
Optimizing the Safety and Efficacy of Bio-Radiopharmaceuticals for Cancer Therapy
 , , , ,
, , , , 
Abstract
: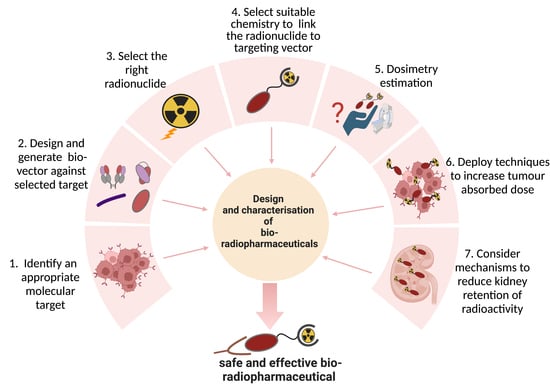
1. Introduction
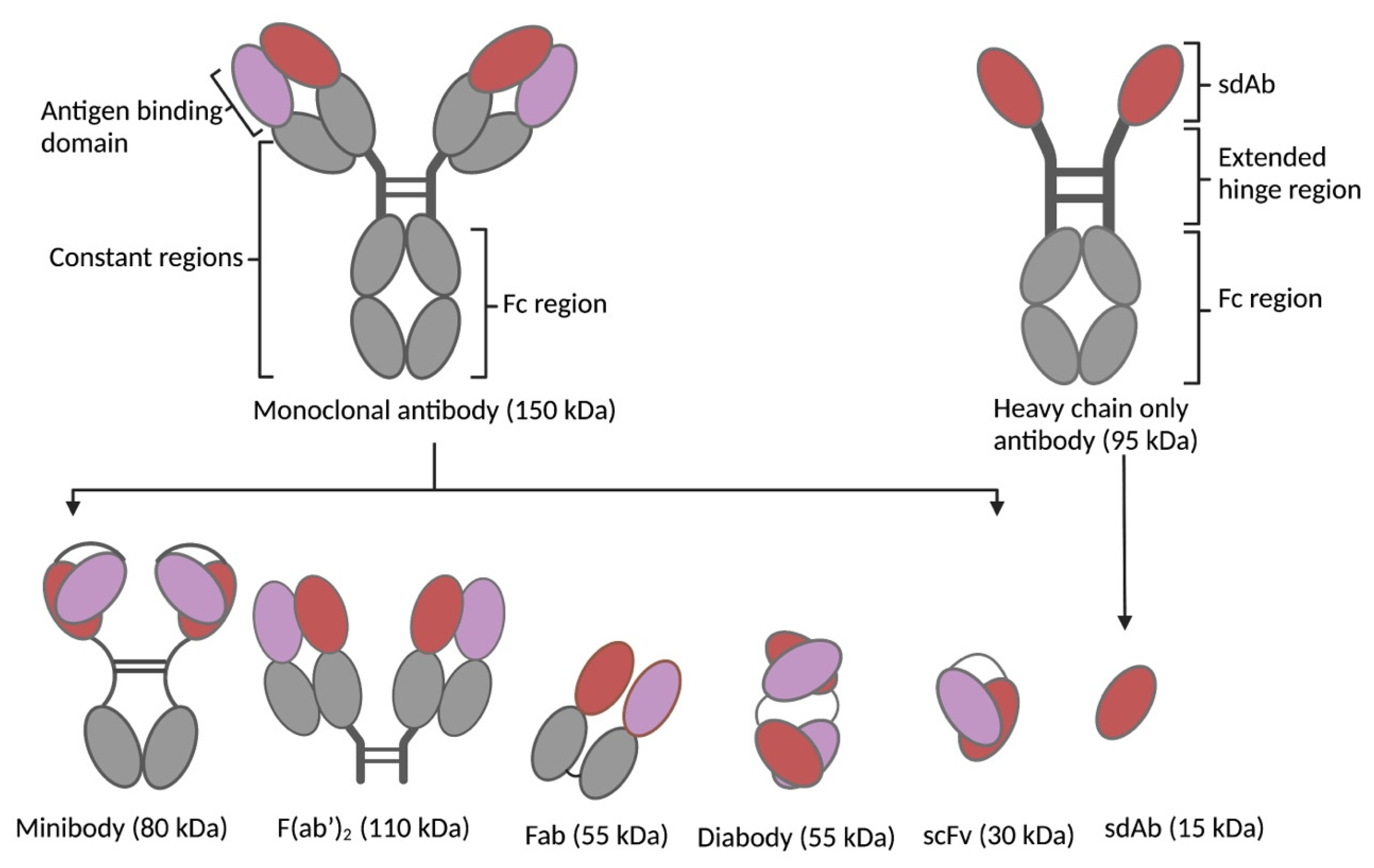
1.1. Types of Biological Vectors in TRT
1.2. Development Status of Biological Vectors in TRT
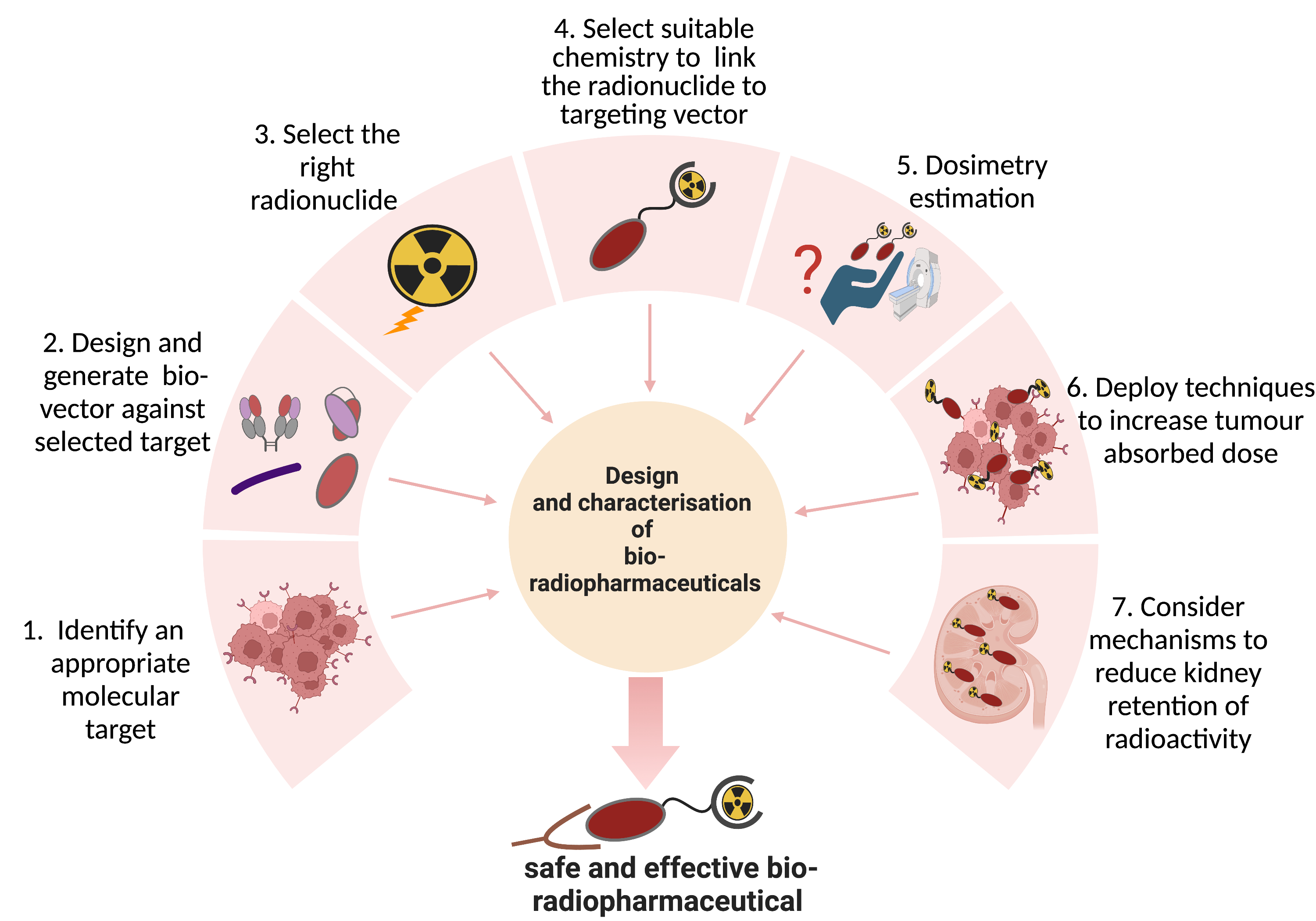
2. Target Selection
2.1. Antigen Specificity and Expression Pattern
2.2. Stability
2.3. Function
3. Vector Design
3.1. Binding Affinity
3.2. Size of Targeting Vector
3.3. Epitope and Functional Effect
3.4. Stability
3.5. Immunogenicity
3.6. Production Process
3.7. Chemical Modification
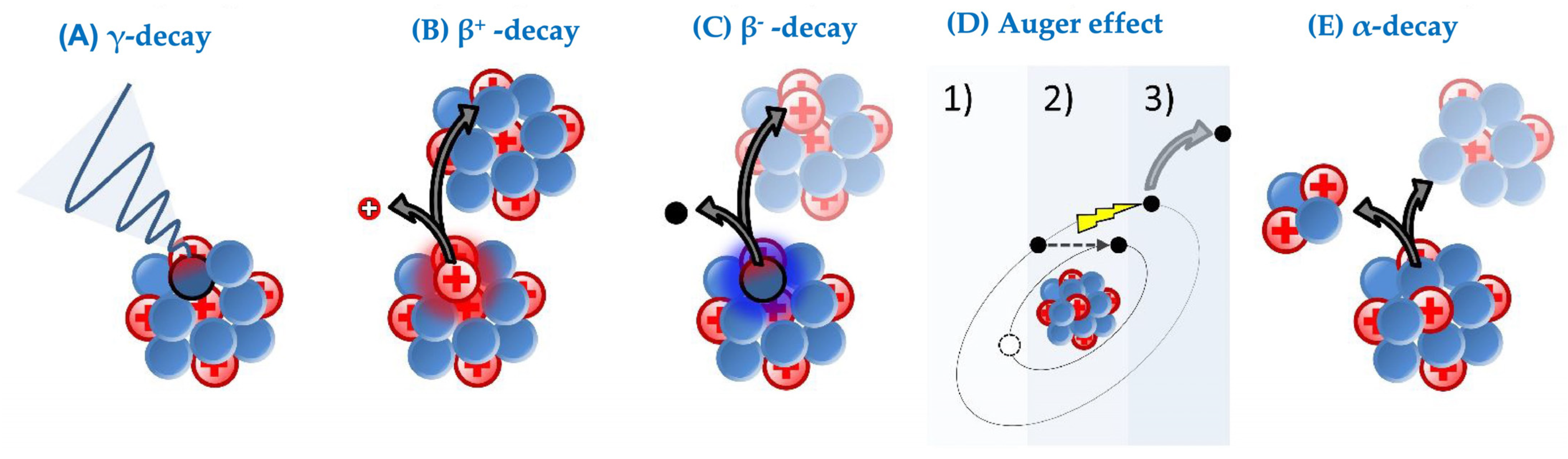
4. Choice of Radionuclide

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Particles/Waves | Application in Oncology | Penetration Range in Tissue | Particle/Wave Consistency | Commonly Used Isotopes |
|---|---|---|---|---|
| Alpha, α | Therapy | 20 to 100 µm [55] | 2 protons and 2 neutrons () | 227Th, 225Ac, 224Ra, 223Ra, 213Bi, 212Pb, 211At, and 149Tb |
| Beta−, β− | Therapy | 0.5 to 12 mm [53,54] | Electron () | 177Lu, 161Tb, 131I, and 90Y |
| Auger, AE | Therapy | <0.5 μm [65] | Electron () | 201TI, 161Tb, 111In, 99mTc, 67Ga, and 64Cu |
| Beta+, β+ | Imaging | 0.6 mm [74] | Positron () | 89Zr, 68Ga, 18F, 124I, and 64Cu |
| Gamma, γ | Imaging | Requires inches of lead to be stopped | Electromagnetic wave () | 131I, 123I, 111In, 99mTc and 67Ga |
5. Bioconjugation Strategies
- 1.
- The label should be compatible with the vector. The half-life of the radionuclide should be correctly matched with the biological half-life of the vector to ensure that the intended activity is delivered to the targeted tissues. The nature of the radionuclide and chemical bonds involved also have an impact on the radiolabeling efficiency, radiolabeling conditions, shelf-life, and in vivo stability of the final radiopharmaceutical.
- 2.
- The conditions of the radiolabeling reaction necessary to couple the radionuclide should not denature the vector nor impact the vector’s integrity. To this end, the modification of a vector in its framework structure or complementarity determining region (CDR) has the potential to negatively affect the affinity or in vivo behavior of the compound due to steric hindrance that might impede binding to the target. Ideally, the radiolabeling strategy should not alter the vector’s affinity and should have a minimal effect on the pharmacokinetics, bio-distribution, and immunogenicity.
- 3.
- When applying high radioactive amounts, radiolysis can likely occur. This is due to direct radiation damage emanating from the direct ionization of the surrounding molecules by the emitted radiation [76]. More specifically, therapeutic radionuclides have an increased propensity to cause radiolysis since they have different emission properties, and higher dosages are often used compared to diagnostic radionuclides and thus may cause more damage to the vector.
5.1. Site-Specific versus Random Radiolabeling Approaches
5.2. Radiolabeling and Functionalization Strategies: Direct versus Indirect
5.3. Radiohalogen Chemistry
5.4. Radiometal Chemistry
6. Dosimetry
7. Optimizing Radioactivity Delivery to Tumor Cells
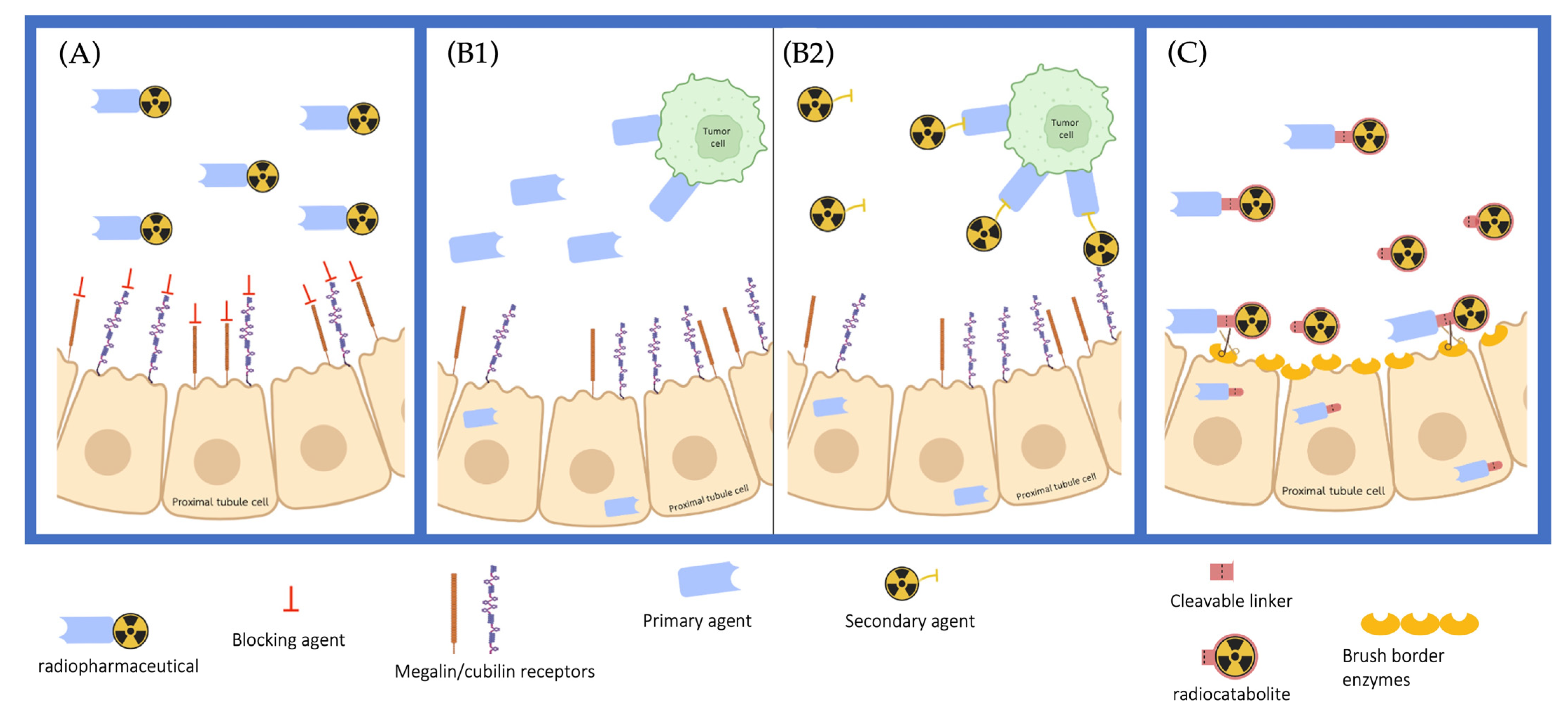
8. Reducing Kidney Retention
8.1. Kidney Retention
8.2. Co-Administration of Compounds Limiting the Re-Uptake of LMW Radiopharmaceuticals
8.3. Physicochemical Properties Influencing Kidney Retention
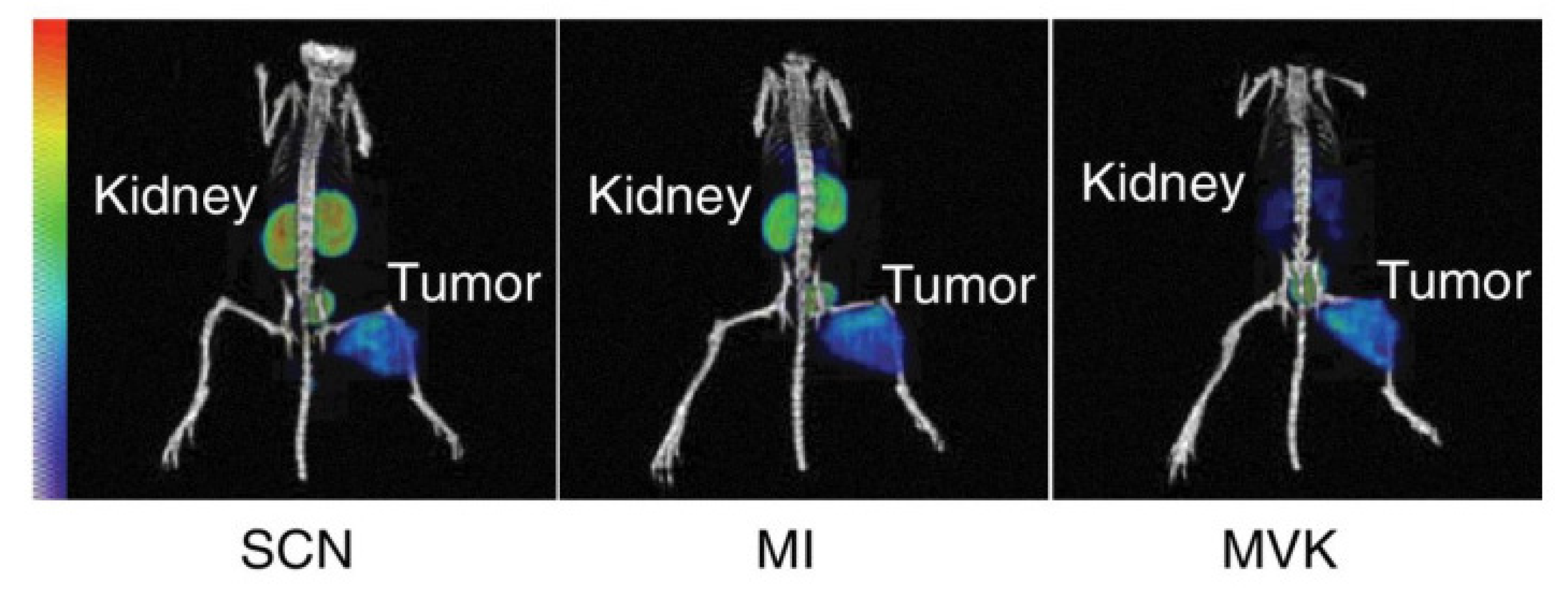
8.4. Cleavable Linkers
9. Expert Opinion and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Carter, L.; Poty, S.; Sharma, S.K.; Lewis, J.S. Preclinical optimization of antibody-based radiopharmaceuticals for cancer imaging and radionuclide therapy-Model, vector, and radionuclide selection. J. Label. Compd. Radiopharm. 2018, 61, 611–635. [Google Scholar] [CrossRef] [PubMed]
- Dash, A.; Knapp, F.F.R.; Pillai, M.R.A. Targeted Radionuclide Therapy—An Overview. Curr. Radiopharm. 2013, 6, 152–180. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.-N.; Mi, L.; Xu, J.; Song, F.; Zhang, Q.; Zhang, Z.; Xing, J.-L.; Bian, H.-J.; Jiang, J.-L.; Wang, X.-H.; et al. Targeting radioimmunotherapy of hepatocellular carcinoma with iodine (131I) metuximab injection: Clinical Phase I/II trials. Int. J. Radiat. Oncol. 2006, 65, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Emmett, L.; Willowson, K.; Violet, J.; Shin, J.; Blanksby, A.; Lee, J. Lutetium177PSMA radionuclide therapy for men with prostate cancer: A review of the current literature and discussion of practical aspects of therapy. J. Med. Radiat. Sci. 2017, 64, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Hennrich, U.; Kopka, K. Lutathera®: The First FDA- and EMA-Approved Radiopharmaceutical for Peptide Receptor Radionuclide Therapy. Pharmaceuticals 2019, 12, 114. [Google Scholar] [CrossRef]
- Rondon, A.; Rouanet, J.; Degoul, F. Radioimmunotherapy in Oncology: Overview of the Last Decade Clinical Trials. Cancers 2021, 13, 5570. [Google Scholar] [CrossRef]
- Krasniqi, A.; D’Huyvetter, M.; Devoogdt, N.; Frejd, F.Y.; Sörensen, J.; Orlova, A.; Keyaerts, M.; Tolmachev, V. Same-day imaging using small proteins: Clinical experience and translational prospects in oncology. J. Nucl. Med. 2018, 59, 885–891. [Google Scholar] [CrossRef]
- Vegt, E.; de Jong, M.; Wetzels, J.F.; Masereeuw, R.; Melis, M.; Oyen, W.J.; Gotthardt, M.; Boerman, O.C. Renal toxicity of radiolabeled peptides and an-tibody fragments: Mechanisms, impact on radionuclide therapy, and strategies for prevention. J. Nucl. Med. 2010, 51, 1049–1058. [Google Scholar] [CrossRef]
- Chigoho, D.M.; Bridoux, J.; Hernot, S. Reducing the renal retention of low- to moderate-molecular-weight radiopharmaceuticals. Curr. Opin. Chem. Biol. 2021, 63, 219–228. [Google Scholar] [CrossRef]
- Garousi, J.; Orlova, A.; Frejd, F.Y.; Tolmachev, V. Imaging using radiolabelled targeted proteins: Radioimmunodetection and beyond. EJNMMI Radiopharm. Chem. 2020, 5, 16. [Google Scholar] [CrossRef]
- Al-Lazikani, B.; Workman, P. Minimizing bias in target selection by exploiting multidisciplinary Big Data and the protein interactome. Futur. Med. Chem. 2016, 8, 1711–1716. [Google Scholar] [CrossRef] [PubMed]
- Failli, M.; Paananen, J.; Fortino, V. Prioritizing target-disease associations with novel safety and efficacy scoring methods. Sci. Rep. 2019, 9, 9852. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.T.; Burvenich, I.; Scott, A. Novel Target Selection for Nuclear Medicine Studies. Semin. Nucl. Med. 2019, 49, 357–368. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Sgouros, G. Radioimmunotherapy of solid tumors: Searching for the right target. Curr. Drug Deliv. 2011, 8, 26–44. [Google Scholar] [CrossRef]
- Gafita, A.; Wang, H.; Robertson, A.; Armstrong, W.R.; Zaum, R.; Weber, M.; Yagubbayli, F.; Kratochwil, C.; Grogan, T.R.; Nguyen, K.; et al. Tumor Sink Effect in 68Ga-PSMA-11 PET: Myth or Reality? J. Nucl. Med. 2022, 63, 226–232. [Google Scholar] [CrossRef]
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody therapy of cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef]
- Dalm, S.U.; Nonnekens, J.; Doeswijk, G.N.; de Blois, E.; van Gent, D.C.; Konijnenberg, M.W.; de Jong, M. Comparison of the Therapeutic Response to Treatment with a 177Lu-Labeled Somatostatin Receptor Agonist and Antagonist in Preclinical Models. J. Nucl. Med. 2015, 57, 260–265. [Google Scholar] [CrossRef]
- Wild, D.; Fani, M.; Fischer, R.; Del Pozzo, L.; Kaul, F.; Krebs, S.; Rivier, J.E.F.; Reubi, J.-C.; Maecke, H.R.; Weber, W.A. Comparison of Somatostatin Receptor Agonist and Antagonist for Peptide Receptor Radionuclide Therapy: A Pilot Study. J. Nucl. Med. 2014, 55, 1248–1252. [Google Scholar] [CrossRef]
- Gudkov, S.V.; Shilyagina, N.Y.; Vodeneev, V.A.; Zvyagin, A.V. Targeted radionuclide therapy of human tumors. Int. J. Mol. Sci. 2016, 17, 33. [Google Scholar] [CrossRef]
- Hofland, J.; Brabander, T.; Verburg, F.A.; Feelders, R.A.; de Herder, W.W. Peptide Receptor Radionuclide Therapy. J. Clin. Endocrinol. Metab. 2022, 107, 3199–3208. [Google Scholar] [CrossRef]
- Velikyan, I.; Eriksson, O. Advances in GLP-1 receptor targeting radiolabeled agent development and prospective of theranostics. Theranostics 2020, 10, 437–461. [Google Scholar] [CrossRef] [PubMed]
- Klingler, M.; Hnn, A.A.; Guggenberg, E.V. Cholecystokinin-2 Receptor Targeting with Radiolabeled Peptides: Current Status and Future Directions. Curr. Med. Chem. 2020, 27, 7112–7132. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Quinn, T.P. Advances in Receptor-Targeted Radiolabeled Peptides for Melanoma Imaging and Therapy. J. Nucl. Med. 2020, 62, 313–318. [Google Scholar] [CrossRef] [PubMed]
- de Groof, T.W.M.; Bobkov, V.; Heukers, R.; Smit, M.J. Nanobodies: New avenues for imaging, stabilizing and modulating GPCRs. Mol. Cell Endocrinol. 2019, 484, 15–24. [Google Scholar] [CrossRef]
- Ingram, J.R.; Schmidt, F.I.; Ploegh, H.L. Exploiting Nanobodies’ Singular Traits. Annu. Rev. Immunol. 2018, 36, 695–715. [Google Scholar] [CrossRef]
- Frenzel, A.; Schirrmann, T.; Hust, M. Phage display-derived human antibodies in clinical development and therapy. mAbs 2016, 8, 1177–1194. [Google Scholar] [CrossRef]
- Löfblom, J.; Feldwisch, J.; Tolmachev, V.; Carlsson, J.; Ståhl, S.; Frejd, F.Y. Affibody molecules: Engineered proteins for therapeutic, diagnostic and biotechnological applications. FEBS Lett. 2010, 584, 2670–2680. [Google Scholar] [CrossRef]
- Davidson, E.; Doranz, B.J. A high-throughput shotgun mutagenesis approach to mapping B-cell antibody epitopes. Immunology 2014, 143, 13–20. [Google Scholar] [CrossRef]
- Ryman, J.T.; Meibohm, B. Pharmacokinetics of Monoclonal Antibodies. CPT Pharmacomet. Syst. Pharmacol. 2017, 6, 576–588. [Google Scholar] [CrossRef]
- Puttemans, J.; Dekempeneer, Y.; Eersels, J.L.; Hanssens, H.; Debie, P.; Keyaerts, M.; Windhorst, A.D.; van der Aa, F.; Lecocq, Q.; Breckpot, K.; et al. Preclinical Targeted α- and β−-Radionuclide Therapy in HER2-Positive Brain Metastasis Using Camelid Single-Domain Antibodies. Cancers 2020, 12, 1017. [Google Scholar] [CrossRef]
- De Genst, E.; Silence, K.; Decanniere, K.; Conrath, K.; Loris, R.; Kinne, J.; Muyldermans, S.; Wyns, L. Molecular basis for the preferential cleft recog-nition by dromedary heavy-chain antibodies. Proc. Natl. Acad. Sci. USA 2006, 103, 4586–4591. [Google Scholar] [CrossRef] [PubMed]
- Muyldermans, S. Nanobodies: Natural single-domain antibodies. Annu. Rev. Biochem. 2013, 82, 775–797. [Google Scholar] [CrossRef] [PubMed]
- Vaneycken, I.; Devoogdt, N.; Van Gassen, N.; Vincke, C.; Xavier, C.; Wernery, U.; Muyldermans, S.; Lahoutte, T.; Caveliers, V. Preclinical screening of anti-HER2 nano-bodies for molecular imaging of breast cancer. FASEB J. 2011, 25, 2433–2446. [Google Scholar] [CrossRef] [PubMed]
- Najar, T.A.; Khare, S.; Pandey, R.; Gupta, S.K.; Varadarajan, R. Mapping Protein Binding Sites and Conformational Epitopes Using Cysteine Labeling and Yeast Surface Display. Structure 2017, 25, 395–406. [Google Scholar] [CrossRef]
- Casina, V.C.; Hu, W.; Mao, J.-H.; Lu, R.-N.; Hanby, H.A.; Pickens, B.; Kan, Z.-Y.; Lim, W.K.; Mayne, L.; Ostertag, E.M.; et al. High-resolution epitope mapping by HX MS reveals the pathogenic mechanism and a possible therapy for autoimmune TTP syndrome. Proc. Natl. Acad. Sci. USA 2015, 112, 9620–9625. [Google Scholar] [CrossRef]
- Dash, A.; Chakraborty, S.; Pillai, M.R.A.; Knapp, F.F. Peptide Receptor Radionuclide Therapy: An Overview. Cancer Biother. Radiopharm. 2015, 30, 47–71. [Google Scholar] [CrossRef]
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: Current applications and future directions. Signal Transduct. Target. Ther. 2022, 7, 48. [Google Scholar] [CrossRef]
- Malucchi, S.; Bertolotto, A. Clinical Aspects of Immunogenicity to Biopharmaceuticals. Immunogenicity Biopharm. 2008, 27–56. [Google Scholar] [CrossRef]
- Vaisman-Mentesh, A.; Gutierrez-Gonzalez, M.; DeKosky, B.J.; Wine, Y. The Molecular Mechanisms That Underlie the Immune Biology of Anti-drug Antibody Formation Following Treatment With Monoclonal Antibodies. Front. Immunol. 2020, 11, 1951. [Google Scholar] [CrossRef]
- Jawa, V.; Cousens, L.P.; Awwad, M.; Wakshull, E.; Kropshofer, H.; De Groot, A.S. T-cell dependent immunogenicity of protein therapeutics: Preclinical assessment and mitigation. Clin. Immunol. 2013, 149, 534–555. [Google Scholar] [CrossRef]
- Davda, J.; Declerck, P.; Hu-Lieskovan, S.; Hickling, T.P.; Jacobs, I.A.; Chou, J.; Salek-Ardakani, S.; Kraynov, E. Immunogenicity of immunomodulatory, antibody-based, oncology therapeutics. J. Immunother. Cancer 2019, 7, 105. [Google Scholar] [CrossRef] [PubMed]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, N.K.; Shrivastava, A. Recent Developments in Bioprocessing of Recombinant Proteins: Expression Hosts and Process Development. Front. Bioeng. Biotechnol. 2019, 7, 420. [Google Scholar] [CrossRef]
- Goel, M.; Mackeyev, Y.; Krishnan, S. Radiolabeled nanomaterial for cancer diagnostics and therapeutics: Principles and con-cepts. Cancer Nanotechnol. 2023, 14, 15. [Google Scholar] [CrossRef] [PubMed]
- Peltek, O.O.; Muslimov, A.R.; Zyuzin, M.V.; Timin, A.S. Current outlook on radionuclide delivery systems: From design consid-eration to translation into clinics. J. Nanobiotechnology 2019, 17, 90. [Google Scholar] [CrossRef] [PubMed]
- Poletto, G.; Cecchin, D.; Bartoletti, P.; Venturini, F.; Realdon, N.; Evangelista, L. Radionuclide Delivery Strategies in Tumor Treatment: A Systematic Review. Curr. Issues Mol. Biol. 2022, 44, 3267–3282. [Google Scholar] [CrossRef] [PubMed]
- Lebtahi, R.; Moreau, S.; Marchand-Adam, S.; Debray, M.-P.; Brauner, M.; Soler, P.; Marchal, J.; Raguin, O.; Gruaz-Guyon, A.; Reubi, J.-C.; et al. Increased uptake of 111In-octreotide in idiopathic pulmonary fibrosis. J. Nucl. Med. 2006, 47, 1281–1287. [Google Scholar]
- Tharp, K.; Israel, O.; Hausmann, J.; Bettman, L.; Martin, W.H.; Daitzchman, M.; Sandler, M.P.; Delbeke, D. Impact of 131I-SPECT/CT images obtained with an integrated system in the follow-up of patients with thyroid carcinoma. Eur. J. Nucl. Med. 2004, 31, 1435–1442. [Google Scholar] [CrossRef]
- Kemerink, G.J.; Liem, H.; Hofstra, L.; Boersma, H.H.; Buijs, W.C.; Reutelingsperger, C.P.; Heidendal, G.A. Patient dosimetry of intrave-nously administered 99mTc-annexin V. J. Nucl. Med. 2001, 42, 382–387. [Google Scholar]
- Keyaerts, M.; Xavier, C.; Heemskerk, J.; Devoogdt, N.; Everaert, H.; Ackaert, C.; Vanhoeij, M.; Duhoux, F.P.; Gevaert, T.; Simon, P.; et al. Phase I Study of 68Ga-HER2-Nanobody for PET/CT Assessment of HER2 Expression in Breast Carcinoma. J. Nucl. Med. 2015, 57, 27–33. [Google Scholar] [CrossRef]
- Farwell, M.D.; Gamache, R.F.; Babazada, H.; Hellmann, M.D.; Harding, J.J.; Korn, R.; Mascioni, A.; Le, W.; Wilson, I.; Gordon, M.S.; et al. CD8-Targeted PET Imaging of Tu-mor-Infiltrating T Cells in Patients with Cancer: A Phase i First-in-Humans Study of 89Zr-Df-IAB22M2C, a Radiolabeled Anti-CD8 Minibody. J. Nucl. Med. 2022, 63, 720–726. [Google Scholar] [PubMed]
- Christensen, T.N.; Langer, S.W.; Persson, G.F.; Larsen, K.R.; Loft, A.; Amtoft, A.G.; Berthelsen, A.K.; Johannesen, H.H.; Keller, S.H.; Kjaer, A.; et al. 18F-FLT PET/CT Adds Value to 18F-FDG PET/CT for Diagnosing Relapse After Definitive Radiotherapy in Patients with Lung Cancer: Results of a Prospective Clinical Trial. J. Nucl. Med. 2020, 62, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Marcu, L.; Bezak, E.; Allen, B.J. Global comparison of targeted alpha vs targeted beta therapy for cancer: In vitro, in vivo and clinical trials. Crit. Rev. Oncol. Hematol. 2018, 123, 7–20. [Google Scholar] [CrossRef]
- Gill, M.R.; Falzone, N.; Du, Y.; Vallis, K.A. Targeted radionuclide therapy in combined-modality regimens. Lancet Oncol. 2017, 18, e414–e423. [Google Scholar] [CrossRef] [PubMed]
- Aghevlian, S.; Boyle, A.; Reilly, R.M. Radioimmunotherapy of cancer with high linear energy transfer (LET) radiation delivered by radionuclides emitting α-particles or Auger electrons. Adv. Drug Deliv. Rev. 2017, 109, 102–118. [Google Scholar] [CrossRef] [PubMed]
- Pouget, J.-P.; Lozza, C.; Deshayes, E.; Boudousq, V.; Navarro-Teulon, I. Introduction to Radiobiology of Targeted Radionuclide Therapy. Front. Med. 2015, 2, 12. [Google Scholar] [CrossRef]
- Quintiliani, M. The Oxygen Effect in Radiation Inactivation of DNA and Enzymes. Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med. 1986, 50, 573–594. [Google Scholar] [CrossRef] [PubMed]
- Rockwell, S.; Dobrucki, I.T.; Kim, E.Y.; Marrison, S.T.; Vu, V.T. Hypoxia and Radiation Therapy: Past History, Ongoing Research, and Future Promise. Curr. Mol. Med. 2009, 9, 442–458. [Google Scholar] [CrossRef] [PubMed]
- Deb, N.; Goris, M.; Trisler, K.; Fowler, S.; Saal, J.; Ning, S.; Becker, M.; Marquez, C.; Knox, S. Treatment of hormone-refractory prostate cancer with 90Y-CYT-356 monoclonal antibody. Clin. Cancer Res. 1996, 2, 1289–1297. [Google Scholar]
- Tagawa, S.T.; Milowsky, M.I.; Morris, M.; Vallabhajosula, S.; Christos, P.; Akhtar, N.H.; Osborne, J.; Goldsmith, S.J.; Larson, S.; Taskar, N.P.; et al. Phase II Study of Lutetium-177-Labeled Anti-Prostate-Specific Membrane Antigen Monoclonal Antibody J591 for Metastatic Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2013, 19, 5182–5191. [Google Scholar] [CrossRef]
- Strosberg, J.; El-Haddad, G.; Wolin, E.; Hendifar, A.; Yao, J.; Chasen, B.; Mittra, E.; Kunz, P.L.; Kulke, M.H.; Jacene, H.; et al. Phase 3 Trial of 177 Lu-Dotatate for Midgut Neuro-endocrine Tumors. N. Engl. J. Med. 2017, 376, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Paillas, S.; Ladjohounlou, R.; Lozza, C.; Pichard, A.; Boudousq, V.; Jarlier, M.; Sevestre, S.; Le Blay, M.; Deshayes, E.; Sosabowski, J.; et al. Localized Irradiation of Cell Membrane by Auger Electrons Is Cytotoxic Through Oxidative Stress-Mediated Nontargeted Effects. Antioxid. Redox Signal. 2016, 25, 467–484. [Google Scholar] [CrossRef] [PubMed]
- Pouget, J.-P.; Santoro, L.; Raymond, L.; Chouin, N.; Bardiès, M.; Bascoul-Mollevi, C.; Huguet, H.; Azria, D.; Kotzki, P.-O.; Pèlegrin, M.; et al. Cell Membrane is a More Sensitive Target than Cytoplasm to Dense Ionization Produced by Auger Electrons. Radiat. Res. 2008, 170, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Buchegger, F.; Perillo-Adamer, F.; Dupertuis, Y.M.; Bischof Delaloye, A. Auger radiation targeted into DNA: A therapy perspec-tive. Eur. J. Nucl. Med. Mol. Imaging 2006, 33, 1352–1363. [Google Scholar] [CrossRef]
- Rigby, A.; Blower, J.E.; Blower, P.J.; Terry, S.Y.; Abbate, V. Targeted Auger electron-emitter therapy: Radiochemical approaches for thallium-201 radiopharmaceuticals. Nucl. Med. Biol. 2021, 98–99, 1–7. [Google Scholar] [CrossRef]
- Pouget, J.-P.; Constanzo, J. Revisiting the Radiobiology of Targeted Alpha Therapy. Front. Med. 2021, 8, 692436. [Google Scholar] [CrossRef]
- Wulbrand, C.; Seidl, C.; Gaertner, F.C.; Bruchertseifer, F.; Morgenstern, A.; Essler, M.; Senekowitsch-Schmidtke, R. Alpha-Particle Emitting 213Bi-Anti-EGFR Immunoconjugates Eradicate Tumor Cells Independent of Oxygenation. PLoS ONE 2013, 8, e64730. [Google Scholar] [CrossRef]
- Pouget, J.-P.; Navarro-Teulon, I.; Bardies, M.; Chouin, N.; Cartron, G.; Pèlegrin, A.; Azria, D. Clinical radioimmunotherapy—The role of radiobiology. Nat. Rev. Clin. Oncol. 2011, 8, 720–734. [Google Scholar] [CrossRef]
- Dekempeneer, Y.; Keyaerts, M.; Krasniqi, A.; Puttemans, J.; Muyldermans, S.; Lahoutte, T.; D’Huyvetter, M.; Devoogdt, N. Targeted alpha therapy using short-lived alpha-particles and the promise of nanobodies as targeting vehicle. Expert Opin. Biol. Ther. 2016, 16, 1035–1047. [Google Scholar] [CrossRef]
- Kratochwil, C.; Bruchertseifer, F.; Giesel, F.L.; Weis, M.; Verburg, F.A.; Mottaghy, F.; Kopka, K.; Apostolidis, C.; Haberkorn, U.; Morgenstern, A. 225Ac-PSMA-617 for PSMA-Targeted α-Radiation Therapy of Metastatic Castration-Resistant Prostate Cancer. J. Nucl. Med. 2016, 57, 1941–1944. [Google Scholar] [CrossRef]
- National Institutes of Health. Search of: Alpha Targeted Radionuclide Therapy—List Results—ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/ct2/resultscond=&term=alpha+targeted+radionclide+theapy&cntry=&state=&city=&dist= (accessed on 11 December 2022).
- Morgenstern, A.; Apostolidis, C.; Bruchertseifer, F. Supply and Clinical Application of Actinium-225 and Bismuth-Semin. Nucl. Med. 2020, 50, 119–123. [Google Scholar] [CrossRef]
- Nelson, B.J.B.; Andersson, J.D.; Wuest, F. Targeted Alpha Therapy: Progress in Radionuclide Production, Radiochemistry, and Applications. Pharmaceutics 2020, 13, 49. [Google Scholar] [CrossRef] [PubMed]
- Schrader, D.M.; Jean, Y.C. Positron and Positronium Chemistry; Elsevier: Amsterdam, The Netherlands, 1988. [Google Scholar]
- Tolmachev, V.; Stone-Elander, S. Radiolabelled proteins for positron emission tomography: Pros and cons of labelling meth-ods. Biochim. Et Biophys. Acta BBA-Gen. Subj. 2010, 1800, 487–510. [Google Scholar] [CrossRef] [PubMed]
- Baudhuin, H.; Cousaert, J.; Vanwolleghem, P.; Raes, G.; Caveliers, V.; Keyaerts, M.; Lahoutte, T.; Xavier, C. 68Ga-labeling: Laying the foundation for an anti-radiolytic formulation for nota-sdab pet tracers. Pharmaceuticals 2021, 14, 448. [Google Scholar] [CrossRef]
- Zeglis, B.M.; Davis, C.B.; Aggeler, R.; Kang, H.C.; Chen, A.; Agnew, B.J.; Lewis, J.S. Enzyme-Mediated Methodology for the Site-Specific Radiolabeling of Antibodies Based on Catalyst-Free Click Chemistry. Bioconjugate Chem. 2013, 24, 1057–1067. [Google Scholar] [CrossRef]
- D’huyvetter, M.; De Vos, J.; Caveliers, V.; Vaneycken, I.; Heemskerk, J.; Duhoux, F.P.; Fontaine, C.; Vanhoeij, M.; Windhorst, A.D.; van der Aa, F.; et al. Phase I Trial of 131I-GMIB-Anti-HER2-VHH1, a New Promising Candidate for HER2-Targeted Radionuclide Therapy in Breast Cancer Patients. J. Nucl. Med. 2020, 62, 1097–1105. [Google Scholar] [CrossRef]
- Debie, P.; Van Quathem, J.; Hansen, I.; Bala, G.; Massa, S.; Devoogdt, N.; Xavier, C.; Hernot, S. Effect of Dye and Conjugation Chemistry on the Biodistribution Profile of Near-Infrared-Labeled Nanobodies as Tracers for Image-Guided Surgery. Mol. Pharm. 2017, 14, 1145–1153. [Google Scholar] [CrossRef]
- Adumeau, P.; Sharma, S.K.; Brent, C.; Zeglis, B.M. Site-Specifically Labeled Immunoconjugates for Molecular Imaging—Part 2: Peptide Tags and Unnatural Amino Acids. Mol. Imaging Biol. 2016, 18, 153–165. [Google Scholar] [CrossRef]
- Feng, Y.; Zhou, Z.; McDougald, D.; Meshaw, R.L.; Vaidyanathan, G.; Zalutsky, M.R. Site-specific radioiodination of an anti-HER2 single domain antibody fragment with a residualizing prosthetic agent. Nucl. Med. Biol. 2020, 92, 171–183. [Google Scholar] [CrossRef]
- SMAC—NBE Therapeutics. Cited 2022 Dec. Available online: https://www.nbe-therapeutics.com/technology/smac (accessed on 14 November 2020).
- Knall, A.C.; Slugovc, C. Inverse electron demand Diels-Alder (iEDDA)-initiated conjugation: A (high) potential click chemistry scheme. Chem. Soc. Rev. 2013, 42, 5131. [Google Scholar] [CrossRef]
- Sletten, E.M.; Bertozzi, C.R. Bioorthogonal Chemistry: Fishing for Selectivity in a Sea of Functionality. Angew. Chem. Int. Ed. 2009, 48, 6974–6998. [Google Scholar] [CrossRef]
- Hennrich, U.; Eder, M. [177Lu]Lu-PSMA-617 (PluvictoTM): The First FDA-Approved Radiotherapeutical for Treatment of Prostate Cancer. Pharmaceuticals 2022, 15, 1292. [Google Scholar] [CrossRef] [PubMed]
- Velikyan, I.; Maecke, H.; Langstrom, B. Convenient Preparation of 68Ga-Based PET-Radiopharmaceuticals at Room Temperature. Bioconjugate Chem. 2008, 19, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.K.; Doss, M.; Shaller, C.; Narayanan, D.; Marks, J.D.; Adler, L.P.; Gonza, D.E.; Adams, G.P. Quantitative immuno-positron emission to-mography imaging of HER2-positlve tumor xenografts with an iodine-124 labeled anti-HER2 diabody. Cancer Res. 2005, 65, 1471–1478. [Google Scholar] [CrossRef] [PubMed]
- Pruszynski, M.; Koumarianou, E.; Vaidyanathan, G.; Revets, H.; Devoogdt, N.; Lahoutte, T.; Lyerly, H.K.; Zalutsky, M.R. Improved Tumor Targeting of Anti-HER2 Nanobody Through N-Succinimidyl 4-Guanidinomethyl-3-Iodobenzoate Radiolabeling. J. Nucl. Med. 2014, 55, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Adam, M.J.; Wilbur, D.S. Radiohalogens for imaging and therapy. Chem. Soc. Rev. 2005, 34, 153–163. [Google Scholar] [CrossRef]
- Vaughan, A.; Fremlin, J. The preparation of astatine labelled proteins using an electrophilic reaction. Int. J. Nucl. Med. Biol. 1978, 5, 229–230. [Google Scholar] [CrossRef]
- Guérard, F.; Maingueneau, C.; Liu, L.; Eychenne, R.; Gestin, J.F.; Montavon, G.; Galland, N. Advances in the Chemistry of Astatine and Implications for the Development of Radiopharmaceuticals. Acc. Chem. Res. 2021, 54, 3264–3275. [Google Scholar] [CrossRef]
- Zalutsky, M.R.; Reardon, D.A.; Akabani, G.; Coleman, R.E.; Friedman, A.H.; Friedman, H.S.; McLendon, R.E.; Wong, T.Z.; Bigner, D.D. Clinical Experience with a-Particle-Emitting 211 At: Treatment of Recurrent Brain Tumor Patients with 211 At-Labeled Chimeric Antitenascin Mono-clonal Antibody 81C6. J. Nucl. Med. 2008, 49, 30–38. [Google Scholar] [CrossRef]
- Maingueneau, C.; Berdal, M.; Eychenne, R.; Gaschet, J.; Chérel, M.; Gestin, J.F.; Guérard, F. 211 At and 125 I-Labeling of (Hetero)Aryliodonium Ylides: Astatine Wins Again. Chemistry 2022, 28, e202104169. [Google Scholar] [CrossRef]
- Navarro, L.; Berdal, M.; Chérel, M.; Pecorari, F.; Gestin, J.-F.; Guérard, F. Prosthetic groups for radioiodination and astatination of peptides and proteins: A comparative study of five potential bioorthogonal labeling strategies. Bioorganic Med. Chem. 2018, 27, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Herrero Álvarez, N.; Bauer, D.; Hernández-Gil, J.; Lewis, J.S. Recent Advances in Radiometals for Combined Imaging and Therapy in Cancer. ChemMedChem 2021, 16, 2909–2941. [Google Scholar] [CrossRef] [PubMed]
- Vogel, W.V.; van der Marck, S.C.; Versleijen, M.W.J. Challenges and future options for the production of lutetium-177. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 2329–2335. [Google Scholar] [CrossRef] [PubMed]
- Holik, H.A.; Ibrahim, F.M.; Elaine, A.A.; Putra, B.D.; Achmad, A.; Kartamihardja, A.H.S. The Chemical Scaffold of Theranostic Radi-opharmaceuticals: Radionuclide, Bifunctional Chelator, and Pharmacokinetics Modifying Linker. Molecules 2022, 27, 3062. [Google Scholar] [CrossRef]
- Pretze, M.; Kunkel, F.; Runge, R.; Freudenberg, R.; Braune, A.; Hartmann, H.; Schwarz, U.; Brogsitter, C.; Kotzerke, J. Ac-EAZY! Towards GMP-Compliant Module Syntheses of 225Ac-Labeled Peptides for Clinical Application. Pharmaceuticals 2021, 14, 652. [Google Scholar] [CrossRef]
- Kelly, J.M.; Amor-Coarasa, A.; Sweeney, E.; Wilson, J.J.; Causey, P.W.; Babich, J.W. A suitable time point for quantifying the radiochemical purity of 225Ac-labeled radiopharmaceuticals. EJNMMI Radiopharm. Chem. 2021, 6, 38. [Google Scholar] [CrossRef]
- Thiele, N.A.; Brown, V.; Kelly, J.M.; Amor-Coarasa, A.; Jermilova, U.; MacMillan, S.N.; Nikolopoulou, A.; Ponnala, S.; Ramogida, C.F.; Robertson, A.K.H. Radiopharmaceuticals Hot Paper An Eighteen-Membered Macrocyclic Ligand for Actinium-225 Targeted Alpha Therapy. Angew. Chem. Int. Ed. 2017, 56, 14712–14717. [Google Scholar] [CrossRef]
- Karlsson, J.; Schatz, C.A.; Wengner, A.M.; Hammer, S.; Scholz, A.; Cuthbertson, A.; Wagner, V.; Hennekes, H.; Jardine, V.; Hagemann, U.B. Targeted thorium-227 conjugates as treatment options in oncology. Front. Med. 2023, 9. [Google Scholar] [CrossRef]
- Kluetz, P.G.; Pierce, W.; Maher, V.E.; Zhang, H.; Tang, S.; Song, P.; Liu, Q.; Haber, M.T.; Leutzinger, E.E.; Al-Hakim, A.; et al. Radium Ra 223 Dichloride Injection: U.S. Food and Drug Administration Drug Approval Summary. Clin. Cancer Res. 2014, 20, 9–14. [Google Scholar] [CrossRef]
- Abou, D.S.; Thiele, N.A.; Gutsche, N.T.; Villmer, A.; Zhang, H.; Woods, J.J.; Baidoo, K.E.; Escorcia, F.E.; Wilson, J.J.; Thorek, D.L.J. Towards the stable chelation of radium for biomedical applications with an 18-membered macrocyclic ligand. Chem. Sci. 2021, 12, 3733–3742. [Google Scholar] [CrossRef]
- Ahenkorah, S.; Murce, E.; Cawthorne, C.; Ketchemen, J.P.; Deroose, C.M.; Cardinaels, T.; Seimbille, Y.; Fonge, H.; Gsell, W.; Bormans, G.; et al. 3p-C-NETA: A versatile and effective chelator for development of Al18F-labeled and therapeutic radiopharmaceuticals. Theranostics 2022, 12, 5971–5985. [Google Scholar] [CrossRef] [PubMed]
- Huizing, D.M.V.; de Wit-van der Veen, B.J.; Verheij, M.; Stokkel, M.P.M. Dosimetry methods and clinical applications in peptide receptor radionuclide therapy for neuroendocrine tumours: A literature review. EJNMMI Res. 2018, 8, 89. [Google Scholar] [CrossRef] [PubMed]
- Bodei, L.; Mueller-Brand, J.; Baum, R.P.; Pavel, M.E.; Hörsch, D.; O’Dorisio, M.S.; O’Dorisiol, T.M.; Howe, J.R.; Cremonesi, M.; Kwekkeboom, D.J.; et al. The joint IAEA, EANM, and SNMMI practical guidance on peptide receptor radionuclide therapy (PRRNT) in neuroendocrine tumours. Eur. J. Nucl. Med. Mol. Imaging 2013, 40, 800–816. [Google Scholar] [CrossRef] [PubMed]
- Bolch, W.E.; Eckerman, K.F.; Sgouros, G.; Thomas, S.R. MIRD pamphlet No. 21: A generalized schema for radiopharmaceutical dosimetry-standardization of nomenclature. J. Nucl. Med. 2009, 50, 477–484. [Google Scholar] [CrossRef]
- Vargas, C.S.; Struelens, L.; D’huyvetter, M.; Caveliers, V.; Covens, P. Assessment of mouse-specific pharmacokinetics in kidneys based on 131I activity measurements using micro-SPECT. EJNMMI Phys. 2022, 9, 13. [Google Scholar] [CrossRef]
- Tamborino, G.; De Saint-Hubert, M.; Struelens, L.; Seoane, D.C.; Ruigrok, E.A.M.; Aerts, A.; van Cappellen, W.A.; de Jong, M.; Konijnenberg, M.W.; Nonnekens, J. Cellular dosimetry of [177Lu]Lu-DOTA-[Tyr3]octreotate radionuclide therapy: The impact of modeling assumptions on the correlation with in vitro cytotoxicity. EJNMMI Phys. 2020, 7, 8. [Google Scholar] [CrossRef]
- Spoormans, K.; Crabbé, M.; Struelens, L.; De Saint-Hubert, M.; Koole, M. A Review on Tumor Control Probability (TCP) and Preclinical Dosimetry in Targeted Radionuclide Therapy (TRT). Pharmaceutics 2022, 14, 2007. [Google Scholar] [CrossRef]
- Bates, A.; Power, C.A. David vs. Goliath: The Structure, Function, and Clinical Prospects of Antibody Fragments. Antibodies 2019, 8, 28. [Google Scholar] [CrossRef]
- Liu, L. Pharmacokinetics of monoclonal antibodies and Fc-fusion proteins. Protein Cell 2017, 9, 15–32. [Google Scholar] [CrossRef]
- Rondon, A.; Ty, N.; Bequignat, J.B.; Quintana, M.; Briat, A.; Witkowski, T.; Bouchon, B.; Boucheix, C.; Miot-Noirault, E.; Pouget, J.-P.; et al. Antibody PEGylation in bioorthogonal pretargeting with trans-cyclooctene/tetrazine cycloaddition: In vitro and in vivo evaluation in colorectal cancer models. Sci. Rep. 2017, 7, 14918. [Google Scholar] [CrossRef]
- Suzuki, H.; Kise, S.; Kaizuka, Y.; Watanabe, R.; Sugawa, T.; Furukawa, T.; Fujii, H.; Uehara, T. Copper-64-Labeled Antibody Fragments for Im-muno-PET/ Radioimmunotherapy with Low Renal Radioactivity Levels and Amplified Tumor-Kidney Ratios. ACS Omega 2021, 6, 21556–21562. [Google Scholar] [CrossRef]
- Krasniqi, A.; Bialkowska, M.; Xavier, C.; Van der Jeught, K.; Muyldermans, S.; Devoogdt, N.; D’huyvetter, M. Pharmacokinetics of radiolabeled dimeric sdAbs constructs targeting human CD20. N. Biotechnol. 2018, 45, 69–79. [Google Scholar] [CrossRef]
- Rondon, A.; Mahri, S.; Morales-Yanez, F.; Dumoulin, M.; Vanbever, R. Protein Engineering Strategies for Improved Pharmaco-kinetics. Adv. Funct. Mater. 2021, 31, 2101633. [Google Scholar] [CrossRef]
- Tchouate Gainkam, L.O.; Caveliers, V.; Devoogdt, N.; Vanhove, C.; Xavier, C.; Boerman, O.; Muyldermans, S.; Bossuyt, A.; Lahoutte, T. Localization, mechanism and reduction of renal retention of technetium-99m labeled epidermal growth factor receptor-specific nanobody in mice. Contrast Media Mol. Imaging 2011, 6, 85–92. [Google Scholar] [CrossRef]
- Grana, C.M.; Chinol, M.; Robertson, C.; Mazzetta, C.; Bartolomei, M.S.; De Cicco, C.; Fiorenza, M.T.; Gatti, M.; Caliceti, P.; Paganelli, G. Pretargeted adjuvant radioimmunotherapy with Yttrium-90-biotin in malignant glioma patients: A pilot study. Br. J. Cancer 2002, 86, 207–212. [Google Scholar] [CrossRef]
- Rossin, R.; Läppchen, T.; Bosch, S.M.V.D.; Laforest, R.; Robillard, M.S. Diels–Alder Reaction for Tumor Pretargeting: In Vivo Chemistry Can Boost Tumor Radiation Dose Compared with Directly Labeled Antibody. J. Nucl. Med. 2013, 54, 1989–1995. [Google Scholar] [CrossRef]
- Van Duijnhoven, S.M.J.; Rossin, R.; Van Den Bosch, S.M.; Wheatcroft, M.P.; Hudson, P.J.; Robillard, M.S. Diabody pretargeting with click chemistry in vivo. J. Nucl. Med. 2015, 56, 1422–1428. [Google Scholar] [CrossRef]
- Myrhammar, A.; Vorobyeva, A.; Westerlund, K.; Yoneoka, S.; Orlova, A.; Tsukahara, T.; Tolmachev, V.; Karlström, A.E.; Altai, M. Evaluation of an antibody-PNA conjugate as a clearing agent for antibody-based PNA-mediated radionuclide pretargeting. Sci. Rep. 2020, 10, 20777. [Google Scholar] [CrossRef]
- Heskamp, S.; Hernandez, R.; Molkenboer-Kuenen, J.D.; Essler, M.; Bruchertseifer, F.; Morgenstern, A.; Steenbergen, E.J.; Cai, W.; Seidl, C.; McBride, W.J.; et al. α- Versus β-Emitting Radionuclides for Pretargeted Radioimmunotherapy of Carcinoembryonic Antigen–Expressing Human Colon Cancer Xenografts. J. Nucl. Med. 2017, 58, 926–933. [Google Scholar] [CrossRef]
- Aarts, F.; Boerman, O.C.; Sharkey, R.M.; Hendriks, T.; Chang, C.H.; McBride, W.J.; Bleichrodt, R.P.; Oyen, W.J.G.; Goldenberg, D.M. Pretargeted radioimmunoscintigraphy in patients with primary colorectal cancer using a bispecific anticarcinoembryonic antigen CEA X an-ti-di-diethylenetriaminepentaacetic acid F(ab’)2 antibody. Cancers 2010, 116, 1111–1117. [Google Scholar] [CrossRef]
- Bodet-Milin, C.; Ferrer, L.; Rauscher, A.; Masson, D.; Rbah-Vidal, L.; Faivre-Chauvet, A.; Cerato, E.; Rousseau, C.; Hureaux, J.; Couturier, O. Pharmacokinetics and dosimetry studies for optimization of pretargeted radioimmunotherapy in CEA-expressing advanced lung cancer patients. Front. Med. 2015, 2, 84. [Google Scholar] [CrossRef]
- Stéen, J.; Jørgensen, J.T.; Christoph, D.; Battisti, U.M.; Nørregaard, K.; Edem, P.; Bratteby, K.; Shalgunov, V.; Martin, W.; Svatunek, D.; et al. Lipophilicity and Click Reactivity Determine the Performance of Bioorthogonal Tetrazine Tools in Pretargeted in Vivo Chemistry. ACS Pharmacol. Transl. Sci. 2021, 4, 824–833. [Google Scholar] [CrossRef]
- Verhoeven, M.; Seimbille, Y.; Dalm, S.U. Therapeutic Applications of Pretargeting. Pharmaceutics 2019, 11, 434. [Google Scholar] [CrossRef]
- Kramer, K.; Pandit-Taskar, N.; Kushner, B.H.; Zanzonico, P.; Humm, J.L.; Tomlinson, U.; Donzelli, M.; Wolden, S.L.; Haque, S.; Dunkel, I.; et al. Phase 1 study of intraventricular 131I-omburtamab targeting B7H3 (CD276)-expressing CNS malignancies. J. Hematol. Oncol. 2022, 15, 165. [Google Scholar] [CrossRef]
- Christensen, E.I.; Birn, H.; Verroust, P.; Moestrup, S.K. Megalin-Mediated endocytosis in renal proximal tubule. Ren. Fail. 1998, 20, 191–199. [Google Scholar] [CrossRef]
- Vegt, E.; Wetzels, J.F.M.; Russel, F.G.M.; Masereeuw, R.; Boerman, O.C.; Van Eerd, J.E.; Corstens, F.H.M.; Oyen, W.J.G. Renal uptake of radiolabeled octreotide in human subjects is efficiently inhibited by succinylated gelatin. J. Nucl. Med. 2006, 47, 432–436. [Google Scholar]
- de Jong, M.; Barone, R.; Krenning, E.; Bernard, B.; Melis, M.; Visser, T.; Gekle, M.; Willnow, T.E.; Walrand, S.; Jamar, F.; et al. Megalin Is Essential for Renal Proximal Tubule Re-absorption of <sup>111</sup>In-DTPA-Octreotide. J. Nucl. Med. 2005, 46, 1696–1700. [Google Scholar]
- Jia, L.; Zhang, L.; Shao, C.; Song, E.; Sun, W.; Li, M.; Gao, Y. An attempt to understand kidney’s protein handling function by com-paring plasma and urine proteomes. PLoS ONE 2009, 4, e5146. [Google Scholar] [CrossRef]
- Christensen, E.I.; Verroust, P.J.; Nielsen, R. Receptor-mediated endocytosis in renal proximal tubule. Pflügers Arch.-Eur. J. Physiol. 2009, 458, 1039–1048. [Google Scholar] [CrossRef]
- Negri, A.L. Proximal tubule endocytic apparatus as the specific renal uptake mechanism for vitamin D-binding pro-tein/25-(OH)D3 complex. Nephrology 2006, 11, 510–515. [Google Scholar] [CrossRef]
- Kozyraki, R.; Fyfe, J.; Verroust, P.J.; Jacobsen, C.; Dautry-Varsat, A.; Gburek, J.; Willnow, T.E.; Christensen, E.I.; Moestrup, S.K. Megalin-dependent cubilin-mediated endo-cytosis is a major pathway for the apical uptake of transferrin in polarized epithelia. Proc. Natl. Acad. Sci. USA 2001, 98, 12491–12496. [Google Scholar] [CrossRef] [PubMed]
- Farooque, S.; Kenny, M.; Marshall, S.D. Anaphylaxis to intravenous gelatin-based solutions: A case series examining clinical features and severity. Anaesthesia 2018, 74, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Melis, M.; Bijster, M.; de Visser, M.; Konijnenberg, M.W.; de Swart, J.; Rolleman, E.J.; Boerman, O.C.; Krenning, E.P.; de Jong, M. Dose-response effect of Gelofusine on renal uptake and retention of radiolabelled octreotate in rats with CA20948 tumours. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 1968–1976. [Google Scholar] [CrossRef]
- Rolleman, E.J.; Valkema, R.; De Jong, M.; Kooij, P.P.M.; Krenning, E.P. Safe and effective inhibition of renal uptake of radiolabelled octreotide by a combination of lysine and arginine. Eur. J. Nucl. Med. 2003, 30, 9–15. [Google Scholar] [CrossRef]
- Chan, H.S.; Konijnenberg, M.W.; Daniels, T.; Nysus, M.; Makvandi, M.; De Blois, E.; Breeman, W.A.; Atcher, R.W.; De Jong, M.; Norenberg, J.P. Improved safety and efficacy of 213Bi-DOTATATE-targeted alpha therapy of somatostatin receptor-expressing neuroendocrine tumors in mice pre-treated with l-lysine. EJNMMI Res. 2016, 6, 83. [Google Scholar] [CrossRef]
- Ring, J.; Messmer, K. Incidence and severity of anaphylactoid reactions to colloid volume substitutes. Lancet 1977, 1, 466–469. [Google Scholar] [CrossRef]
- D’Huyvetter, M.; Vincke, C.; Xavier, C.; Aerts, A.; Impens, N.; Baatout, S.; De Raeve, H.; Muyldermans, S.; Caveliers, V.; Devoogdt, N.; et al. Targeted Radionuclide Therapy with A 177Lu-labeled Anti-HER2 Nanobody. Theranostics 2014, 4, 708–720. [Google Scholar] [CrossRef]
- Bergsma, H.; Konijnenberg, M.W.; Kam, B.L.R.; Teunissen, J.J.M.; Kooij, P.P.; de Herder, W.W.; Franssen, G.J.H.; van Eijck, C.H.J.; Krenning, E.P.; Kwekkeboom, D.J. Subacute haematotoxicity after PRRT with 177Lu-DOTA-octreotate: Prognostic factors, incidence and course. Eur. J. Nucl. Med. 2015, 43, 453–463. [Google Scholar] [CrossRef]
- Barone, R.; Pauwels, S.; De Camps, J.; Krenning, E.P.; Kvols, L.K.; Smith, M.C.; Bouterfa, H.; Devuyst, O.; Jamar, F. Metabolic effects of amino acid solutions infused for renal protection during therapy with radiolabelled somatostatin analogues. Nephrol. Dial. Transplant. 2004, 19, 2275–2281. [Google Scholar] [CrossRef]
- Thomson, L.A. Significance of Amino Acid Solution with Lutetium Lu 177 Dotatate—Oncology Nurse Advisor. cited 2023 Jan. Available online: https://www.oncologynurseadvisor.com/home/departments/advisor-forum/significance-of-amino-acid-solution-with-lutetium-lu-177-dotatate/ (accessed on 7 February 2019).
- Xiong, C.; Yin, D.; Li, J.; Huang, Q.; Ravoori, M.K.; Kundra, V.; Zhu, H.; Yang, Z.; Lu, Y.; Li, C. Metformin Reduces Renal Uptake of Radiotracers and Protects Kidneys from Radiation-Induced Damage. Mol. Pharm. 2019, 16, 808–815. [Google Scholar] [CrossRef]
- Chen, K.; Li, Y.; Guo, Z.; Zeng, Y.; Zhang, W.; Wang, H. Metformin: Current clinical applications in nondiabetic patients with cancer. Aging 2020, 12, 3993–4009. [Google Scholar] [CrossRef]
- Gong, L.; Goswami, S.; Giacomini, K.M.; Altman, R.B.; Klein, T.E. Metformin pathways: Pharmacokinetics and pharmacodynamics. Pharmacogenet 2012, 22, 820–827. [Google Scholar] [CrossRef]
- Yang, K.; Liu, Z.; Thong, M.S.Y.; Doege, D.; Arndt, V. Higher Incidence of Diabetes in Cancer Patients Compared to Cancer-Free Population Controls: A Systematic Review and Meta-Analysis. Cancers 2022, 14, 1808. [Google Scholar] [CrossRef]
- Fowler, H.; Belot, A.; Ellis, L.; Maringe, C.; Luque-Fernandez, M.A.; Njagi, E.N.; Navani, N.; Sarfati, D.; Rachet, B. Comorbidity prevalence among cancer patients: A population-based cohort study of four cancers. BMC Cancer 2020, 20, 2. [Google Scholar] [CrossRef]
- Irizarry, L.; Li, Q.E.; Duncan, I.; Thurston, A.L.; Fitzner, K.A.; Edwards, B.J.; McKoy-Bent, J.M.; Tulas, K.M.; McKoy, J.M. Effects of cancer comorbidity on disease manage-ment: Making the case for diabetes education (a report from the SOAR program. Popul. Health Manag. 2013, 16, 53–57. [Google Scholar] [CrossRef]
- Matteucci, F.; Mezzenga, E.; Caroli, P.; Di Iorio, V.; Sarnelli, A.; Celli, M.; Fantini, L.; Moretti, A.; Galassi, R.; De Giorgi, U.; et al. Reduction of 68Ga-PSMA renal uptake with mannitol infusion: Preliminary results. Eur. J. Nucl. Med. 2017, 44, 2189–2194. [Google Scholar] [CrossRef]
- Baranski, A.-C.; Schäfer, M.; Bauder-Wüst, U.; Wacker, A.; Schmidt, J.; Liolios, C.; Mier, W.; Haberkorn, U.; Eisenhut, M.; Kopka, K.; et al. Improving the Imaging Contrast of 68Ga-PSMA-11 by Targeted Linker Design: Charged Spacer Moieties Enhance the Pharmacokinetic Properties. Bioconjugate Chem. 2017, 28, 2485–2492. [Google Scholar] [CrossRef]
- Flook, A.M.; Yang, J.; Miao, Y. Substitution of the Lys linker with the β-Ala linker dramatically decreased the renal uptake of 99mTc-labeled Arg-X-Asp-conjugated and X-Ala-Asp-conjugated α-melanocyte stimulating hormone peptides. J. Med. Chem. 2014, 57, 9010–9018. [Google Scholar] [CrossRef]
- Hofström, C.; Altai, M.; Honarvar, H.; Strand, J.; Malmberg, J.; Hosseinimehr, S.J.; Orlova, A.; Gräslund, T.; Tolmachev, V. Influence of Position and Composition of Histidine Containing Tags on Biodistribution of [99mTc(CO)3]+-Labeled Affibody Molecules. J. Med. Chem. 2013, 56, 4966–4974. [Google Scholar] [CrossRef]
- Bapst, J.P.; Eberle, A.N. Receptor-Mediated Melanoma Targeting with Radiolabeled α-Melanocyte-Stimulating Hormone: Rel-evance of the Net Charge of the Ligand. Front. Endocrinol. 2017, 8, 93. [Google Scholar] [CrossRef]
- Ekblad, T.; Tran, T.; Orlova, A.; Widström, C.; Feldwisch, J.; Abrahmsén, L.; Wennborg, A.; Karlström, A.E.; Tolmachev, V. Development and preclinical characterisation of 99mTc-labelled Affibody molecules with reduced renal uptake. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 2245–2255. [Google Scholar] [CrossRef] [PubMed]
- Potemkin, R.; Strauch, B.; Kuwert, T.; Prante, O.; Maschauer, S. Development of 18F-Fluoroglycosylated PSMA-Ligands with Improved Renal Clearance Behavior. Mol. Pharm. 2020, 17, 933–943. [Google Scholar] [CrossRef] [PubMed]
- Bouvet, V.; Wuest, M.; Bailey, J.J.; Bergman, C.; Janzen, N.; Valliant, J.F.; Wuest, F. Targeting Prostate-Specific Membrane Antigen (PSMA) with F-18-Labeled Compounds: The Influence of Prosthetic Groups on Tumor Uptake and Clearance Profile. Mol. Imaging Biol. 2017, 19, 923–932. [Google Scholar] [CrossRef]
- Maschauer, S.; Haubner, R.; Kuwert, T.; Prante, O. 18F-Glyco-RGD Peptides for PET Imaging of Integrin Expression: Efficient Radiosynthesis by Click Chemistry and Modulation of Biodistribution by Glycosylation. Mol. Pharm. 2014, 11, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Läppchen, T.; Tönnesmann, R.; Eersels, J.; Meyer, P.T.; Maecke, H.R.; Rylova, S.N. Radioiodinated Exendin-4 Is Superior to the Ra-diometal-Labelled Glucagon-Like Peptide-1 Receptor Probes Overcoming Their High Kidney Uptake. PLoS ONE 2017, 12, e0170435. [Google Scholar] [CrossRef]
- Strand, J.; Nordeman, P.; Honarvar, H.; Altai, M.; Orlova, A.; Larhed, M.; Tolmachev, V. Site-Specific Radioiodination of HER2-Targeting Affibody Molecules using 4-Iodophenethylmaleimide Decreases Renal Uptake of Radioactivity. Chemistryopen 2015, 4, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Bala, G.; Crauwels, M.; Blykers, A.; Remory, I.; Marschall, A.L.; Dübel, S.; Dumas, L.; Broisat, A.; Martin, C.; Ballet, S.; et al. Radiometal-labeled anti-VCAM-1 nanobodies as molecular tracers for atherosclerosis—impact of radiochemistry on pharmacokinetics. Biol. Chem. 2018, 400, 323–332. [Google Scholar] [CrossRef]
- Dietlein, M.; Kobe, C.; Kuhnert, G.; Stockter, S.; Fischer, T.; Schomäcker, K.; Schmidt, M.; Dietlein, F.; Zlatopolskiy, B.D.; Krapf, P.; et al. Comparison of [(18)F]DCFPyL and [ (68)Ga]Ga-PSMA-HBED-CC for PSMA-PET Imaging in Patients with Relapsed Prostate Cancer. Mol. Imaging Biol. 2015, 17, 575–584. [Google Scholar] [CrossRef]
- Duncan, J.R.; Behr, T.M.; DeNardo, S.J. Intracellular Fate of Radiometals. J. Nucl. Med. 1997, 38, 829. [Google Scholar]
- Werner, R.A.; Derlin, T.; Lapa, C.; Sheikbahaei, S.; Higuchi, T.; Giesel, F.L.; Behr, S.; Drzezga, A.; Kimura, H.; Buck, A.K.; et al. 18F-Labeled, PSMA-Targeted Radiotracers: Lever-aging the Advantages of Radiofluorination for Prostate Cancer Molecular Imaging. Theranostics 2020, 10, 1. [Google Scholar] [CrossRef]
- D’Huyvetter, M.; De Vos, J.; Xavier, C.; Pruszynski, M.; Sterckx, Y.G.; Massa, S.; Raes, G.; Caveliers, V.; Zalutsky, M.R.; Lahoutte, T.; et al. 131I-labeled Anti-HER2 Camelid sdAb as a Theranostic Tool in Cancer Treatment. Clin. Cancer Res. 2017, 23, 6616–6628. [Google Scholar] [CrossRef] [PubMed]
- Arano, Y. Renal brush border strategy: A developing procedure to reduce renal radioactivity levels of radiolabeled polypep-tides. Nucl. Med. Biol. 2021, 92, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Yim, C.-B.; Mikkola, K.; Fagerholm, V.; Elomaa, V.-V.; Ishizu, T.; Rajander, J.; Schlesinger, J.; Roivainen, A.; Nuutila, P.; Solin, O. Synthesis and preclinical characterization of [64Cu]NODAGA-MAL-exendin-4 with a Nε-maleoyl-l-lysyl-glycine linkage. Nucl. Med. Biol. 2013, 40, 1006–1012. [Google Scholar] [CrossRef] [PubMed]
- Bendre, S.; Zhang, Z.; Kuo, H.-T.; Rousseau, J.; Zhang, C.; Merkens, H.; Roxin, Á.; Bénard, F.; Lin, K.-S. Evaluation of Met-Val-Lys as a Renal Brush Border Enzyme-Cleavable Linker to Reduce Kidney Uptake of 68Ga-Labeled DOTA-Conjugated Peptides and Peptidomimetics. Molecules 2020, 25, 3854. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, C.; Uehara, T.; Kanazawa, N.; Wada, S.; Suzuki, H.; Arano, Y. Preferential Cleavage of a Tripeptide Linkage by Enzymes on Renal Brush Border Membrane To Reduce Renal Radioactivity Levels of Radiolabeled Antibody Fragments. J. Med. Chem. 2018, 61, 5257–5268. [Google Scholar] [CrossRef] [PubMed]
- Uehara, T.; Kanazawa, N.; Suzuki, C.; Mizuno, Y.; Suzuki, H.; Hanaoka, H.; Arano, Y. Renal Handling of 99mTc-Labeled Antibody Fab Fragments with a Linkage Cleavable by Enzymes on Brush Border Membrane. Bioconjugate Chem. 2020, 31, 2618–2627. [Google Scholar] [CrossRef]
- Valpreda, G.; Trachsel, B.; Vogel, V.; Schibli, R.; Mu, L.; Behe, M. Dual MVK cleavable linkers effectively reduce renal retention of 111In-fibronectin-binding peptides. Bioorganic Med. Chem. 2022, 73, 117040. [Google Scholar] [CrossRef]
- Roscher, M.; Hormann, I.; Leib, O.; Marx, S.; Moreno, J.; Miltner, E.; Friesen, C. Targeted alpha-therapy using [Bi-213]anti-CD20 as novel treatment option for radio- and chemoresistant non-Hodgkin lymphoma cells. Oncotarget 2013, 4, 218–230. [Google Scholar] [CrossRef]
- Patel, R.B.; Hernandez, R.; Carlson, P.; Grudzinski, J.; Bates, A.M.; Jagodinsky, J.C.; Erbe, A.; Marsh, I.R.; Arthur, I.; Aluicio-Sarduy, E.; et al. Low-dose targeted radionuclide therapy renders immunologically cold tumors responsive to immune checkpoint blockade. Sci. Transl. Med. 2021, 13, eabb3631. [Google Scholar] [CrossRef]
- Ertveldt, T.; De Beck, L.; De Ridder, K.; Locy, H.; De Mey, W.; Goyvaerts, C.; Lecocq, Q.; Ceuppens, H.; De Vlaeminck, Y.; Awad, R.M.; et al. Targeted Radionuclide Therapy with Low and High-Dose Lutetium-177-Labeled Single Domain Antibodies Induces Distinct Immune Signatures in a Mouse Melanoma Model. Mol. Cancer Ther. 2022, 21, 1136–1148. [Google Scholar] [CrossRef]
- Ertveldt, T.; Krasniqi, A.; Ceuppens, H.; Puttemans, J.; Dekempeneer, Y.; De Jonghe, K.; de Mey, W.; Lecocq, Q.; De Vlaeminck, Y.; Awad, R.M.; et al. Targeted α-Therapy Using 225Ac Radiolabeled Single-Domain Antibodies Induces Antigen-Specific Immune Responses and Instills Immunomodula-tion Both systemically and at the Tumor Microenvironment. J. Nucl. Med. 2023, 264752. [Google Scholar] [CrossRef]
- Läppchen, T.; Rossin, R.; van Mourik, T.R.; Gruntz, G.; Hoeben, F.J.; Versteegen, R.; Janssen, H.M.; Lub, J.; Robillard, M.S. DOTA-tetrazine probes with modified linkers for tumor pretargeting. Nucl. Med. Biol. 2017, 55, 19–26. [Google Scholar] [CrossRef] [PubMed]
- McBride, W.H.; Schaue, D. Radiation-induced tissue damage and response. J. Pathol. 2020, 250, 647–655. [Google Scholar] [CrossRef] [PubMed]





| Vector Characteristics | Peptides | Scaffold Proteins | Antibody Fragments | Monoclonal Antibodies |
|---|---|---|---|---|
| Size | 0.5–5 kDa | 2–20 kDa | 12–110 kDa | 150 kDa |
| Affinity | pM–μM range | pM–μM range | pM–nM range | pM–nM range |
| Stability | Variable | + | + | + |
| Tissue penetration | + | + | Low to high | - |
| Blood clearance Elimination route | Fast Kidneys | Fast Kidneys | Fast to slow Kidneys/liver (depending on size) | Slow Liver |
| Immunogenicity | - | ± | - | ± |
| Production cost | Low | High | High | Very high |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Funeh, C.N.; Bridoux, J.; Ertveldt, T.; De Groof, T.W.M.; Chigoho, D.M.; Asiabi, P.; Covens, P.; D’Huyvetter, M.; Devoogdt, N. Optimizing the Safety and Efficacy of Bio-Radiopharmaceuticals for Cancer Therapy. Pharmaceutics 2023, 15, 1378. https://doi.org/10.3390/pharmaceutics15051378
Funeh CN, Bridoux J, Ertveldt T, De Groof TWM, Chigoho DM, Asiabi P, Covens P, D’Huyvetter M, Devoogdt N. Optimizing the Safety and Efficacy of Bio-Radiopharmaceuticals for Cancer Therapy. Pharmaceutics. 2023; 15(5):1378. https://doi.org/10.3390/pharmaceutics15051378
Chicago/Turabian StyleFuneh, Cyprine Neba, Jessica Bridoux, Thomas Ertveldt, Timo W. M. De Groof, Dora Mugoli Chigoho, Parinaz Asiabi, Peter Covens, Matthias D’Huyvetter, and Nick Devoogdt. 2023. "Optimizing the Safety and Efficacy of Bio-Radiopharmaceuticals for Cancer Therapy" Pharmaceutics 15, no. 5: 1378. https://doi.org/10.3390/pharmaceutics15051378