. DA7R: A 7-Letter Zip Code to Target PDAC. Pharmaceutics. 2023; 15(5):1508. https://doi.org/10.3390/pharmaceutics15051508
Chicago/Turabian StyleParrasia, Sofia, Andrea Rossa, Nicola Roncaglia, Andrea Mattarei, Claudia Honisch, Ildikò Szabò, Paolo Ruzza, and Lucia Biasutto. 2023. "DA7R: A 7-Letter Zip Code to Target PDAC" Pharmaceutics 15, no. 5: 1508. https://doi.org/10.3390/pharmaceutics15051508