
Protein Fitness Prediction Is Impacted by the Interplay of Language Models, Ensemble Learning, and Sampling Methods
Abstract
:1. Introduction
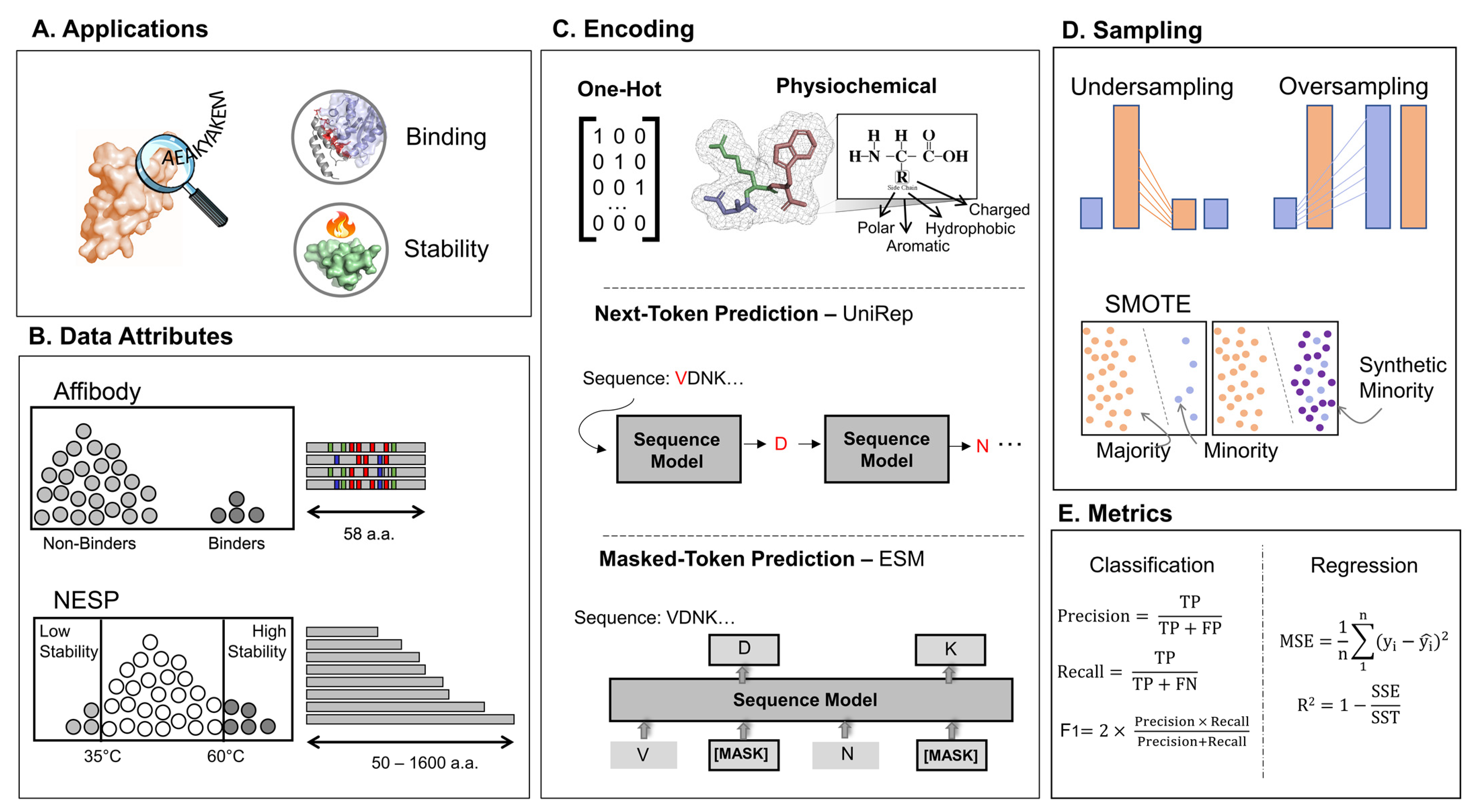
2. Materials and Methods
2.1. Obtaining Experimentally Labeled Sequence Data
2.2. Obtaining the Sequence Representations
2.3. Sampling and Splitting
2.4. Algorithm Selection and Training Details
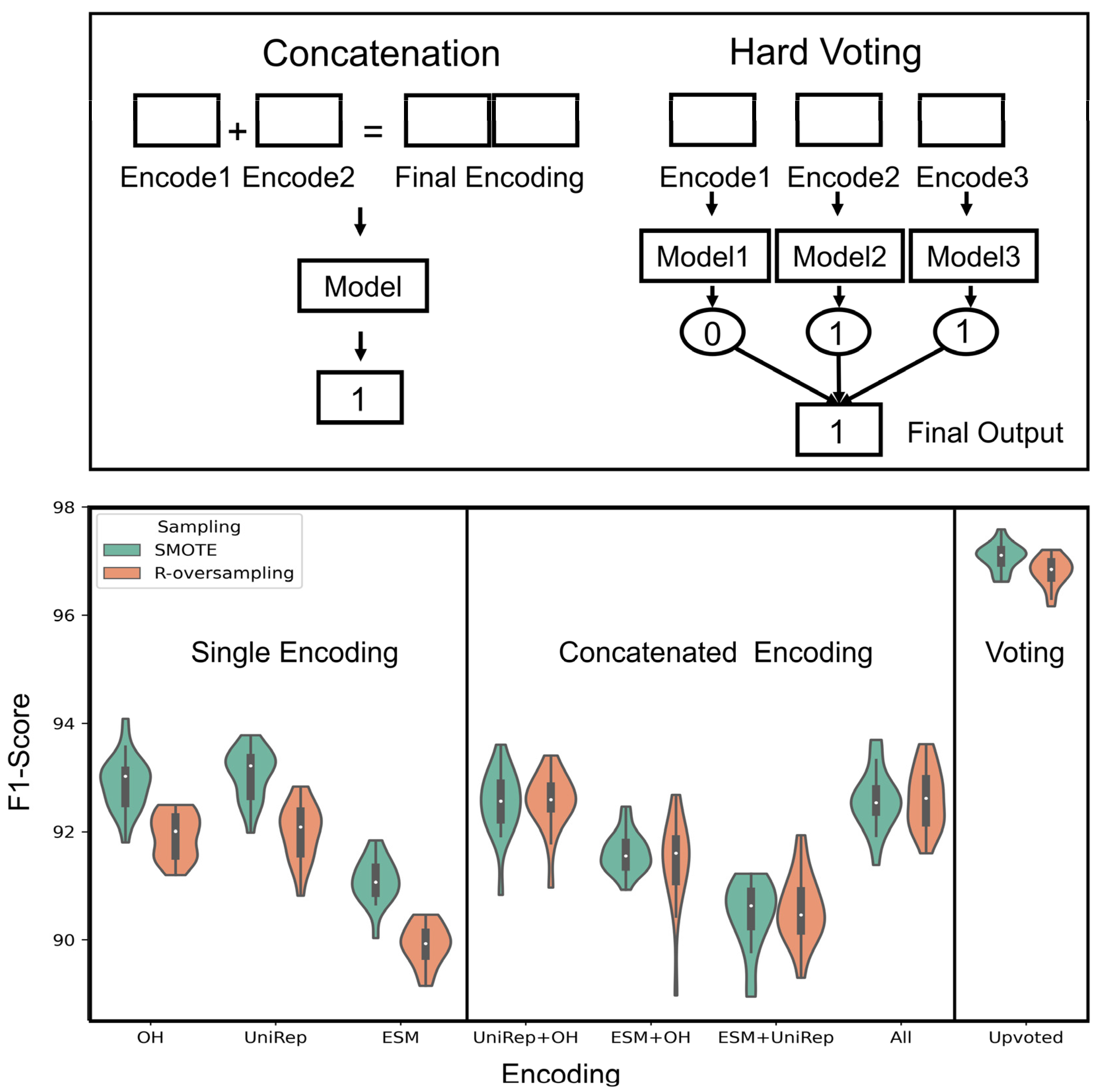
2.5. Ensemble Learning
2.6. Metrics and Statistical Analysis
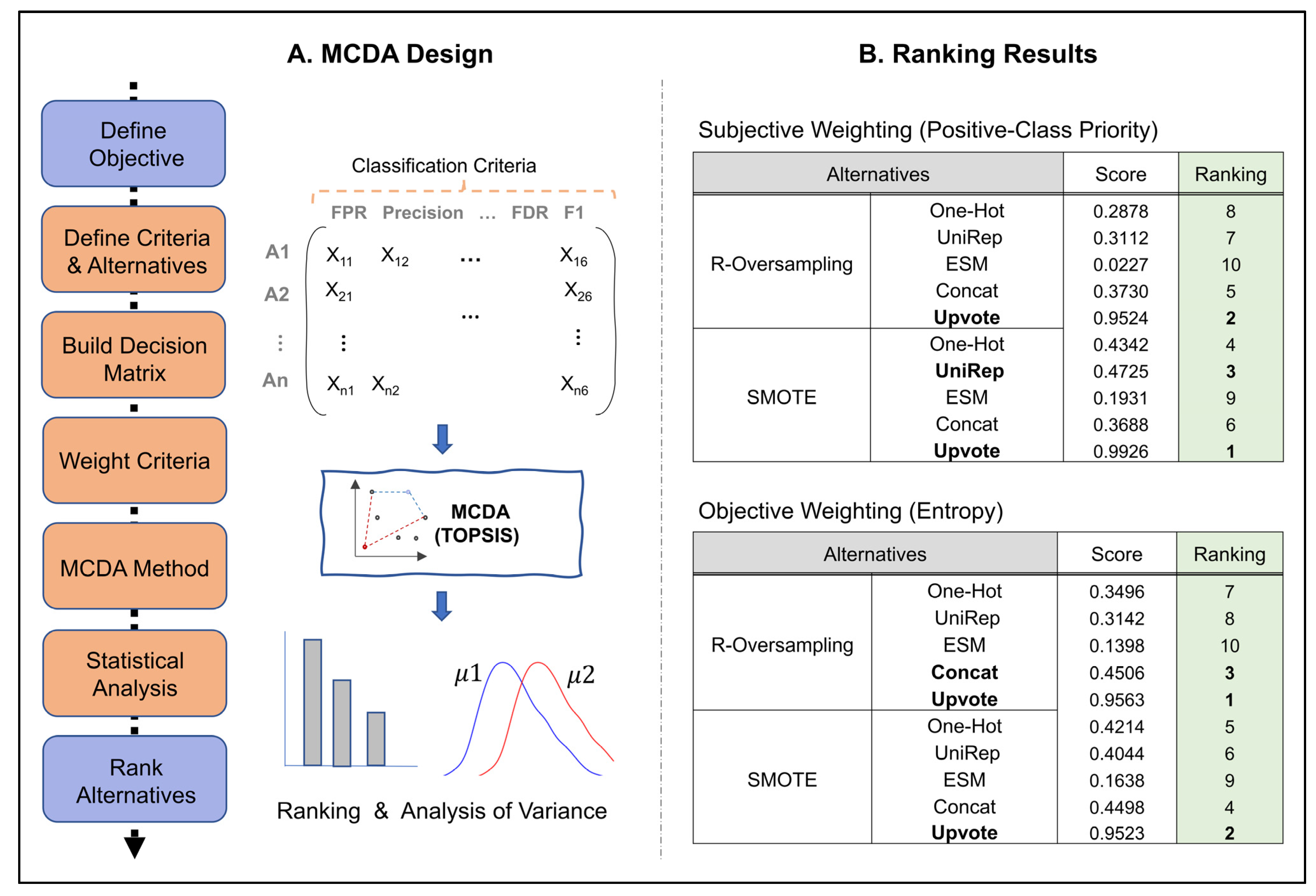
2.7. Multiple Criteria Decision Analysis (MCDA)
3. Results
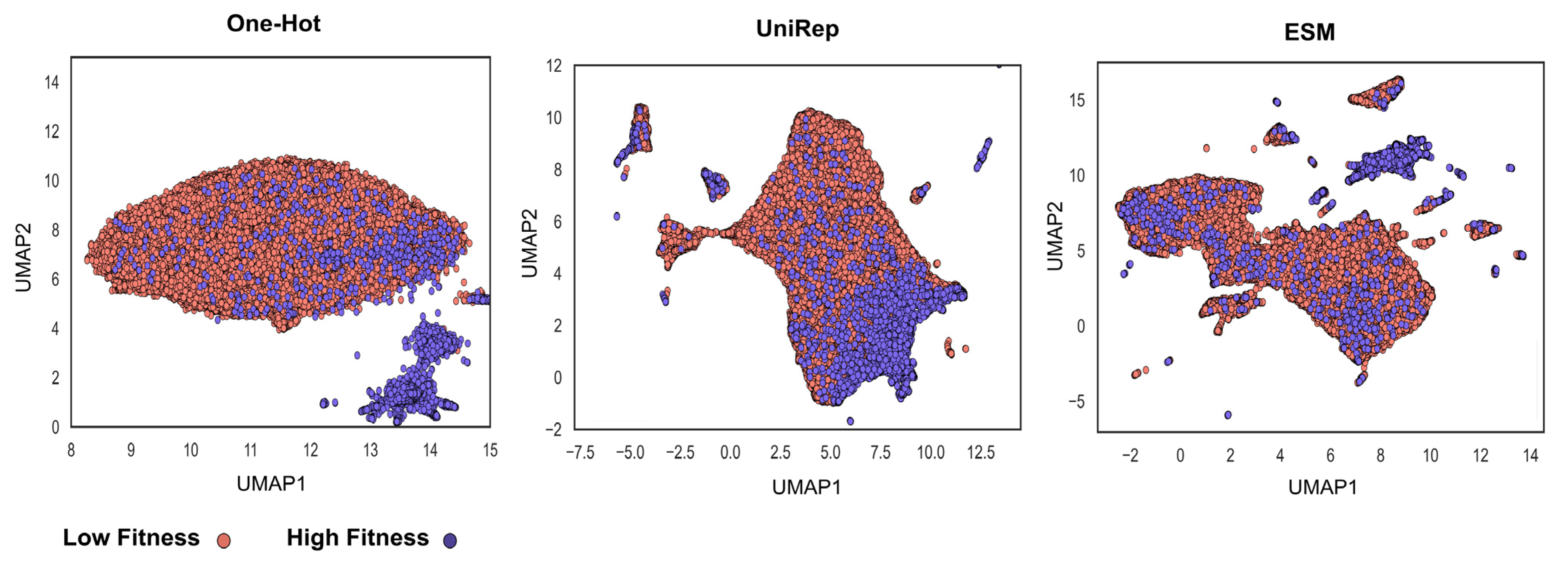
3.1. Sequence-Function Mapping Obtained from High-Throughput Selection Methods and Deep Sequencing Affibody Dataset

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset | Task | Fitness | Model | Attributes |
|---|---|---|---|---|
| Affibody | Classification | Binding Affinity | Logistic Regression | 82,663 non-binders |
| 6077 binders | ||||
| NESP | Classification | Stability | Logistic Regression | 3743 high-stability |
| 1311 low-stability | ||||
| NESP | Regression | Stability | Random Forest Regressor | 18,190 total |
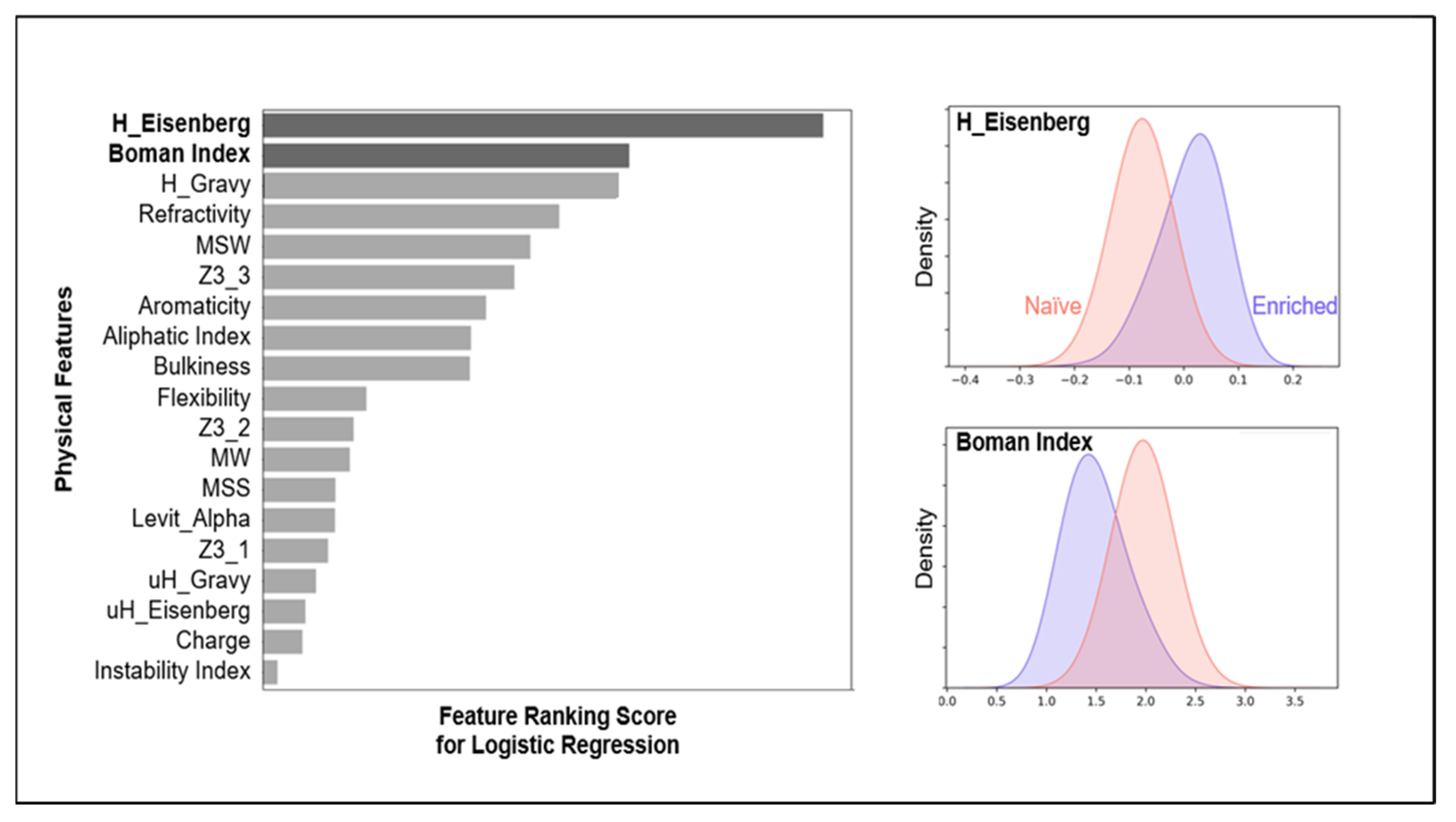
3.2. Physiochemical Feature Encoding, Interpretable Yet Lower Predictive Capacity
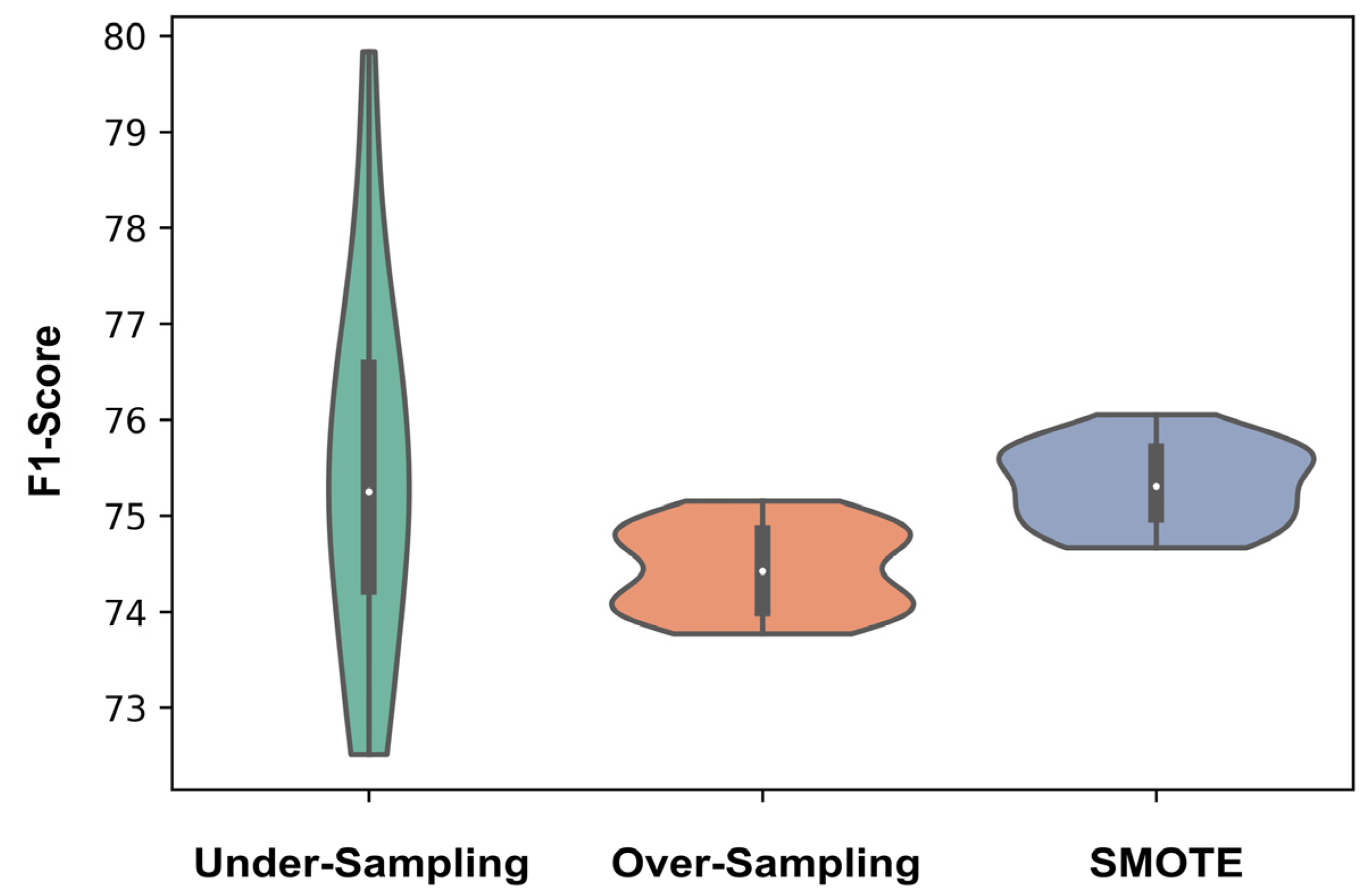
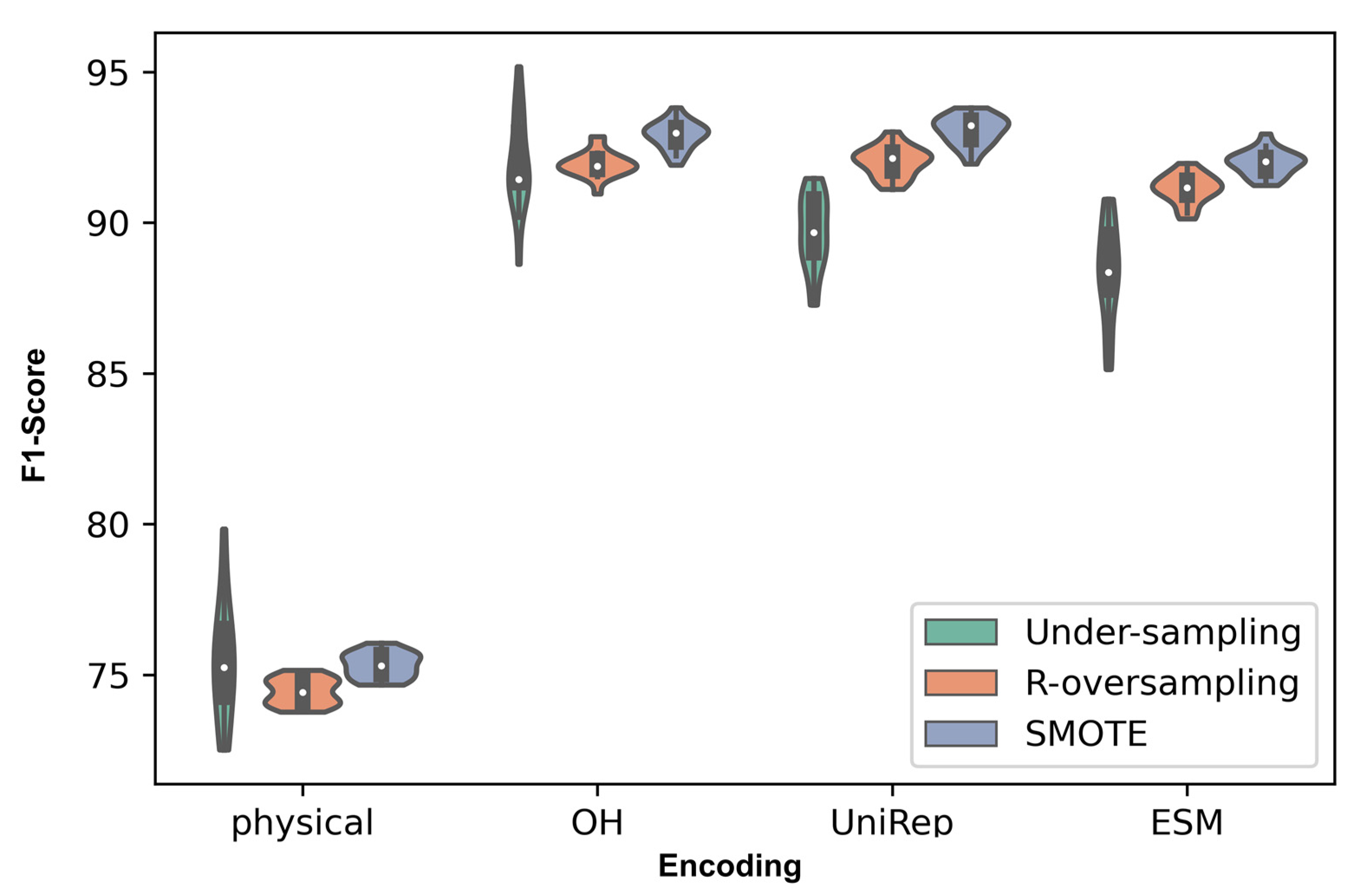
3.3. Comparison over All the Encoding and Sampling Methods
3.4. Increased Generalizability and Predictive Performance via Ensemble Learning
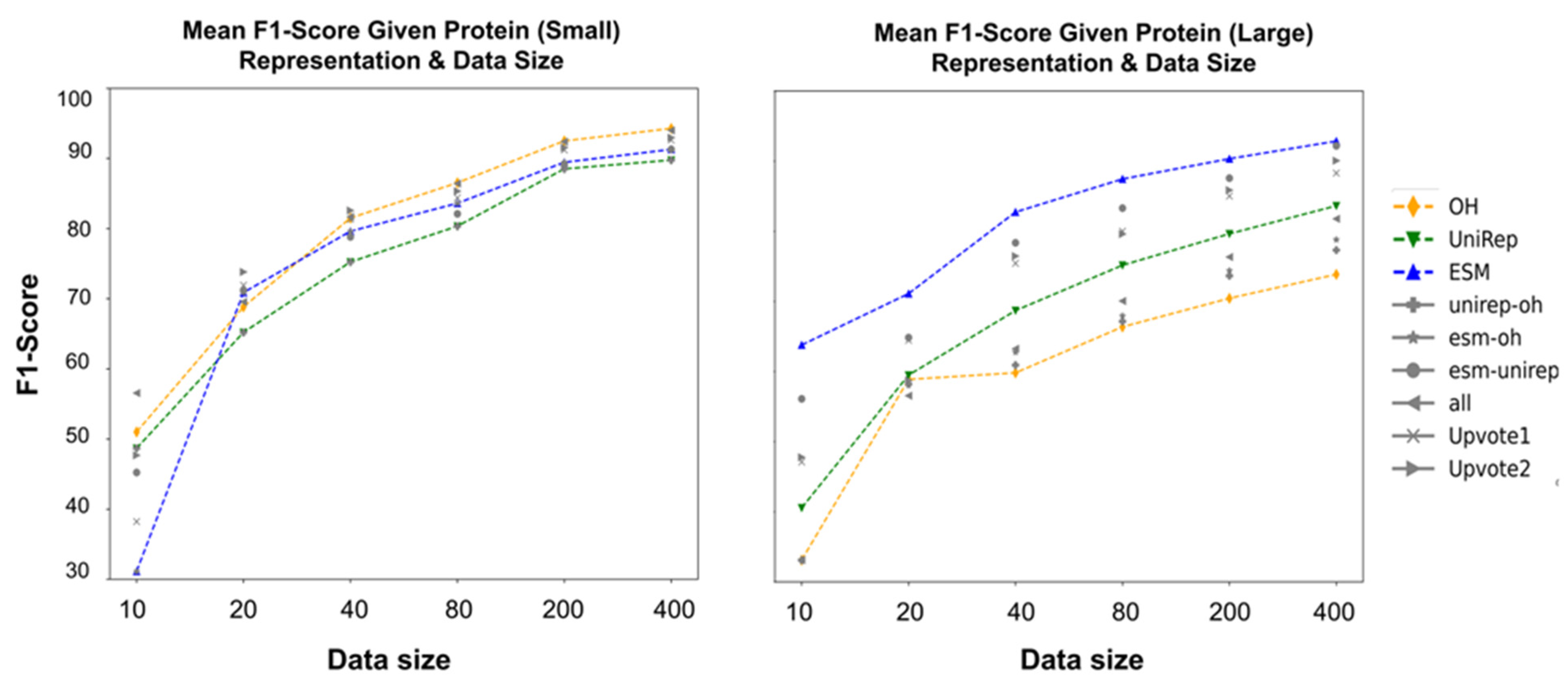
3.5. How Protein Encodings Perform Considering Different Data Attributes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liebermeister, W.; Noor, E.; Flamholz, A.; Davidi, D.; Bernhardt, J.; Milo, R. Visual Account of Protein Investment in Cellular Functions. Proc. Natl. Acad. Sci. USA 2014, 111, 8488–8493. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2000, 103, 211–225. [Google Scholar] [CrossRef] [PubMed]
- Hogan, B.L. Bone Morphogenetic Proteins: Multifunctional Regulators of Vertebrate Development. Genes Dev. 1996, 10, 1580–1594. [Google Scholar] [CrossRef] [PubMed]
- Andrianantoandro, E.; Basu, S.; Karig, D.K.; Weiss, R. Synthetic Biology: New Engineering Rules for an Emerging Discipline. Mol. Syst. Biol. 2006, 2, 2006.0028. [Google Scholar] [CrossRef]
- Heim, M.; Römer, L.; Scheibel, T. Hierarchical Structures Made of Proteins. The Complex Architecture of Spider Webs and Their Constituent Silk Proteins. Chem. Soc. Rev. 2010, 39, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Kolmar, H. Biological Diversity and Therapeutic Potential of Natural and Engineered Cystine Knot Miniproteins. Curr. Opin. Pharmacol. 2009, 9, 608–614. [Google Scholar] [CrossRef]
- Krasniqi, A.; D’Huyvetter, M.; Devoogdt, N.; Frejd, F.Y.; Sörensen, J.; Orlova, A.; Keyaerts, M.; Tolmachev, V. Same-Day Imaging Using Small Proteins: Clinical Experience and Translational Prospects in Oncology. J. Nucl. Med. 2018, 59, 885–891. [Google Scholar] [CrossRef]
- Romero, P.A.; Arnold, F.H. Exploring Protein Fitness Landscapes by Directed Evolution. Nat. Rev. Mol. Cell Biol. 2009, 10, 866–876. [Google Scholar] [CrossRef]
- Hellinga, H.W. Rational Protein Design: Combining Theory and Experiment. Proc. Natl. Acad. Sci. USA 1997, 94, 10015–10017. [Google Scholar] [CrossRef]
- Jäckel, C.; Kast, P.; Hilvert, D. Protein Design by Directed Evolution. Annu. Rev. Biophys. 2008, 37, 153–173. [Google Scholar] [CrossRef]
- Li, G.; Dong, Y.; Reetz, M.T. Can Machine Learning Revolutionize Directed Evolution of Selective Enzymes? Adv. Synth. Catal. 2019, 361, 2377–2386. [Google Scholar] [CrossRef]
- Anand, N.; Eguchi, R.; Mathews, I.I.; Perez, C.P.; Derry, A.; Altman, R.B.; Huang, P.S. Protein Sequence Design with a Learned Potential. Nat. Commun. 2022, 13, 716. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Jennifer Kan, S.B.; Lewis, R.D.; Wittmann, B.J.; Arnold, F.H. Machine Learning-Assisted Directed Protein Evolution with Combinatorial Libraries. Proc. Natl. Acad. Sci. USA 2019, 116, 8852–8858. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Oikawa, M.; Sato, T.; Nakazawa, H.; Ito, T.; Kameda, T.; Tsuda, K.; Umetsu, M. Machine-Learning-Guided Library Design Cycle for Directed Evolution of Enzymes: The Effects of Training Data Composition on Sequence Space Exploration. ACS Catal. 2021, 11, 14615–14624. [Google Scholar] [CrossRef]
- Golinski, A.W.; Mischler, K.M.; Laxminarayan, S.; Neurock, N.L.; Fossing, M.; Pichman, H.; Martiniani, S.; Hackel, B.J. High-Throughput Developability Assays Enable Library-Scale Identification of Producible Protein Scaffold Variants. Proc. Natl. Acad. Sci. USA 2021, 118, e2026658118. [Google Scholar] [CrossRef]
- Chen, J.; Zheng, S.; Zhao, H.; Yang, Y. Structure-Aware Protein Solubility Prediction from Sequence through Graph Convolutional Network and Predicted Contact Map. J. Cheminform. 2021, 13, 1–10. [Google Scholar] [CrossRef]
- Wang, S.; Liu, D.; Ding, M.; Du, Z.; Zhong, Y.; Song, T.; Zhu, J.; Zhao, R. SE-OnionNet: A Convolution Neural Network for Protein–Ligand Binding Affinity Prediction. Front. Genet. 2021, 11, 607824. [Google Scholar] [CrossRef]
- Kuzmin, K.; Adeniyi, A.E.; DaSouza, A.K.; Lim, D.; Nguyen, H.; Molina, N.R.; Xiong, L.; Weber, I.T.; Harrison, R.W. Machine Learning Methods Accurately Predict Host Specificity of Coronaviruses Based on Spike Sequences Alone. Biochem. Biophys. Res. Commun. 2020, 533, 553–558. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Chakrabarti, S. Classification and Prediction of Protein–Protein Interaction Interface Using Machine Learning Algorithm. Sci. Rep. 2021, 11, 1761. [Google Scholar] [CrossRef]
- Vander Meersche, Y.; Cretin, G.; de Brevern, A.G.; Gelly, J.C.; Galochkina, T. MEDUSA: Prediction of Protein Flexibility from Sequence. J. Mol. Biol. 2021, 433, 166882. [Google Scholar] [CrossRef]
- Young, T.; Hazarika, D.; Poria, S.; Cambria, E. Recent Trends in Deep Learning Based Natural Language Processing [Review Article]. IEEE Comput. Intell. Mag. 2018, 13, 55–75. [Google Scholar] [CrossRef]
- Collobert, R.; Weston, J.; Bottou, L.; Karlen, M.; Kavukcuoglu, K.; Kuksa, P. Natural Language Processing (Almost) from Scratch. J. Mach. Learn. Res. 2011, 12, 2493–2537. [Google Scholar]
- Mnasri, M. Recent Advances in Conversational NLP: Towards the Standardization of Chatbot Building. arXiv 2019, arXiv:1903.09025. [Google Scholar]
- Campagna, G.; Xu, S.; Moradshahi, M.; Socher, R.; Lam, M.S. Genie: A Generator of Natural Language Semantic Parsers for Virtual Assistant Commands. In Proceedings of the 40th ACM SIGPLAN Conference on Programming Language Design and Implementation (PLDI ’19), Phoenix, AZ, USA, 22–26 June 2019; pp. 394–410. [Google Scholar] [CrossRef]
- Heinzinger, M.; Elnaggar, A.; Wang, Y.; Dallago, C.; Nechaev, D.; Matthes, F.; Rost, B. Modeling Aspects of the Language of Life through Transfer-Learning Protein Sequences. BMC Bioinform. 2019, 20, 723. [Google Scholar] [CrossRef] [PubMed]
- Ofer, D.; Brandes, N.; Linial, M. The Language of Proteins: NLP, Machine Learning & Protein Sequences. Comput. Struct. Biotechnol. J. 2021, 19, 1750–1758. [Google Scholar] [CrossRef]
- Bateman, A.; Martin, M.J.; O’Donovan, C.; Magrane, M.; Apweiler, R.; Alpi, E.; Antunes, R.; Arganiska, J.; Bely, B.; Bingley, M.; et al. UniProt: A Hub for Protein Information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar] [CrossRef]
- Katz, K.; Shutov, O.; Lapoint, R.; Kimelman, M.; Rodney Brister, J.; O’Sullivan, C. The Sequence Read Archive: A Decade More of Explosive Growth. Nucleic Acids Res. 2022, 50, D387–D390. [Google Scholar] [CrossRef]
- Torrisi, M.; Pollastri, G.; Le, Q. Deep Learning Methods in Protein Structure Prediction. Comput. Struct. Biotechnol. J. 2020, 18, 1301–1310. [Google Scholar] [CrossRef]
- Elnaggar, A.; Heinzinger, M.; Dallago, C.; Rehawi, G.; Yu, W.; Jones, L.; Gibbs, T.; Feher, T.; Angerer, C.; Steinegger, M.; et al. ProtTrans: Towards Cracking the Language of Lifes Code Through Self-Supervised Deep Learning and High Performance Computing. IEEE Trans. Pattern Anal. Mach. Intell. 2021, 14, 7112–7127. [Google Scholar] [CrossRef]
- Ferruz, N.; Schmidt, S.; Höcker, B. ProtGPT2 Is a Deep Unsupervised Language Model for Protein Design. Nat. Commun. 2022, 13, 4348. [Google Scholar] [CrossRef]
- Alley, E.C.; Khimulya, G.; Biswas, S.; AlQuraishi, M.; Church, G.M. Unified Rational Protein Engineering with Sequence-Based Deep Representation Learning. Nat. Methods 2019, 16, 1315–1322. [Google Scholar] [CrossRef] [PubMed]
- Rives, A.; Meier, J.; Sercu, T.; Goyal, S.; Lin, Z.; Liu, J.; Guo, D.; Ott, M.; Zitnick, C.L.; Ma, J.; et al. Biological Structure and Function Emerge from Scaling Unsupervised Learning to 250 Million Protein Sequences. Proc. Natl. Acad. Sci. USA 2021, 118, e2016239118. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.; Bhattacharya, N.; Thomas, N.; Duan, Y.; Chen, X.; Canny, J.; Abbeel, P.; Song, Y.S. Evaluating Protein Transfer Learning with Tape. Adv. Neural Inf. Process. Syst. 2019, 32, 9689. [Google Scholar]
- Brandes, N.; Ofer, D.; Peleg, Y.; Rappoport, N.; Linial, M. ProteinBERT: A Universal Deep-Learning Model of Protein Sequence and Function. Bioinformatics 2022, 38, 2102–2110. [Google Scholar] [CrossRef]
- Hsu, C.; Nisonoff, H.; Fannjiang, C.; Listgarten, J. Combining Evolutionary and Assay-Labelled Data for Protein Fitness Prediction. bioRxiv 2021. [Google Scholar] [CrossRef]
- Meier, J.; Rao, R.; Verkuil, R.; Liu, J.; Sercu, T.; Rives, A. Language Models Enable Zero-Shot Prediction of the Effects of Mutations on Protein Function. Adv. Neural Inf. Process. Syst. 2021, 35, 29287–29303. [Google Scholar]
- Chu, S.K.S.; Siegel, J. Predicting Single-Point Mutational Effect on Protein Stability. Growth 2021, 16, 35. [Google Scholar]
- Lv, Z.; Wang, P.; Zou, Q.; Jiang, Q. Identification of Sub-Golgi Protein Localization by Use of Deep Representation Learning Features. Bioinformatics 2020, 36, 5600–5609. [Google Scholar] [CrossRef]
- Li, K.; Zhong, Y.; Lin, X.; Quan, Z. Predicting the Disease Risk of Protein Mutation Sequences with Pre-Training Model. Front. Genet. 2020, 11, 605620. [Google Scholar] [CrossRef]
- Min, S.; Kim, H.G.; Lee, B.; Yoon, S. Protein Transfer Learning Improves Identification of Heat Shock Protein Families. PLoS ONE 2021, 16, e0251865. [Google Scholar] [CrossRef]
- Woldring, D.R.; Holec, P.V.; Stern, L.A.; Du, Y.; Hackel, B.J. A Gradient of Sitewise Diversity Promotes Evolutionary Fitness for Binder Discovery in a Three-Helix Bundle Protein Scaffold. Biochemistry 2017, 56, 1656–1671. [Google Scholar] [CrossRef] [PubMed]
- Pultz, D.; Friis, E.; Salomon, J.; Maggie; Fischer Hallin, P.; Baagøe Jørgensen, S. Novozymes Enzyme Stability Prediction; Kaggle: San Francisco, CA, USA, 2022. [Google Scholar]
- Keeney, R.L.; Raiffa, H.; Rajala, D.W. Decisions with Multiple Objectives: Preferences and Value Trade-Offs. IEEE Trans. Syst. Man. Cybern. 1977, 9, 403. [Google Scholar] [CrossRef]
- Müller, A.T.; Gabernet, G.; Hiss, J.A.; Schneider, G. ModlAMP: Python for Antimicrobial Peptides. Bioinformatics 2017, 33, 2753–2755. [Google Scholar] [CrossRef]
- Yang, K.K.; Wu, Z.; Bedbrook, C.N.; Arnold, F.H. Learned Protein Embeddings for Machine Learning. Bioinformatics 2018, 34, 2642–2648. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; Verkuil, R.; Kabeli, O.; Shmueli, Y.; et al. Evolutionary-Scale Prediction of Atomic Level Protein Structure with a Language Model. Science 2023, 379, 1123–1130. [Google Scholar] [CrossRef]
- Kovács, B.; Tinya, F.; Németh, C.; Ódor, P. SMOTE: Synthetic Minority Over-Sampling Technique Nitesh. Ecol. Appl. 2020, 30, 321–357. [Google Scholar]
- Fernández, A.; García, S.; Herrera, F.; Chawla, N.V. SMOTE for Learning from Imbalanced Data: Progress and Challenges, Marking the 15-Year Anniversary. J. Artif. Intell. Res. 2018, 61, 863–905. [Google Scholar] [CrossRef]
- Mohammed, A.J. Improving Classification Performance for a Novel Imbalanced Medical Dataset Using SMOTE Method. Int. J. Adv. Trends Comput. Sci. Eng. 2020, 9, 3161–3172. [Google Scholar] [CrossRef]
- Rupapara, V.; Rustam, F.; Shahzad, H.F.; Mehmood, A.; Ashraf, I.; Choi, G.S. Impact of SMOTE on Imbalanced Text Features for Toxic Comments Classification Using RVVC Model. IEEE Access 2021, 9, 78621–78634. [Google Scholar] [CrossRef]
- Hasanin, T.; Khoshgoftaar, T.M.; Leevy, J.L.; Bauder, R.A. Severely Imbalanced Big Data Challenges: Investigating Data Sampling Approaches. J. Big Data 2019, 6, 1–25. [Google Scholar] [CrossRef]
- Blagus, R.; Lusa, L. Evaluation of SMOTE for High-Dimensional Class-Imbalanced Microarray Data. In Proceedings of the 2012 11th International Conference on Machine Learning and Applications, Boca Raton, FL, USA, 12–15 December 2012; pp. 89–94. [Google Scholar] [CrossRef]
- van den Goorbergh, R.; van Smeden, M.; Timmerman, D.; Van Calster, B. The Harm of Class Imbalance Corrections for Risk Prediction Models: Illustration and Simulation Using Logistic Regression. J. Am. Med. Inform. Assoc. 2022, 29, 1525–1534. [Google Scholar] [CrossRef]
- Akiba, T.; Sano, S.; Yanase, T.; Ohta, T.; Koyama, M. Optuna: A Next-generation Hyperparameter Optimization Framework. In Proceedings of the 25th ACM SIGKDD International Conference on Knowledge Discovery & Data Mining, Anchorage, AK, USA, 4–8 August 2019; pp. 2623–2631. [Google Scholar] [CrossRef]
- McHugh, M.L. Multiple Comparison Analysis Testing in ANOVA. Biochem. Med. 2011, 21, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, R.A. When to Use the Bonferroni Correction. Ophthalmic Physiol. Opt. 2014, 34, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Tukey, J. The Problem of Multiple Comparisons. Department of Statistics; Department of Statistics, Princeton University: Princeton, NJ, USA, 1953. [Google Scholar]
- Branco, P.; Torgo, L.; Ribeiro, R. A Survey of Predictive Modelling under Imbalanced Distributions. ACM Comput. Surv. (CSUR) 2015, 49, 1–50. [Google Scholar] [CrossRef]
- Borowska, K.; Stepaniuk, J. Imbalanced Data Classification: A Novel Re-Sampling Approach Combining Versatile Improved SMOTE and Rough Sets. In Proceedings of the Computer Information Systems and Industrial Management: 15th IFIP TC8 International Conference, CISIM 2016, Vilnius, Lithuania, 14–16 September 2016; Volume 9842, pp. 31–42. [Google Scholar] [CrossRef]
- Hwang, C.-L.; Yoon, K. Methods for Multiple Attribute Decision Making BT—Multiple Attribute Decision Making: Methods and Applications A State-of-the-Art Survey; Hwang, C.-L., Yoon, K., Eds.; Springer: Berlin/Heidelberg, Germany, 1981; pp. 58–191. ISBN 978-3-642-48318-9. [Google Scholar]
- Shannon, C.E. A Mathematical Theory of Communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef]
- Johnson, R.A.; Wichern, D.W. Applied Multivariate Statistical Analysis; Pearson Prentice Hall: Hoboken, NJ, USA, 2007; ISBN 0-13-187715-1. [Google Scholar]
- McInnes, L.; Healy, J.; Melville, J. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv 2018, arXiv:1802.03426. [Google Scholar]
- Jolliffe, I.T. Principal Component Analysis: A Beginner’s Guide—I. Introduction and Application. Weather 1990, 45, 375–382. [Google Scholar] [CrossRef]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Costa, A.d.S.; Fazel-Zarandi, M.; Sercu, T.; Candido, S.; et al. Language Models of Protein Sequences at the Scale of Evolution Enable Accurate Structure Prediction. bioRxiv 2022. [Google Scholar] [CrossRef]
- Marquet, C.; Heinzinger, M.; Olenyi, T.; Dallago, C.; Erckert, K.; Bernhofer, M.; Nechaev, D.; Rost, B. Embeddings from Protein Language Models Predict Conservation and Variant Effects. Hum. Genet. 2021, 141, 1629–1647. [Google Scholar] [CrossRef]
- Biswas, S. Low-N Protein Engineering with Data-Efficient Deep Learning A Paradigm for Low-N Protein Engineering. Nat. Methods 2020, 18, 389–396. [Google Scholar] [CrossRef]
- Rao, R.M.; Liu, J.; Verkuil, R.; Meier, J.; Canny, J.; Abbeel, P.; Sercu, T.; Rives, A. MSA Transformer. In Proceedings of the 38th International Conference on Machine Learning, Virtual Event, 18–24 July 2021; Meila, M., Zhang, T., Eds.; Volume 139, pp. 8844–8856. [Google Scholar]
- Shanehsazzadeh, A.; Belanger, D.; Dohan, D. Is Transfer Learning Necessary for Protein Landscape Prediction? arXiv 2020, arXiv:2011.03443. [Google Scholar]
- Wittmann, B.J.; Yue, Y.; Arnold, F.H. Informed Training Set Design Enables Efficient Machine Learning-Assisted Directed Protein Evolution. Cell Syst. 2021, 12, 1026–1045.e7. [Google Scholar] [CrossRef] [PubMed]
- Wettig, A.; Gao, T.; Zhong, Z.; Chen, D. Should You Mask 15% in Masked Language Modeling? arXiv 2022, arXiv:2202.08005. [Google Scholar]
- Lupo, U.; Sgarbossa, D.; Bitbol, A.F. Protein Language Models Trained on Multiple Sequence Alignments Learn Phylogenetic Relationships. Nat. Commun. 2022, 13, 6298. [Google Scholar] [CrossRef]
- Nourani, E.; Asgari, E.; Mc Hardy, A.; Mofrad, M. TripletProt: Deep Representation Learning of Proteins Based on Siamese Networks. IEEE/ACM Trans. Comput. Biol. Bioinform. 2021, 19, 3744–3753. [Google Scholar] [CrossRef]
- Hua, H.; Li, X.; Dou, D.; Xu, C.-Z.; Luo, J. Fine-Tuning Pre-Trained Language Models with Noise Stability Regularization. arXiv 2022, arXiv:2206.05658. [Google Scholar]
- Wang, B.; Member, S.; Wang, A.; Chen, F.; Member, S.; Wang, Y.; Kuo, C.J. Evaluating Word Embedding Models: Methods and Experimental Results. APSIPA Trans. Signal Inf. Process. 2019, 8, e19. [Google Scholar] [CrossRef]








| Comparison | Mean GP1 | Mean GP2 | Metrics | Reject Null |
|---|---|---|---|---|
| 0.9712 | 0.9682 | F1 | FALSE | |
| Upvote_SM | 0.0187 | 0.0258 | FDR | FALSE |
| vs. | 0.9614 | 0.9622 | TPR | FALSE |
| Upvote_RO | 0.9813 | 0.9742 | Precision | FALSE |
| 0.9972 | 0.9972 | NPV | FALSE | |
| 0.0019 | 0.0013 | FPR | FALSE | |
| 0.9712 | 0.9307 | F1 | TRUE | |
| Upvote_SM | 0.0187 | 0.0930 | FDR | TRUE |
| vs. | 0.9614 | 0.9557 | TPR | TRUE |
| UniRep_SM | 0.9813 | 0.9070 | Precision | TRUE |
| 0.9972 | 0.9967 | NPV | TRUE | |
| 0.0019 | 0.0072 | FPR | TRUE | |
| 0.9712 | 0.9261 | F1 | TRUE | |
| Upvote_SM | 0.0187 | 0.1061 | FDR | TRUE |
| vs. | 0.9614 | 0.9607 | TPR | FALSE |
| Concat_RO | 0.9813 | 0.8939 | Precision | TRUE |
| 0.9972 | 0.9971 | NPV | FALSE | |
| 0.0019 | 0.0084 | FPR | TRUE | |
| 0.9682 | 0.9307 | F1 | TRUE | |
| Upvote_RO | 0.0258 | 0.0930 | FDR | TRUE |
| vs. | 0.9622 | 0.9557 | TPR | TRUE |
| UniRep_SM | 0.9742 | 0.9070 | Precision | TRUE |
| 0.9972 | 0.9967 | NPV | TRUE | |
| 0.0013 | 0.0072 | FPR | TRUE | |
| 0.9682 | 0.9261 | F1 | TRUE | |
| Upvote_RO | 0.0258 | 0.1061 | FDR | TRUE |
| vs. | 0.9622 | 0.9607 | TPR | FALSE |
| Concat_RO | 0.9742 | 0.8939 | Precision | TRUE |
| 0.9972 | 0.9971 | NPV | FALSE | |
| 0.0013 | 0.0084 | FPR | TRUE | |
| 0.9261 | 0.9307 | F1 | TRUE | |
| Concat_RO | 0.1061 | 0.0930 | FDR | TRUE |
| vs. | 0.9607 | 0.9557 | TPR | TRUE |
| UniRep_SM | 0.8939 | 0.9070 | Precision | TRUE |
| 0.9971 | 0.9967 | NPV | TRUE | |
| 0.0084 | 0.0072 | FPR | TRUE |
| Encoding | Validation | Test | ||
|---|---|---|---|---|
| R² | MSE | R² | MSE | |
| One-Hot | 0.21 | 141 | 0.24 | 130 |
| UniRep | 0.49 | 108 | 0.40 | 102 |
| ESM | 0.65 | 63 | 0.65 | 60 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mardikoraem, M.; Woldring, D. Protein Fitness Prediction Is Impacted by the Interplay of Language Models, Ensemble Learning, and Sampling Methods. Pharmaceutics 2023, 15, 1337. https://doi.org/10.3390/pharmaceutics15051337
Mardikoraem M, Woldring D. Protein Fitness Prediction Is Impacted by the Interplay of Language Models, Ensemble Learning, and Sampling Methods. Pharmaceutics. 2023; 15(5):1337. https://doi.org/10.3390/pharmaceutics15051337
Chicago/Turabian StyleMardikoraem, Mehrsa, and Daniel Woldring. 2023. "Protein Fitness Prediction Is Impacted by the Interplay of Language Models, Ensemble Learning, and Sampling Methods" Pharmaceutics 15, no. 5: 1337. https://doi.org/10.3390/pharmaceutics15051337