1. Introduction
Statistics from the National Cancer Institute show that cervical cancer was persistently the second leading cause of cancer death in women aged 20–39 years in 2019, despite the popularization of HPV vaccination [
1]. Compared to conventional therapies (surgery, chemotherapy, and/or radiotherapy), nanotechnology offers many possibilities in the delivery of chemotherapeutic drugs for cancer and has achieved considerable progress with the initial success of Doxil
® (PEGylated liposomal doxorubicin) [
2].
Paclitaxel (PTX) is one of the most widely used broad-spectrum anti-cancer agents together with doxorubicin and cisplatin; PTX is considered one of the most successful natural anti-cancer drugs available and has been approved by the Food and Drug Administration for the treatment of breast, ovarian, and lung cancer, as well as Kaposi’s sarcoma [
3,
4]. As a highly lipophilic compound, the application of paclitaxel is restricted by its limited solubility, low bioavailability, and cosolvent-induced toxicity. Various nanocarrier systems, including micelles, liposomes, nanoparticles, bioconjugates, transfersomes, and polymeric dendrimers, have been employed to improve PTX’s solubility and reduce its undesired side effects [
5,
6].
Charge-reversal nanocarriers are neutral/negatively charged at various physiological conditions, such as in the blood circulation, to achieve long circulation time, but could be converted to positively charged by a certain stimulus, either exogenous (variations in temperature, ultrasound intensity, magnetic field, light, or electric pulses) or endogenous (changes in pH, redox gradients, or enzyme concentration) when they arrive at micro-vessels around the tumor [
7,
8,
9]. Nanoparticles with cationic charge surfaces can much more easily transverse across the blood capillaries into the tumor [
10] and also have stronger affinities to negatively charged tumor cell membranes; thus, charge-reversal can provide nanocarriers with both long blood circulation and deep tumor penetration [
11], both of which are essential for tumor-targeted drug delivery. In contrast, although nanoparticles with an anionic charge surface can avoid protein adsorption, thus prolonging their blood circulation time, their weak interaction with negatively charged cell membranes leads to inefficient internalization by tumor cells [
12]. Based on these, γ-Glutamyl transpeptidase (GGT)-triggered charge reversal drug-loading nanoparticles were constructed in this paper to achieve better anti-tumor therapeutic efficacy for PTX.
GGT is usually regarded as a nonspecific marker of liver dysfunction and has a vital role in the extracellular catabolism of GSH (the major antioxidant in mammalian cells). The serum levels of GGT may be associated with a wide range of disease outcomes, including cancer [
13]. Increased expression of GGT has been found in several human tumors (including HEK293 [
14], Hela [
15], HepG2 [
16], MCF-7 [
17], and U87MG cell lines [
14]) for cleaving extracellular glutathione and providing cells with amino acids necessary for intracellular biosynthesis to meet the requirements of the vigorous metabolism of tumor cells [
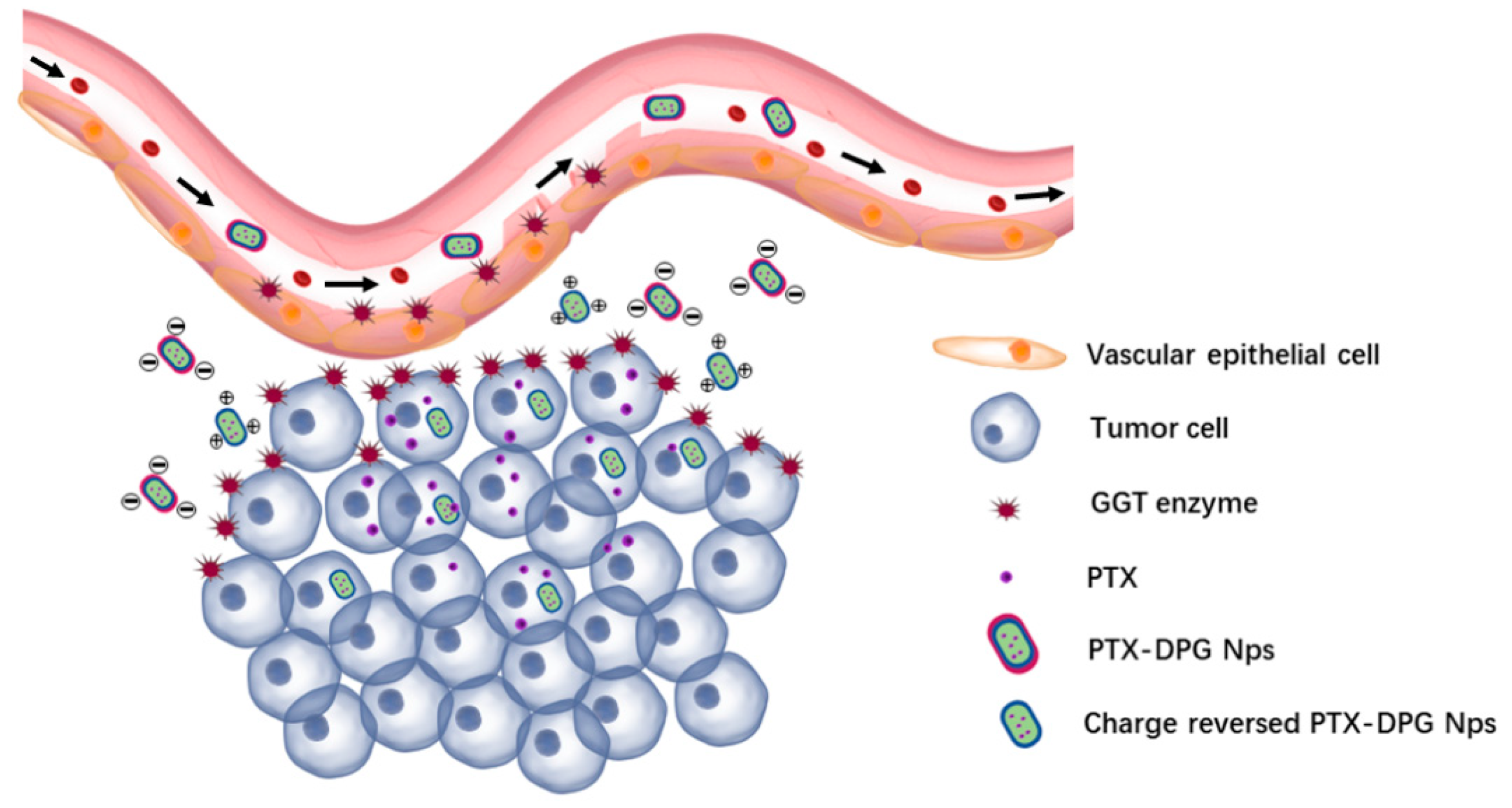
18]. GGT is overexpressed on the external surface of the endothelial cells and metabolically active tumor cells at the periphery of the blood vessels (
Figure 1), and the activity of GGT on the tumor cellular membrane surface is ten-fold higher than that of GGT in the blood [
19]. It has been proven that the overexpressed GGT on the vascular endothelial cell or tumor cell triggers the γ-glutamyl transfer reactions of glutathione to achieve deep penetration of nanocarriers modified with glutathione [
20] or containing γ-glutamyl moieties [
17]. The near-infrared (NIR) fluorescent probe with the GGT-recognizable substrate and c-RGD can effectively differentiate the tumor cells from GGT-positive and αvβ3-deficient normal cells [
14].
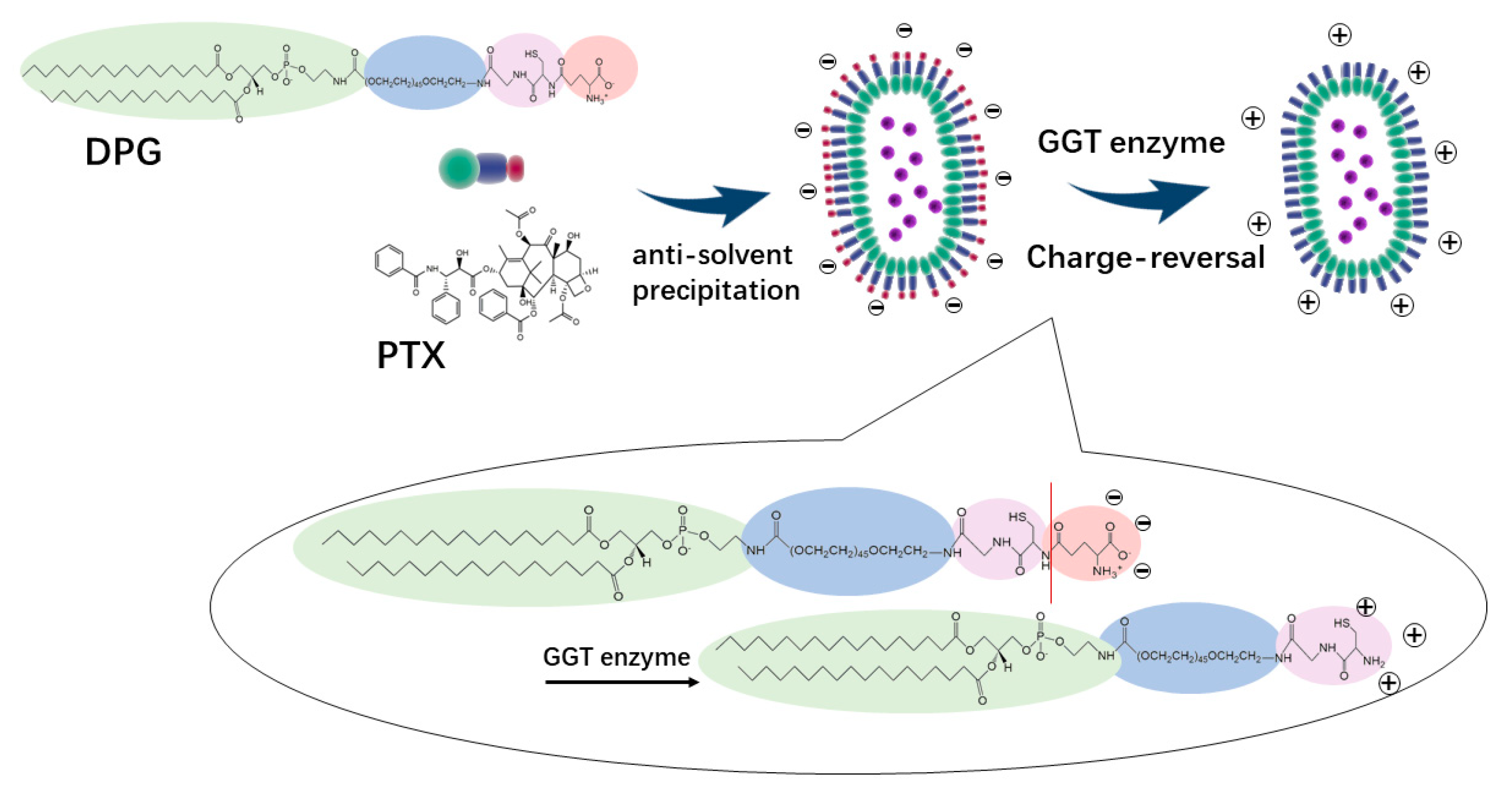
In this work, DSPE-mPEG2000-NH2 was conjugated with the −COOH group of the protected GSH tripeptide by amide condensation, followed by the removal of the protective groups. The DSPE-PEG2000-GSH (DPG) is nearly neutral, but its terminal GSH peptide contains γ-glutamyl amide that is sensitive to and can be hydrolyzed by GGT enzyme, which is highly expressed in the tumor, leading to the exposure of the amino group and positive charge of the whole system.
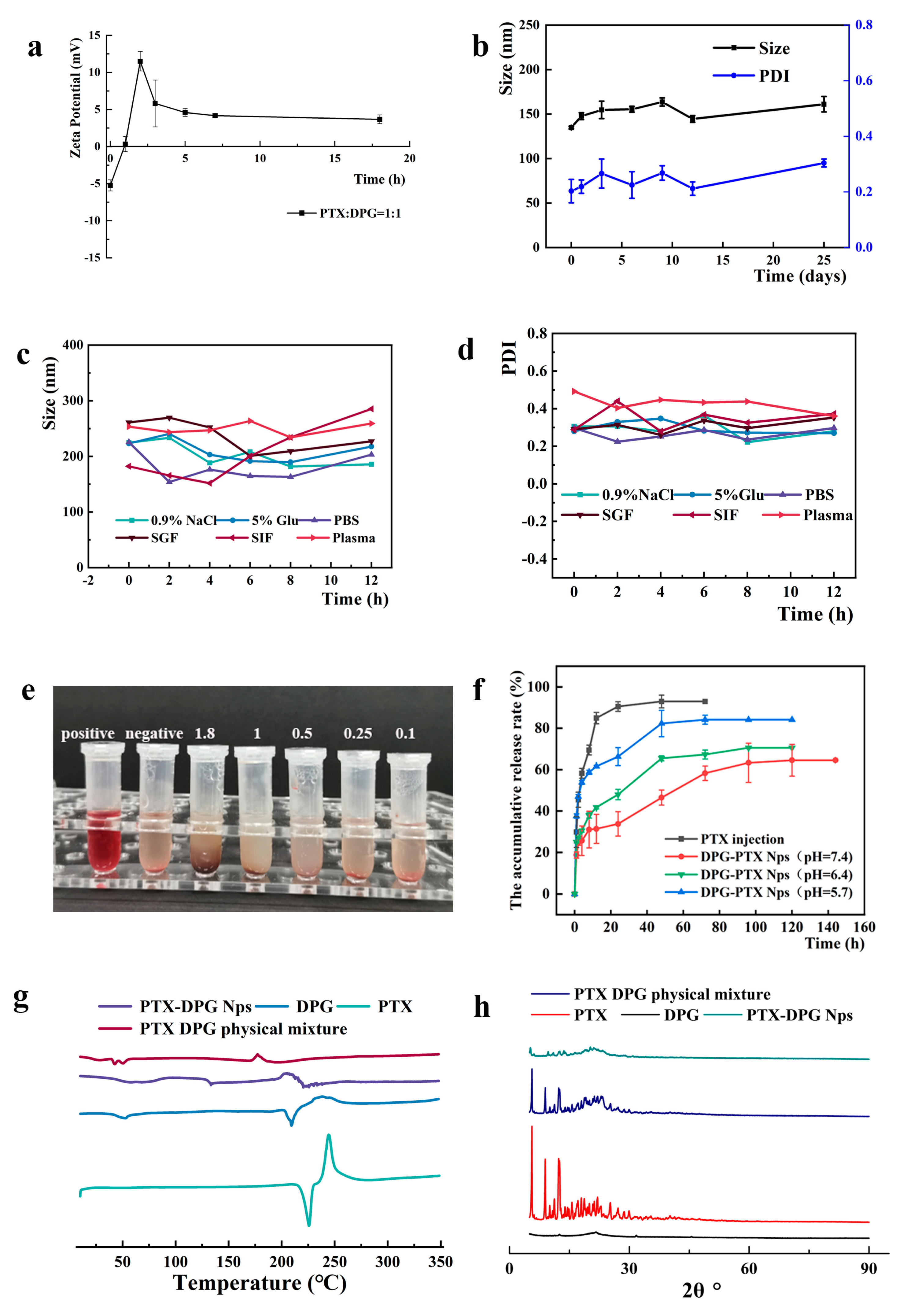
DPG and PTX were made into nanoparticles via the anti-solvent precipitation method. The resultant rod-like PTX-DPG nanoparticles are characteristic of negative charge. However, in the presence of the GGT enzyme, the zeta potential of PTX-DPG NPs changed from −5.24 mV to neutral within 1 h and converted to be cationic in the subsequent hours, demonstrating the charge reversal property of PTX-DPG NPs. It is reported that cationization could effectively induce adsorption-mediated transcytosis of nanoparticles and realize the deep penetration into multilayered cell culture. This property enables the PTX-DPG NPs to be widely and deeply distributed in tumors and leads to remarkably improved anti-tumor efficacy in vivo.
2. Materials and Methods
2.1. Materials
DSPE-mPEG2000-NH2 was provided by Shanghai ToYong Bio. Tech. Inc. (Shanghai, China). Boc-γ-Glu (OtBu)-Cys (Trt)-Gly [Boc-γ-GSH] was from Nanjing TG peptide Biotechnology Co., LTD (Nanjing, China). Paclitaxel (PTX) was bought from Jiangsu Aikang Biomedical Research and Development Co. Ltd. (Nanjing, China). Trifluoroacetic acid (TFA) was purchased from Shanghai Macklin Biochemical Co., Ltd. (Shanghai, China). Dichloromethane (super dry solvent, with molecular sieves, water ≤ 30 ppm) was bought from J&K Scientific, Inc. (Beijing, China). Triisopropylsilane (97.5%) (Tis) was provided by J&K Scientific, Inc. (Beijing, China). 1-Ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDCI) was from Meryer (Shanghai) Chemical Technology Co., Ltd. (Shanghai, China). N-Hydroxy succinimide (NHS) was purchased from Shanghai Yuanye Bio-Technology Co., Ltd. (Shanghai, China). DiR iodide [1-1-dioctadecyl-3,3,3,3-tetramethlindotricarboc-yanine iodide] (DiR) was purchased from AAT BioQuest (Sunnyvale, CA, USA). Acetonitrile (HPLC grade) was from Fisher Scientific (Pittsburgh, PA, USA). Paclitaxel injection (PTX injection) was obtained from Beijing Union Pharmaceutical Factory (Beijing, China). Deionized water was used during the experiments.
2.2. Animals and Cell Line
The Hela (cervical carcinoma) cell line was purchased from China Infrastructure of Cell Line Resource. The cells were cultured in DMEM medium containing 10% fetal bovine serum (FBS, Gibco, Waltham, MA, USA); 100 U/Ml streptomycin and penicillin (Gibco, Waltham, MA, USA) were added for the prevention of bacterial contamination. The cells were kept in a 5% CO2 atmosphere at 37 °C constantly.
Female nu/nu BALB/c healthy mice weighing 20 ± 2 g (6–8 weeks old) (SPF grade) were purchased from SPF (Beijing, China) Biotechnology Co., Ltd. The experimental animals were adapted to the environment of SPF-class housing of laboratory at least 7 days before experimentation and were provided ad libitum feeding.
All animal experiments were conducted according to the guidelines for Ethical and Regulatory Animal Experiments Stipulated by the Institute of Medicinal Plant Development (IMPLAD).
2.3. Synthesis of DSPE-mPEG2000-GSH(DPG)
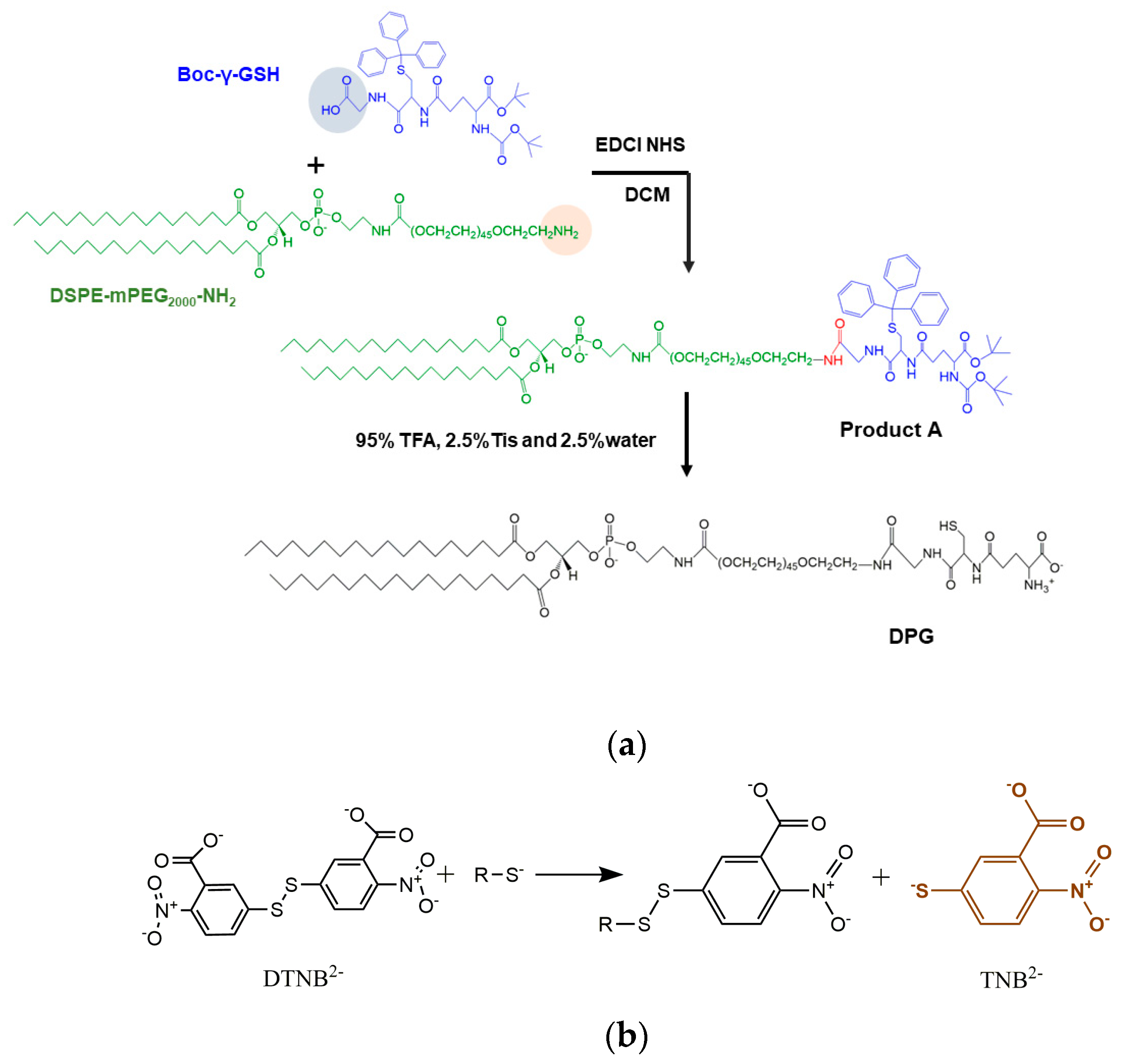
The synthesis of DPG was carried out as shown in
Figure 2a. Firstly, Boc-γ-GSH (26.4 mg, 0.037 mmol), EDCI (14.13 mg, 0.074 mmol), and NHS (8.51 mg, 0.074 mmol) were co-dissolved in 5 mL anhydrous dichloromethane and stirred for 4 h at 0 °C in an ice bath. After activation, DSPE-mPEG
2000-NH
2 (104 mg, 0.037 mmol) and one drop of triethylamine were added and reacted at room temperature for 24 h. When the reaction was completed, DCM was removed by rotary evaporation at 45 °C, and the mixture was washed three times with saturated NaCl solution to generate Product A (
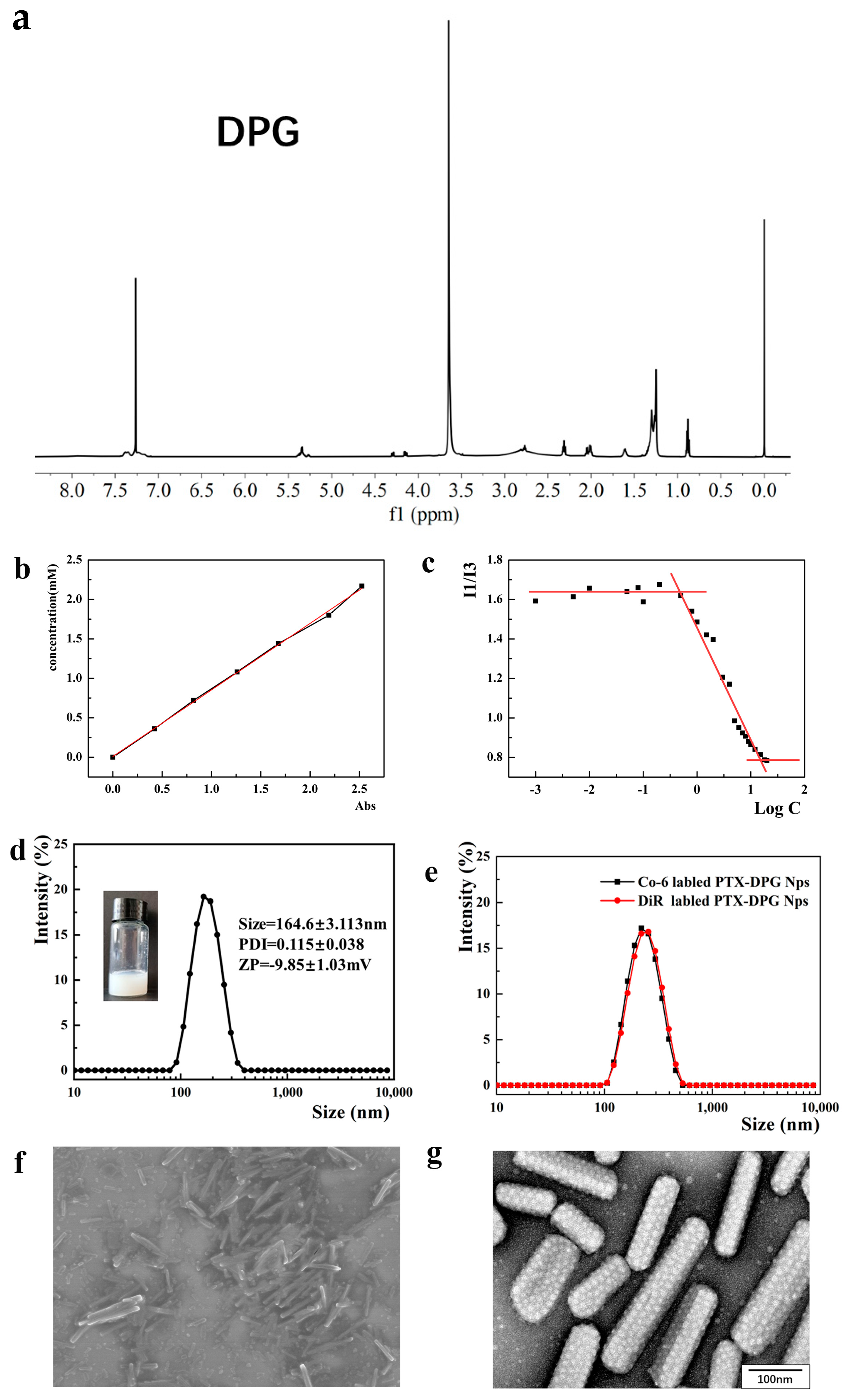
Figure 2a). Then 95% TFA, 2.5% Tis, and 2.5% water were added to deprotect the Boc and Trt groups from Product A. After stirring overnight, the solution was concentrated by rotary evaporation, and then the final product was precipitated by a large amount of ice ether, followed by centrifugation at 8000 rpm for 10 min to collect DPG (yield ~72.58%). The final product was dried under freeze-drying overnight and characterized by
1H NMR.
1H NMR (600 MHz, Chloroform-d): δ0.88 (t, 6 H), 1.26 (s, 56 H), 1.66 (s, 4 H), 2.32 (m, 4 H), 3.16 (s, 1 H), 3.37 (s, 1 H), 3.5–3.95 (m, 184 H), 4.0–4.5 (m, 8 H), 5.26 (s, 1 H), and 6.76 (s, 1 H).
2.4. GSH Grafting Ratio Determination
Ellman introduced 5,5’-dithio-bis-(2-nitrobenzoic acid), also known as DTNB, as a versatile water-soluble compound for quantitating free sulfhydryl groups in a solution [
21]. DTNB solution presents a yellow-colored product that could be used in quantitative analysis when it reacts with sulfhydryl (
Figure 2b). Sulfhydryl groups may be estimated by comparison to a standard curve of L-cysteine, which is composed of known concentrations of a sulfhydryl-containing compound. A series of concentrations of L-cysteine was mixed with Ellman’s reagent, incubated at room temperature for 15 min, and then the absorbance at 412 nm was measured using a UV spectrophotometer to plot a standard curve. Each molecule of DPG contains one molecule of −SH, which is the same as L-cysteine. 52.4 mg DPG was dissolved in 5.24 mL methanol and mixed with 50 μL Ellman’s reagent, incubated for 15 min, and then the absorbance at 412 nm was determined and the -SH concentration was obtained from the standard curve to compare with the DPG concentration to calculate the GSH grafting ratio.
2.5. Critical Micelle Concentration (CMC) Determination of DPG
Pyrene is a commonly used hydrophobic fluorescent probe with a fluorescence spectrum of five peaks, among which the ratio of fluorescence intensity at 373 nm (I1) against that at 384 nm (I3) (I1/I3) is very sensitive to the polarity of the microenvironment. After the formation of micelles, the pyrene probe enters the hydrophobic microregion of micelles from the aqueous phase, and the change in the microenvironment polarity decreases the value of I1/I3 significantly. The corresponding surfactant concentration at this abrupt change is determined as a CMC value [
22].
Different concentrations (20, 18, 15, 12, 10, 9, 8, 7, 6, 5, 4, 3, 2, 1.5, 1, 0.8, 0.5, 0.2, 0.1, 0.08, 0.05, 0.01, 0.005, and 0.001 μg/mL) of DPG micelles were mixed with pyrene acetone solution (0.1 mM) for 2 h to obtain the fluorescence spectra of the mixture with different concentrations (Ex wavelength = 335 nm). The CMC value of DPG was obtained by calculating the fluorescence intensity ratio (I1/I3).
2.6. Preparation of PTX-DPG NPs
The encapsulation of PTX by DSPE-PEG
2000-GSH (DPG) was carried out through the method of anti-solvent precipitation (
Figure 3). PTX and DPG with a mass ratio of 1:1 were co-dissolved in methanol, and then the mixture was injected into an appropriate volume of deionized water under constant ultrasonic conditions, drop by drop (250 W, 25 °C). The organic solvent was removed under vacuum by rotary evaporation at 45 °C. Then, the resultant nanosuspensions were homogenized (~1560 bar) at room temperature 10 times to obtain the final PTX-DPG NPs.
When DiR or coumarin 6 (Co-6) was co-dissolved in methanol together with PTX and DPG (the mass ratio of DiR or Co-6 with PTX being 1:40), the same method as above produced DiR-labeled PTX-DPG NPs or Co-6 labeled PTX-DPG NPs. When DiR was co-dissolved in methanol with DPG (the mass ratio of DiR to DPG as 1:40), in the absence of PTX, the same method as above produced DiR-labeled DPG micelles.
2.7. Morphology of PTX-DPG NPs
The morphology of DPG-PTX NPs was observed by Scanning Electron Microscopy (SEM; S-4800, Hitachi Limited, Tokyo, Japan) and Transmission Electron Microscopy (TEM; JEM-1400, JEOL, Tokyo, Japan). Briefly, diluted DPG-PTX NPs were placed on the matrix and air-dried. After sputter-coating with Au/Pd for 1 min, the samples were observed by SEM at an accelerating potential of 30 mV. Another drop of diluted DPG-PTX NPs was added to the copper grid (300 mesh), air-dried, and then dyed with uranyl acetate for an appropriate time. Then, the morphology was observed under TEM at an accelerating voltage of 120 kV.
2.8. HPLC Determination
The concentration of PTX in nanoparticles was measured by the HPLC system (DIONEX Ultimate 3000, Waltham, MA, USA). A Symmetry C18 column (250 mm × 4.6 mm, 5 μm, Venusil) was used at 25 °C for chromatographic separation. The mobile phase constituted 75% acetonitrile and 25% water (3:1, v/v) at a flow rate of 0.8 mL/min. The detection wavelength UV was 240 nm.
2.9. Drug Loading Content
Lyophilized PTX-DPG NPs were accurately weighed, dissolved in acetonitrile, centrifuged at 5000 rpm for 5 min, and the supernatant was injected in HPLC for the determination of PTX concentration. The drug loading content (
DLC) of PTX-DPG NPs was calculated as follows:
(V: Volume of an acetonitrile solution of lyophilized PTX-DPG NPs powder; C: Concentration of PTX; W: Weight of the lyophilized powder of PTX-DPG NPs).
2.10. Charge-Reversal Examination
As the tumor site highly expresses GGT enzyme up to 10 U/mL [
19], 1.8 mg of GGT (27.2 U/mg) was weighed and dissolved in 5 mL of pre-warmed 37 °C PBS aqueous solution (pH = 7.4) to obtain 10 U/mL of GGT solution (pH = 7.4). 10 U/mL of GGT solution was diluted into 0.05 U/mL to simulate the activity of the GGT enzyme in the blood. 1 mL PTX-DPG nanosuspensions and 1 mL GGT solution of different concentrations were mixed and incubated at 37 °C. Zeta potential changes were determined by DLS at specific time intervals.
2.11. Surface Element Analysis
The surface chemical composition of PTX powder and PTX-DPG NPs were investigated by Scanning electron microscopy (SEM) with Energy Dispersive Spectrometer (SEM-EDS; S-4800, Hitachi Limited, Tokyo, Japan). These samples were sputter-coated with a conductive layer of gold-palladium (Au/Pd) for 1 min. An accelerating potential of 30 mV was used for the observation and analysis.
2.12. Stability of PTX-DPG NPs On-Shelf or in Physiological Media
PTX-DPG NPs were stored at 4 °C for 25 days. Their particle size and PDI value were measured at 0, 1, 3, 5, 7, 14, and 25 days. Each sample was performed in triplicate. PTX-DPG NPs were respectively incubated with the same volume of 1.8% NaCl, 10% glucose, 2 × PBS (pH 7.4) (1:1, v/v), and plasma or four times the volume of simulated gastric fluid (SGF, 1% pepsin in 1 mol/L diluted HCl) and simulated intestinal fluid (SIF, 1% pancreatin in pH 6.8 PBS, 0.01 M) (1:4, v/v) at 37 °C. The size and particle size distribution were determined by DLS at specific time intervals. Each sample was carried out in triplicate.
2.13. Hemolytic Test
Red blood cells (RBCs) were obtained from healthy mice; whole blood was centrifuged, washed several times with normal saline, and diluted to 4% (v/v) RBCs suspensions with normal saline. The 4% RBCs suspensions were mixed with deionized water (as the positive control group), normal saline (as the negative control group), and different concentrations (1.8, 1, 0.5, 0.25, 0.1 mg/mL) of PTX-DPG NPs (as the test groups) at the volume ratio of 1:1. After incubation at 37 °C for 4 h, the mixture was centrifuged at 5000 rpm for 3 min to observe its hemolytic effect.
2.14. Differential Scanning Calorimetry (DSC) Characterization
DSC thermal profile was obtained using a differential scanning calorimeter (DSC Q200 V24.4 Build 116). A suitable amount of PTX powder, DPG powder, lyophilized PTX-DPG NPs powder, and the physical mixture (DPG:PTX = 1:1 w/w, according to the formulation of PTX-DPG NPs) were sealed in a standard aluminum pan and detected from 0 to 350 °C (10 °C/min, a nitrogen atmosphere).
2.15. X-ray Diffraction (XRD) Measurements
X-ray diffraction (XRD) measurements of PTX powder, DPG powder, lyophilized PTX-DPG NPs powder, and the physical mixture (DPG:PTX = 1:1, w/w, according to the formulation of PTX-DPG NPs) were performed using X-ray diffractometer (DX-2700, China) with Cu-Kα radiation generated at 100 mA and 40 kV. Samples were scanned over an angular range of 3–90° of 2θ°, with a step size of 0.02° and a count time of 3 s per step. Samples were kept rotating at 30 rpm during the analysis.
2.16. Drug Release Profile
The in vitro drug release of PTX from PTX-DPG NPs was conducted using the dialysis method (molecular weight cut-off 8 k–14 kDa) at 37 °C under continuous stirring (200 rpm). 1 mL PTX-DPG NPs was sealed in a dialysis tube and then immersed in 50 mL PBS (pH 5.7, 6.4, and 7.4, containing 2% Cremophor EL) mimicking different physiological environments. At the predetermined time intervals, 1 mL of the dialysate was collected for HPLC analysis and replenished with an equivalent amount of fresh-release medium. The release medium was renewed every 24 h. The above experiment was performed in triplicate.
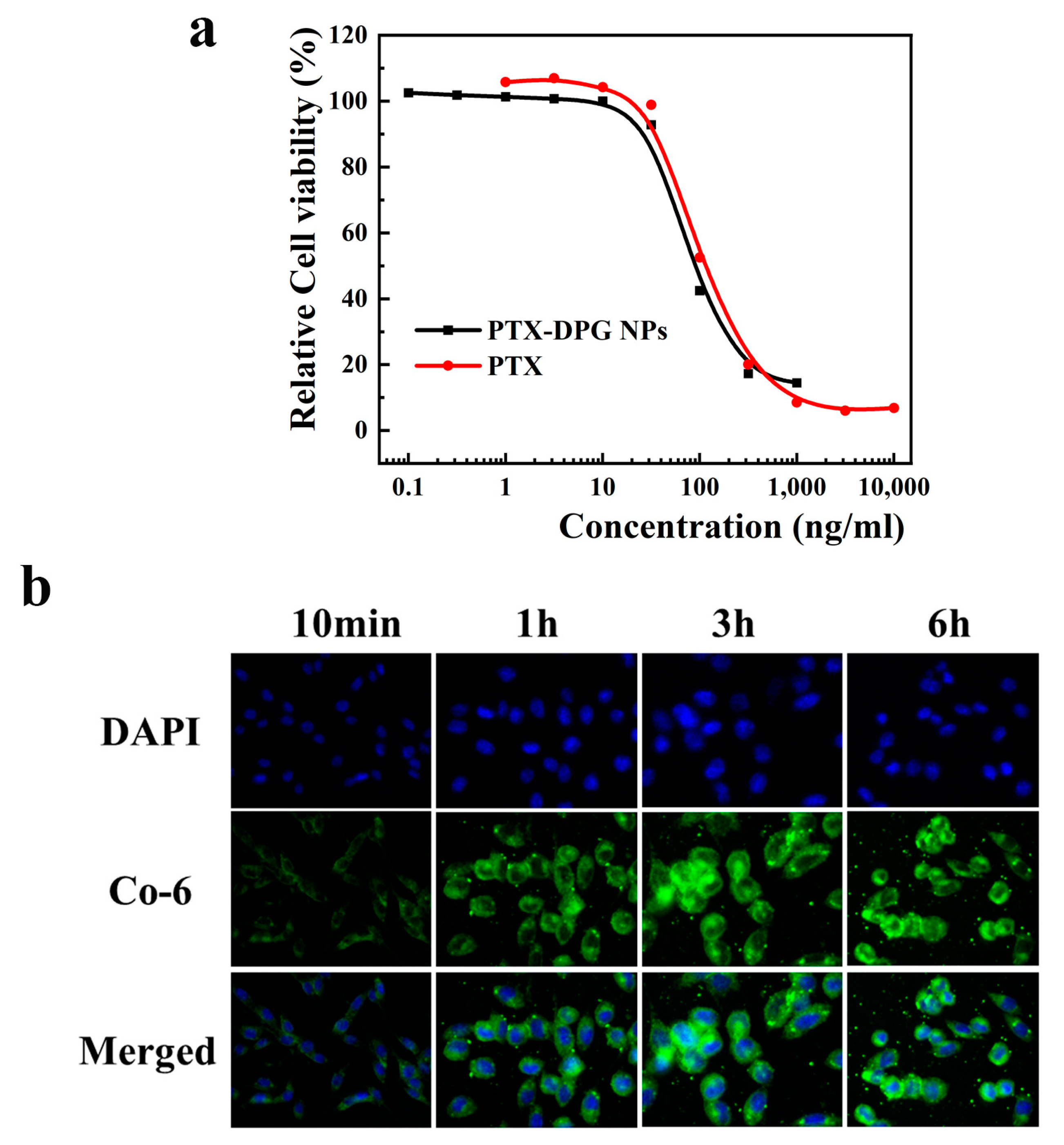
2.17. MTT Assays
Multiple tumor cell lines, including Hela cells, overexpress the γ-GGT enzyme [
14,
15,
16,
23,
24]. MTT assay was carried out to examine the in vitro cytotoxicity of PTX-DPG NPs against the Hela cell line using free PTX as a control. Hela cells (5000 cells/well) were seeded in 96-well plates and incubated at 37 °C and in a 5% CO
2 atmosphere for 24 h. Different concentrations of PTX-DPG NPs (diluted in DMEM medium) were added (150 μL per well) and incubated for 72 h. Then 20 μL of MTT solution (5 mg/mL) was added to each well and incubated for another 4 h. Then, the medium was removed and 150 μL of DMSO was added to dissolve the formazan crystals. The absorbance value of the supernatant in each well was measured at 570 nm using the ELISA plate reader. The
cell viability rate was calculated according to the following formula:
(where ODt means the absorbance value of the test groups, ODn means the absorbance value of the negative control groups).
2.18. Cellular Uptake
Hela cells (5 × 105 cells per well) were inoculated in a 24-well plate and cultured in a DMEM medium for 24 h at 37 °C in the presence of 5% CO2, and then, PTX-DPG NPs (PTX equivalent concentration 10 μg mL−1) were added into the 24-well plate. They were washed, dyed with DAPI, and fixed with 0.4% paraformaldehyde after 0.2, 1, 2, and 6 h, respectively. The results were recorded using an inverted fluorescence microscope.
2.19. In Vivo Anti-Tumor Efficacy and Safety Evaluation
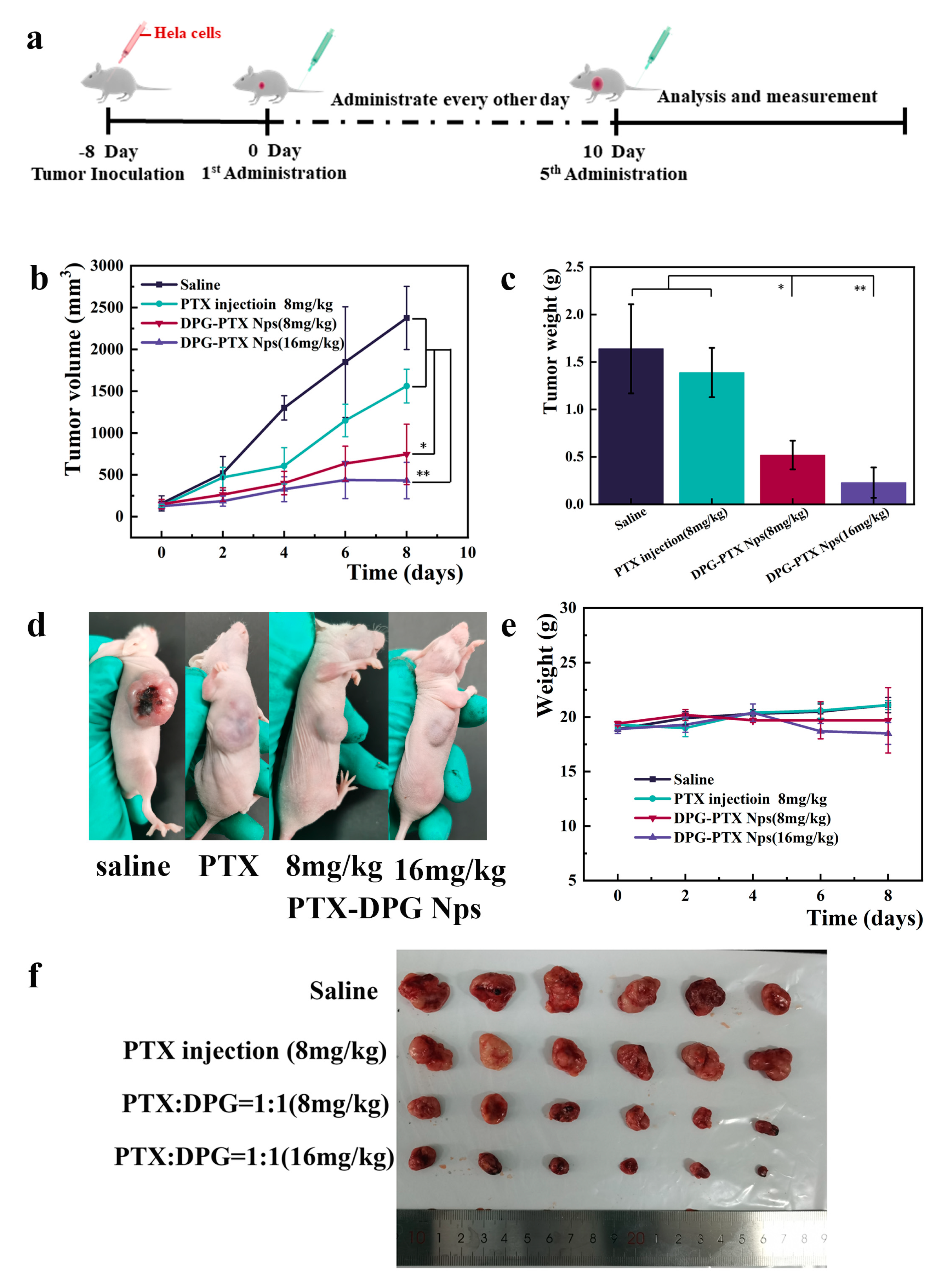
Female nu/nu BALB/c mice weighing 20 ± 2 g bearing Hela tumors were chosen for the in vivo anti-tumor efficacy investigation. 0.2 mL Hela cell suspension (1 × 106 cells) was injected into the right armpit subcutaneously of mice to construct Hela-tumor bearing mice model. When the tumor volume reached 100 mm3, the Hela tumor-bearing mice were randomly divided into 4 groups (6 mice in each group). The negative control group was intravenously injected with 0.2 mL of normal saline, the positive control group was administrated with 0.2 mL of PTX injection (8 mg/kg). PTX injection (8 mg/kg) was diluted from the marked Paclitaxel injection purchased from the Beijing Union pharmaceutical factory by normal saline. The other test groups were intravenously injected with 8 and 16 mg/kg of PTX-DPG NPs, respectively (the equivalent PTX dose). All the above groups were intravenously administrated through the tail vein every 2 days for 5 times. Tumor volume and body weight of mice in each group were measured during the whole experimental process. The tumor volume was calculated by the formula V = (a × b2)/2.
At the end of the experiment, the blood was collected for each mouse, then all the mice were sacrificed, and the tumors, livers, and spleens were dissected and weighed to calculate the tumor inhibition rate (
TIR%), liver index rate (
LIR%), and spleen index rate (
SIR%) according to the following formulas:
(
Wt is the mean tumor weight of mice in test groups and
Wn is the mean tumor weight of mice in the negative control group)
(
Wl is the liver weight and
Wb is the body weight)
(Ws is the spleen weight and Wb is the body weight).
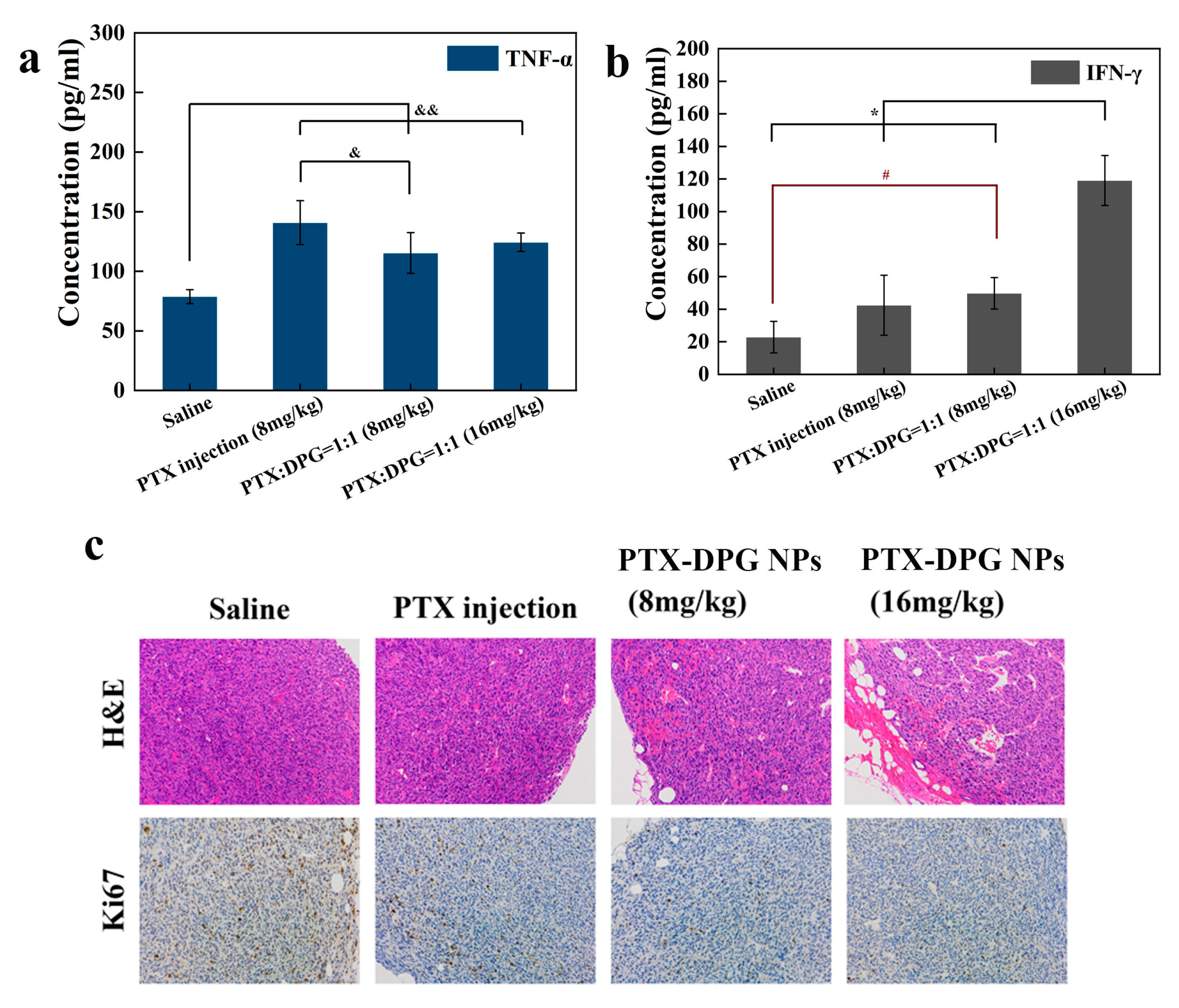
All the tumor tissues were stained with hematoxylin and eosin (H&E) to evaluate the effect of the therapeutic efficacy. The IHC staining of Ki67 was tested to investigate the percentage of proliferous tumor cells positively stained in the examined field. The blood serum was separated to detect the expression levels of tumor cytokines (TNF-α and IFN-γ) by ELISA Kits (Fankewei, Shanghai, China) according to the manufacturer’s protocol.
2.20. Biodistribution Evaluation
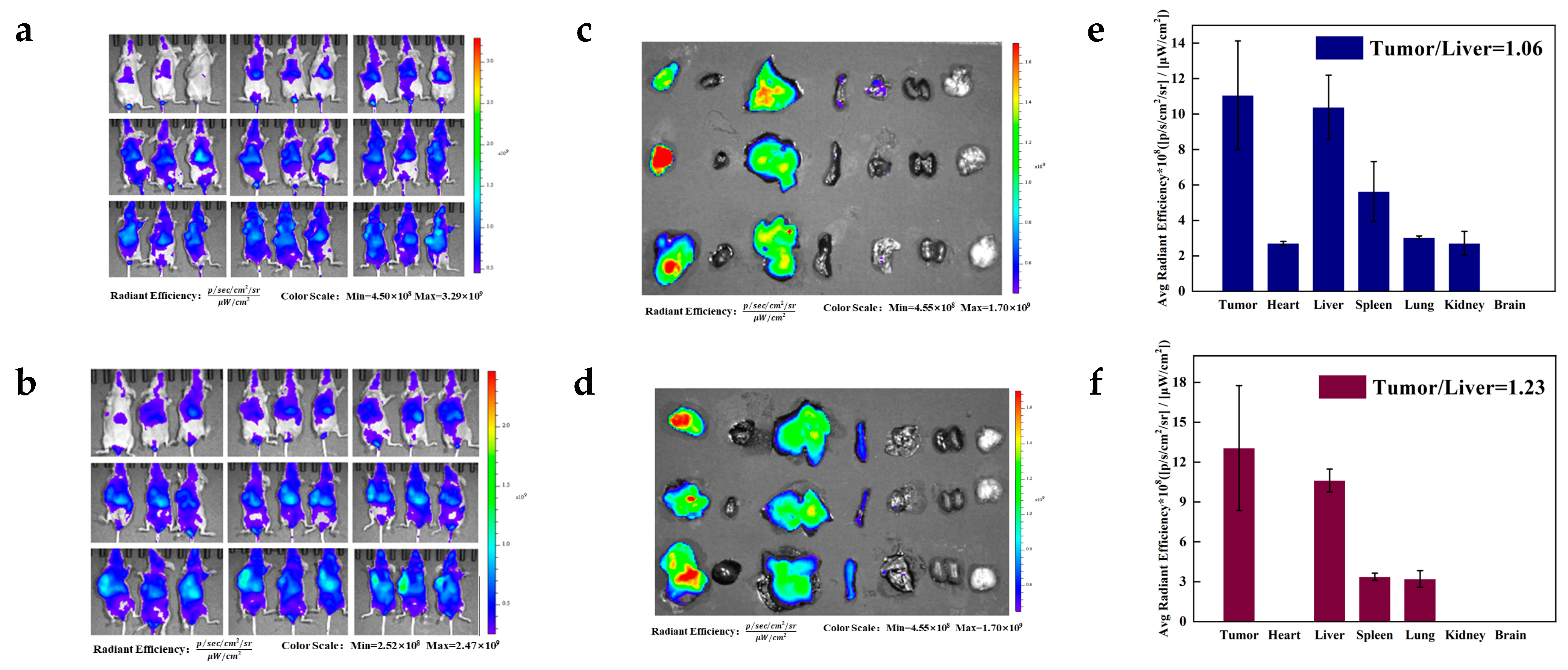
To observe the dynamic biodistribution of DiR-labelled DPG micelles and PTX-DPG NPs, 6 Hela tumor-bearing mice (tumor volume of ~1000 cm3) were injected through the tail vein (3 mice in each group). They were whole-body imaged at 0.2, 0.5, 1, 2, 4, 6, 8, 12, and 24 h post-dose using the IVIS Living Image software (version 4.4, Caliper Life Sciences, Hopkinton, MA, USA). Then the mice were sacrificed, and the tumors and the major organs such as hearts, livers, spleens, lungs, and kidneys, were also imaged to measure the fluorescence intensity in these tissues.
2.21. Statistical Analysis
Statistical analysis of the experimental data was performed using Statistical Package for the Social Sciences software, and IC50 values were calculated by GraphPad Prism software, version 6.01 (GraphPad Software, La Jolla, CA, USA). In vitro and in vivo results were analyzed by t-test and one-way analysis of variance. The value of p < 0.05 was considered statistically significant.


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}