1. Introduction
The development of glycoconjugate vaccines requires the chemical conjugation of glycans to an immunogenic carrier protein. While most of the licensed conjugate vaccines are produced from polysaccharides extracted and purified from natural sources, current vaccine development strategies involve the use of well-defined minimal saccharide epitopes that are synthetized using chemical or enzymatic approaches [
1,
2,
3]. Parallel to the selection of the sugar moieties, the choice of the conjugation strategy is also extremely important since it strongly influences the quality of the final glycoconjugates in terms of site-specificity, glycan loading, and purity/homogeneity of the final product.
The most common approach for the covalent coupling of oligosaccharide epitopes to carrier proteins entails introducing a spacer carrying reactive functional groups at their end terminal. Among the different options, the introduction of an amino group is one of the most convenient since it allows further derivatization with bifunctional or bivalent linkers to form active esters that are able to react with the ε-amino group of lysine residues on the carrier protein surface. The use of disuccinimidyl bivalent linkers (such as disuccinimidyl glutarate (DSG) or disuccinimidyl adipate (DSA)) that are able to react with amino-functionalized oligosaccharides to form reactive esters falls into that category [
4], and several examples showing the application of this synthetic strategy to the development/production of glycovaccines can be found in the literature [
5,
6,
7,
8,
9,
10,
11].
The presence of a linker with a variable number of C units allows desirable conjugation yields to be achieved in terms of the number of sugars covalently loaded on the carrier surface. However, the high tendency of disuccinimidyl-esters for hydrolysis may represent a significant drawback for this conjugation chemistry. This instability can hamper an extensive linker purification, leading to side-reactions between protein lysine residues and residual linkers, to form conjugates of the corresponding carboxylic acid [
12]. Indeed, the reaction of amino-activated oligosaccharides with disuccinimidyl esters requires a high linker molar excess (>5 eq.) to avoid sugar-sugar dimer formation [
13]. A purification step is therefore required to eliminate the linker excess that will preferentially react with the protein and form the corresponding acid in aqueous medium. Most of the protocols reported in the literature mention purification approaches based on liquid–liquid extraction and/or precipitation using different organic solvents, such as ethyl acetate or chloroform [
5,
6,
7], or even water [
13]. The effectiveness of these purification approaches has not been proven since, in most of these experimental works, glycan attachment to the carrier is monitored by SDS-PAGE and/or MALDI-TOF-MS analysis [
6,
7,
9,
11]. The use of these analytical methods only provides the average glycan loading, and no information regarding any side reactions or alterations occurring during conjugation, such as the conjugation with residual linker, can be obtained. Instead, the presence of contaminant conjugates, which alter protein mass, can affect the truthfulness of the determination. Another common approach for glycoconjugate characterization entails the use of phenol-sulfuric acid assay [
5,
7]. This method, exploiting sugar reactivity, is not affected by the presence of contaminant linker/acid conjugates; it still provides the average glycan loading, but there is no assurance of adequate purity.
Interestingly, when performed, a deep insight into the characterization of the glycoconjugates has revealed the presence of linker/acid conjugates [
12]. Their presence not only reduces the glycan loading by occupying reactive sites, but it might strongly influence glycoconjugate properties and biological activity (even in an early development stage).
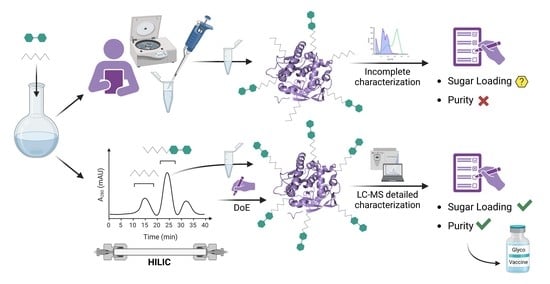
Based on the above, it appears clear that the use of this conjugation chemistry to produce (potential) glycovaccines requires the development of most efficient purification protocols and a detailed analytical characterization of produced glycoconjugates. In this context, the aim of this work is to set up an experimental protocol to use a disuccinimidyl homobifunctional linker, namely DSG, to obtain glycoconjugates with satisfactory sugar loading and adequate purity that is suitable to be used as therapeutic agents.
At first, a commercial model protein, ribonuclease A (RNase A, 13.7 kDa, 10 lysine residues) was considered to set up a suitable purification procedure and optimize conjugation conditions, as well as considering the reactivity of this conjugation chemistry. Accordingly, 3-aminopropyl small-sized (from mono- to tri-) mannose saccharides were ad hoc synthetized and used. Mannose-based saccharides were selected as a model since mannose is one of the main constituents of oligosaccharides used in glycoconjugate vaccines, both for mimicking bacterial surface polysaccharides (e.g.,
Mycobacterium tuberculosis arabinomannans) and/or targeting mannose receptor on antigen presenting cells [
14]. Glycosylation outcomes have been discussed in comparison to an alternative conjugation chemistry since the same protein was used as model in a previous study; that study used an approach which required the activation of oligosaccharides as 2-iminomethoxyethyl (IME) thioglycosides were considered [
15].
Once the suitability of this approach for glycoconjugate synthesis was demonstrated, it was applied to recombinant Ag85B (31.3 kDa, 8 lysine residues), one of the most potent
Mycobacterium tuberculosis antigens [
16,
17], and to its mutated variant, Ag85B K30R-K282R (referred to as Ag85B-dm, 31.3 kDa, 6 lysine residues), developed by our research group [
18].
Ag85B protein was selected and studied by us as a carrier in the development of a novel glycoconjugate vaccine with antitubercular activity [
16]. In our rational design approach, the carrier protein was selected not only to be immunogenic, but also to exert a specific antitubercular activity synergic to the one showed by the glycan antigen. In a previous study, the predominant glycosylation of the two most reactive lysine residues of Ag85B, namely K30 and K282, was demonstrated to reduce intrinsic protein antigenicity [
16,
18]. To address the glycosylation separately from the protein epitope, K30 and K282 were thus substituted with arginine residues. The conservative substitution allowed protein antigenicity to be preserved and to avoid glycosylation at these positions [
16,
18,
19]. The loss of the two most reactive sites had, however, the evident drawback of reducing the glycosylation efficiency in terms of the average number of incorporated saccharides [
18]. Glycosylation via disuccinimidyl linker was herein also considered as a conjugation approach to glycosylate Ag85B-dm, with the aim of achieving adequate antigen loading and making it possible to exploit the potential of this optimized carrier in the future development of a novel antitubercular vaccine.
2. Materials and Methods
2.1. Materials and Instrumentations
Ethyl acetate (EtOAc), dimethylformamide (DMF), tetrahydrofuran (THF), dichloromethane (DCM), acetonitrile (can), di-succinimidyl glutarate (DSG), potassium phosphate, ribonuclease A (RNase A), chymotrypsin, dithiothreitol (DTT), and formic acid (FA) were purchased from Sigma-Aldrich (Milan, Italy). Trifluoroacetic acid (TFA) was purchased from PanReac AppliChem (Monza, Italy). Deionized water was obtained from a Milli-Q® Integral purification system from Merck KGaA (Darmstadt, Germany).
Reactants and chemicals used for the synthesis of sugar derivatives were purchased from commercial sources (Sigma-Aldrich, Burlington, MA, USA (Merck KgaA group), Alfa Aesar Ward Hill, MA, USA (Thermo Fisher Scientific group) and used without further purification. Solvents were purified according to the guidelines described in
Purification of Laboratory Chemicals [
20] and were freshly distilled from the appropriate drying agent. THF was distilled from sodium/benzophenone ketyl, and DCM from CaH
2. Reactions requiring anhydrous conditions were performed under N
2. Compound purification was performed via flash chromatography using Silica Gel high-purity grade, pore size 60 Å 70–230 mesh, 63–200 μm (Sigma-Aldrich). Analytical thin layer chromatography (TLC) was performed on silica gel F254 precoated aluminium sheets (0.2 mm layer, Merck, Darmstadt, Germany), visualized by a 254 nm UV lamp, and stained with 5% H
2SO
4 in ethanol, followed by heating to 150 °C. Characterization of purified compounds was performed by NMR spectroscopy. NMR spectra were recorded on a Bruker Advance III 400 MHz spectrometer (Bruker Corporation, Billerica, MA, USA). High-resolution mass HRMS spectra were acquired using a X500B QTOF System (SCIEX, Framingham, MA 01701, USA) equipped with the Twin Sprayer ESI probe and coupled to an ExionLC™ system (SCIEX). Yields were calculated for compounds purified by flash chromatography and judged homogeneous by thin-layer chromatography, NMR, and mass spectrometry.
Chromatographic separations of 3-aminopropyl saccharides 7, 11, 18, active esters 19, 20, 21 and glycoconjugates were performed on a Dionex UltiMate 3000 HPLC system (Thermo Scientific, San Jose, CA, USA) equipped with mobile-phase online degasser, ternary pump, autosampler, column thermostated compartment, and variable wavelength detector, and controlled by Chromeleon software (6.8 version). MS detection was achieved by using a linear ion trap mass spectrometer (LTQ) equipped with electrospray ion source (ESI) (Thermo Scientific, San Jose, CA, USA) and controlled by X-calibur software (2.0.7 version).
Antigenic proteins Ag85B wild-type (wt) and -dm were produced as recombinant proteins in
Escherichia coli as previously described [
17,
18].
2.2. Synthesis of 3-Aminopropyl Saccharides
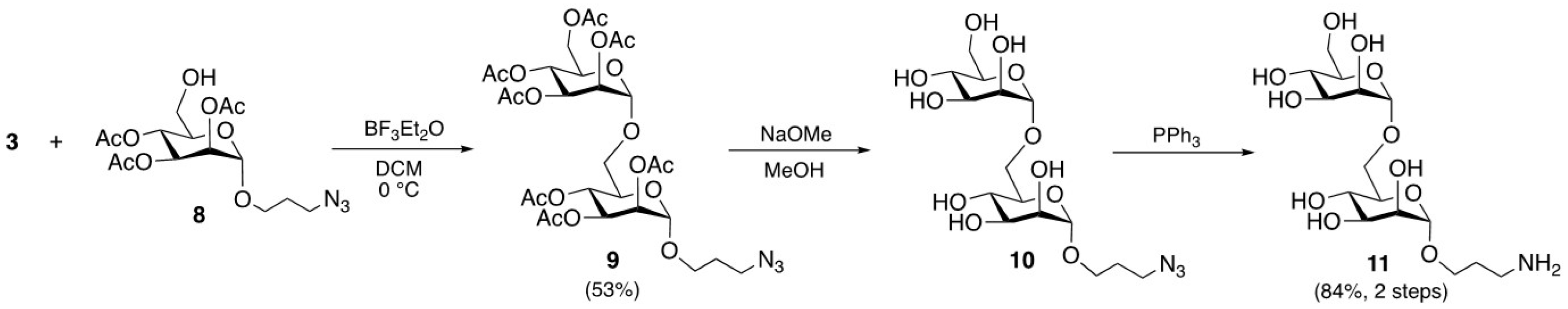
The chemoenzymatic synthesis of 3-aminopropyl monosaccharide and certain intermediate derivatives exploited in the preparation of previously undescribed 3-aminopropyl di/trisaccharides (
vide infra) has been carried out by adapting known synthetic methods [
14,
21,
22,
23]. Detailed experimental procedures, complete characterization data, and copies of
1H- and
13C-NMR spectra for all new compounds are reported in the
Supplementary Material.
2.3. Preparation and Purification of Activated Esters
2.3.1. Functionalization of 3-Aminopropyl Saccharides
Functionalization of 3-aminopropyl saccharides was performed according to Wang et al. [
7]. A mixture of
7,
11, or
18 (2 mg) and DSG (15 equiv) in DMF/phosphate buffer (100 mM, pH 8.0) (4:1
v/v, 0.3 mL) was gently stirred at room temperature. After 4 h, 1 µL of reaction mixture was diluted 1:100 with ACN and analyzed by HPLC-ESI-MS.
2.3.2. HILIC-ESI-MS Monitoring of 3-Aminopropyl Saccharide Activation
HPLC-MS analysis of 3-aminopropyl saccharides and activated derivatives was performed using HILIC mode. The HILIC-ESI-MS method entailed the use of the Waters X-Bridge Amide column (3 × 150 mm; Waters Corporation, Milford, MA, USA) and a mobile phase composed of ACN + 0.1% FA (A) and water + 0.1% FA (B). Gradient elution was performed by linear increase from 15 to 45% B in 10 min. Temperature, flow rate, and injection volume were set to 50 °C, 0.35 mL/min, and 2 µL, respectively. The following MS parameters were applied: positive ion mode, scan range 150–800 m/z in full-scan mode, source voltage 4.6 kV, capillary voltage 49 V, sheath gas flow rate 45 (arbitrary units), auxiliary gas flow rate 20 (arbitrary units), capillary temperature 250 °C, tube lens voltage 250 V.
2.3.3. Purification of Activated Esters by Washing with an Organic Solvent
Activated 3-aminopropyl saccharides were tentatively separated from excessive DSG according to Wang et al. [
7]. The procedure entails washing and precipitation upon addition of 9 volumes of EtOAc to the reaction mixture, followed by further purification by washing with EtOAc 10 times and then drying.
2.3.4. Purification of Activated Esters by HILIC Chromatography
Activated 3-aminopropyl saccharides were purified by HILIC chromatography with a Waters X-Bridge Amide column (3 × 150 mm; Waters Corporation, Milford, MA, USA) and a mobile phase composed of ACN (A) and water (B). Gradient elution was performed by linear increase from 10 to 35% B in 10 min. Temperature, flow rate, and injection volume were set to 35 °C, 0.35 mL/min, and 20–100 µL, respectively. UV absorbance was monitored at 214 nm. Fraction containing activated 3-aminopropyl saccharides was manually collected and dried under nitrogen flow.
2.4. Synthesis and Analytical Characterization of Glycoconjugates
2.4.1. Conjugation Protocol (Optimized Conditions)
Proteins (RNase A, Ag85B wt and -dm) were dissolved in 100 mM phosphate buffer, pH 8 (concentration: 4 mg/mL) in presence of 3-aminopropyl esters
7,
11,
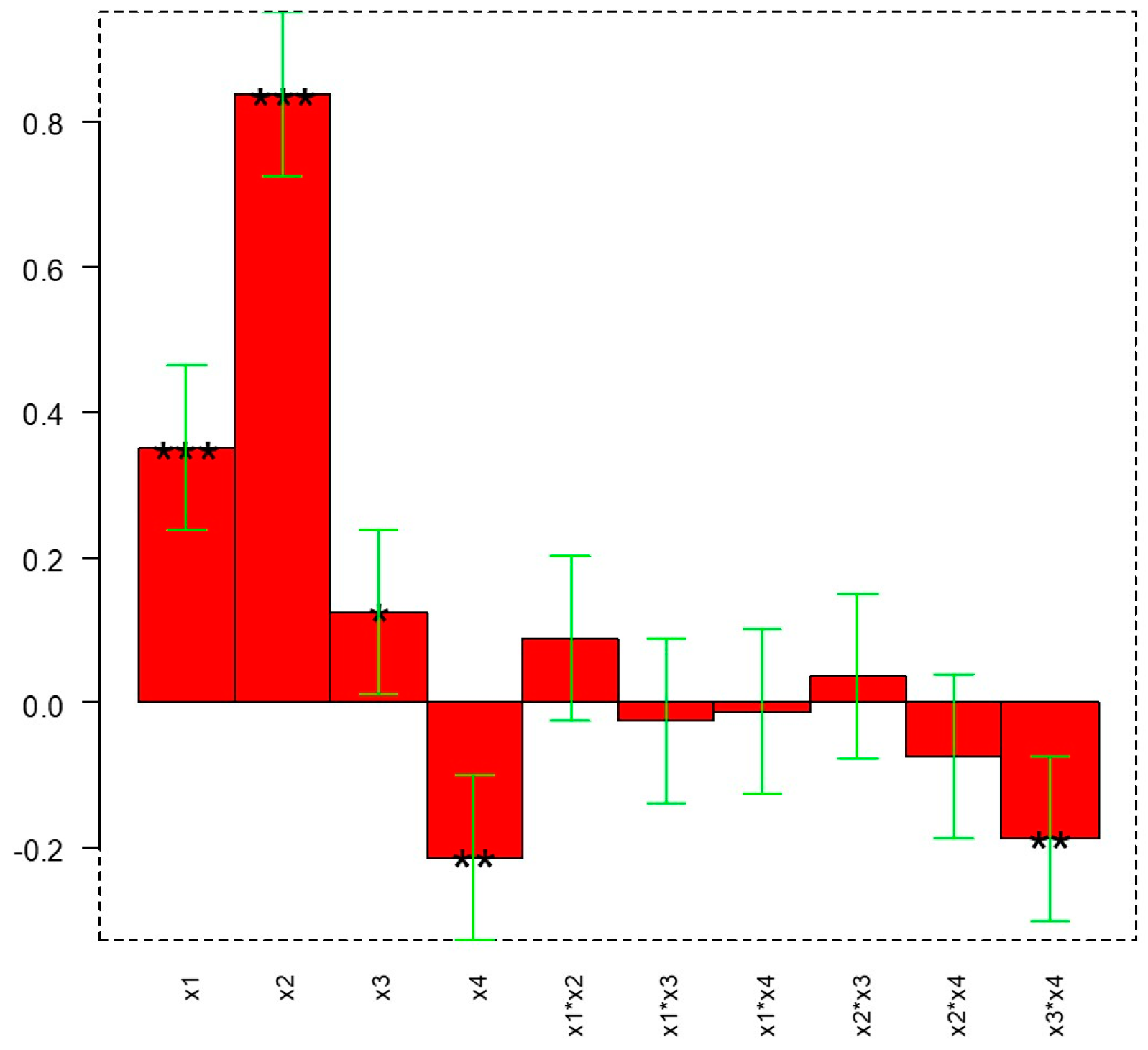
18 to a final glycoside/protein molar ratio of 100/1. The reaction mixtures were incubated for 16 h at 20 °C under continuous stirring. The conditions were set as reported after studying the influence of four different variables (molar ratio, protein concentration, buffer pH, and temperature) on glycosylation outcome by means of a Design of Experiments (DoE) approach (full factorial design, 2
k, k = 4) To this aim, the open-source software Chemometric Agile Tool (CAT) was used (available freely on the site of the Italian Group of Chemometrics,
http://www.gruppochemiometria.it/index.php/software (accessed on 2 March 2023).
2.4.2. Intact Glycoconjugate HILIC-UV-ESI-MS Analysis
HPLC-MS analysis of intact glycoconjugates was performed by using HILIC mode, an AdvanceBio Glycan Mapping column (2.1 × 150 mm; 1.8 µm, Agilent Technologies, Santa Clara, CA, USA), and a mobile phase composed of ACN + 0.1% TFA (A) and water + 0.1% TFA (B). The elution gradient for RNase A glycoconjugates was set as follows: from 20% to 30% B in 1 min and from 30% to 45% B in 15 min. Elution gradient for Ag85B wt and -dm glycoconjugates was set as follows: from 20% to 25% B in 1 min and from 25% to 40% B in 15 min. Temperature, flow rate, and injection volume were set at 50 °C, 0.25 mL/min, and 2 µL, respectively. The following MS parameters were applied: positive ion mode, scan range 700–2000 m/z in full-scan mode, source voltage 4.5 kV, capillary voltage 8 V, sheath gas flow rate 25 (arbitrary units), auxiliary gas flow rate 5 (arbitrary units), capillary temperature 220 °C, and tube lens voltage 220 V.
2.4.3. Chymotryptic Digestion of Glycoconjugate and Peptide Mapping Analysis by HILIC-UV-ESI-MS/MS
For the chymotryptic digestion (cleavage at carboxy-terminal position of methionine, tyrosine, phenylalanine, tryptophan, and leucine residues. Then, 25–100 μM protein solution in 50 mM ammonium bicarbonate (pH 8.5) was added with DTT in 100 mM ammonium bicarbonate (pH 8.5) to obtain a 10 mM final concentration. The solution was heated at 60 °C for 30 min to reduce the protein’s disulfide bonds, and then chymotrypsin was added to a final protein/enzyme ratio of 50:1 (w/w). Then the solution was incubated for 3 h at 37 °C under continuous stirring. The reaction was stopped by adding 2.5% (v/v) TFA.
Glycopeptide mapping was performed by HILIC-UV-ESI-MS3. The column was the AdvanceBio Glycan Mapping column (2.1 × 150 mm; 1.8 µm, Agilent Technologies, Santa Clara, CA, USA) and the mobile phase was composed of ACN + 0.05% TFA (A) and water + 0.05% TFA (B). Elution gradient was set as following: isocratic at 5% B for 0.5 min; linear gradient from 5 to 40% B in 45 min. Temperature, flow rate and injection volume were set at 50 °C, 0.25 mL/min and 15 µL, respectively. UV absorbance was collected at 215 nm. MS detection required the following instrumental conditions: positive ion mode, source voltage 4.5 kV, capillary voltage 31 V, sheath gas flow rate 40 (arbitrary units), auxiliary gas flow rate 10 (arbitrary units), capillary temperature 250 °C, and tube lens voltage 95 V. Full scan mass range was set up from 300 to 2000 Da. MS2 and MS3 spectra were obtained by collision induced dissociation (CID) with normalized collision energy of 35.0. Data processing was performed using Bioworks Browser (Thermo Fisher Scientific (Waltham, MA, USA), revision 3.1).
4. Discussion
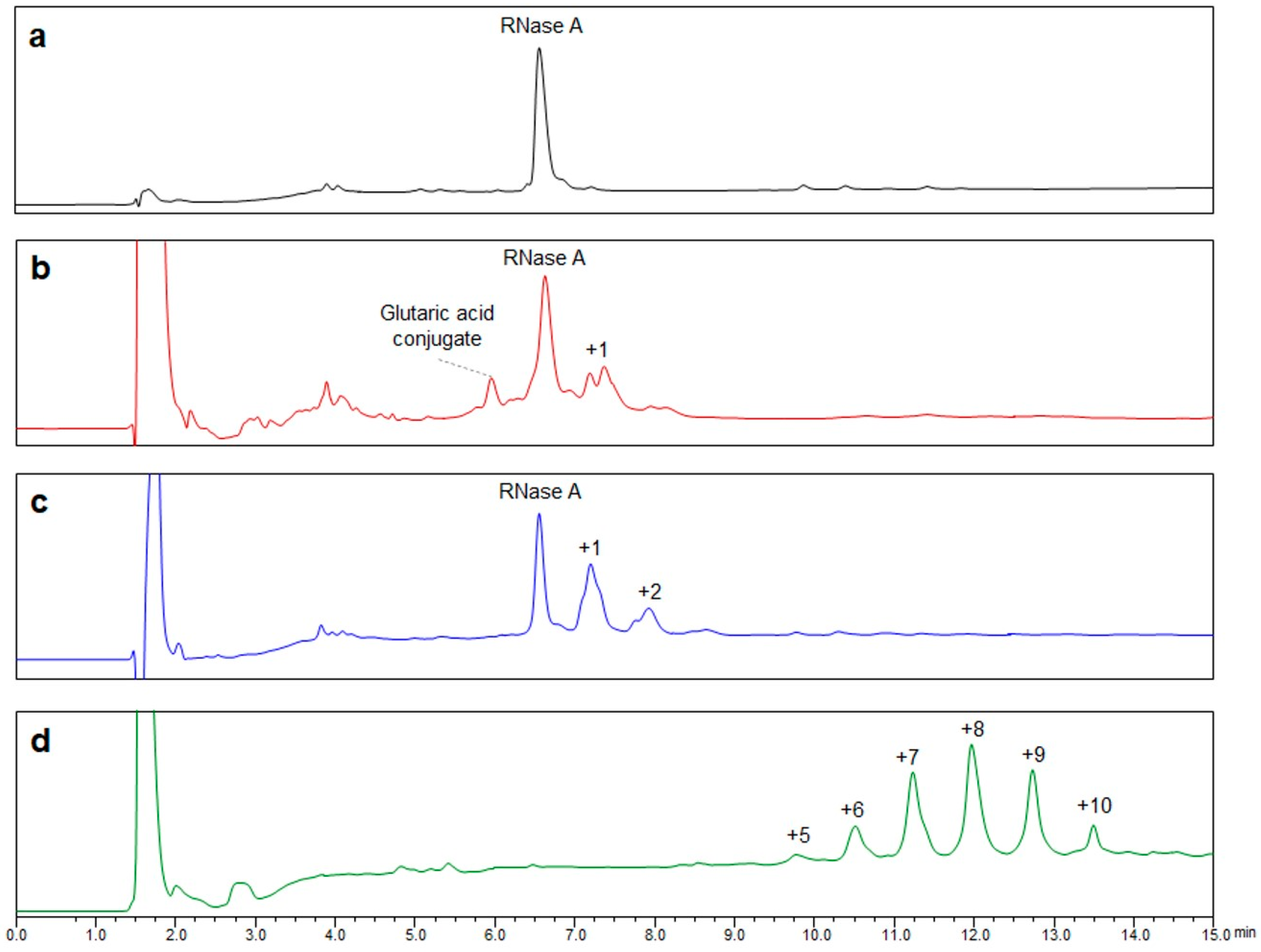
Conjugation via disuccinimidyl homobifunctional linkers is a common method for the synthesis of glycoconjugates. However, the well-known tendency of linkers for hydrolysis reactions represents a chemical limitation for this conjugation which may produce acid conjugates as side products. The application of the most common purification approach, consisting of the removal of excess DSG linker by washing with an organic solvent, such as EtOAc, led us to confirm the presence of a residual amount of linker in the glycosylation mixtures, which led to the formation of glutaric acid conjugates. Thus, we explored an alternative purification method based on HILIC chromatography. This approach allows the quantitative removal of the linker and the production of pure glycoconjugates. It must be underlined that only a detailed characterization, both at intact and peptide level, was allowed to verify the presence/absence of linker conjugates. Failing that, any consideration on glycoconjugate purity and identity is unreliable.
Following the optimization of the purification method, a DoE approach was used to find the reaction conditions that allow the glycan loading to be maximized. In these conditions, we studied the reactivity of mannose small-sized saccharides (
19,
20 and
21) in the glycosylation of RNase A. As expected, the active ester/protein molar ratio has a great influence on the glycosylation yields. In the literature, molar ratio between 30 and 150 (moles of active ester
per mole of protein) are usually considered. In our case, a molar ratio of 100:1 was employed. The same molar ratio was used in a previous work where the RNase A was conjugated with small-sized saccharides and activated as IME thioglycosides [
14]. The conjugation approach considered in this study resulted in significantly higher glycan loadings in comparison to the use of IME-activated saccharides; e.g., with Man (1–6) Man, we obtained an average incorporation number of 7.9 ± 0.1; with the same molar ratio, the average incorporation is reported as 2.3 ± 0.0 [
15] for the IME disaccharide. This evidence can be explained by the high reactivity of the DSG linker and the presence of a longer spacer between the saccharide anomeric position and the reactive group, which then reduces the steric hindrance in the interaction with surface lysine residues of proteins.
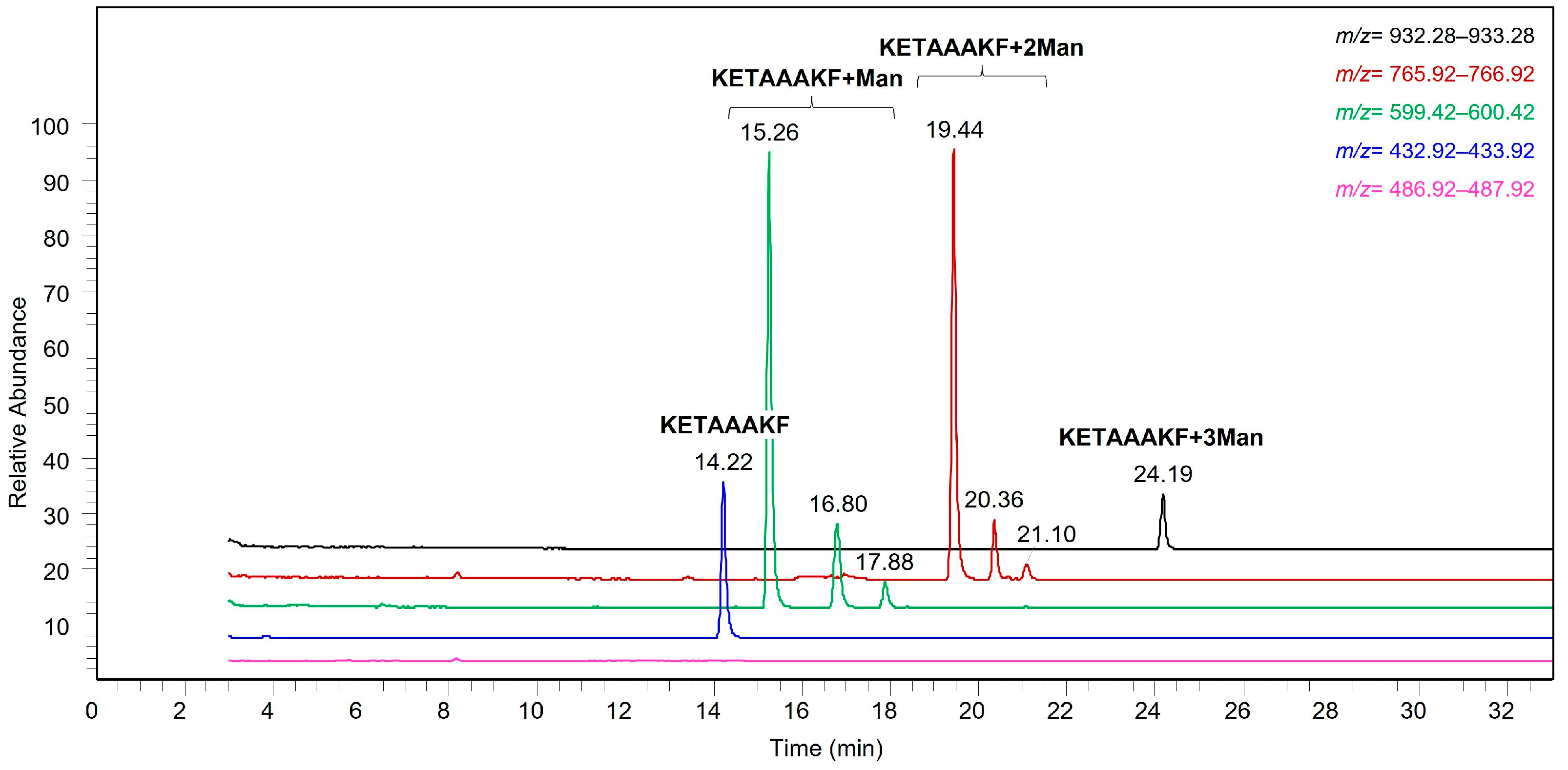
Glycopeptide mapping analysis of the resulting glycoconjugates revealed the same distribution among the potential glycosylation sites for the considered saccharides, with the N-terminal amino acid of RNase A as the predominant conjugation site. In RNase A, the N-terminal amino acid is a lysine residue, making it impossible, in principle, to discriminate between the reactivity of the main amino group and that of the amino group onto the side chain. However, it can be assumed that the reactivity of the main amino group is higher at pH 8.0. Indeed, its pKa value is lower than that of the lysine ε-amino group, thus resulting in a larger fraction of non-protonated form being available for the conjugation.
In the case of antigenic proteins, the use of a molar ratio of 100:1, ester/protein, was able to achieve satisfactory yields with <0.5% of unconjugated protein for both Ag85B wt and dm. Interestingly, no significant differences in terms of sugar loading were observed between the native and the variant protein, even if the latter is missing in the two most reactive reaction sites. In a previous work [
18], the glycosylation of the same proteins via IME-saccharides showed a reduced reactivity for Ag85B-dm with a glycosylation yield of 91.5% and an average number of incorporate saccharides of 2.0 by using the disaccharide Man(1-6)Man with a molar ratio of 200:1. The glycosylation via DSG linker of the same protein with the same disaccharide and at half the molar ratio resulted in a 100% yield and in a more satisfying average number of bond sugars, i.e., 3.4.
In the case of Ag85B, we also did not observe a reduction of reactivity by increasing sugar length from di- to trisaccharide. This result is extremely relevant for the future synthesis of glycoconjugate vaccines, which require longer carbohydrate antigens. Considering that the use of more complex antigens will presumably require the sugar/protein molar ratio to be reconsidered, an accurate balance between the use of a sustainable saccharide molar excess and the achievement of an adequate glycosylation degree should be sought; this should also consider the specific antigen potency.
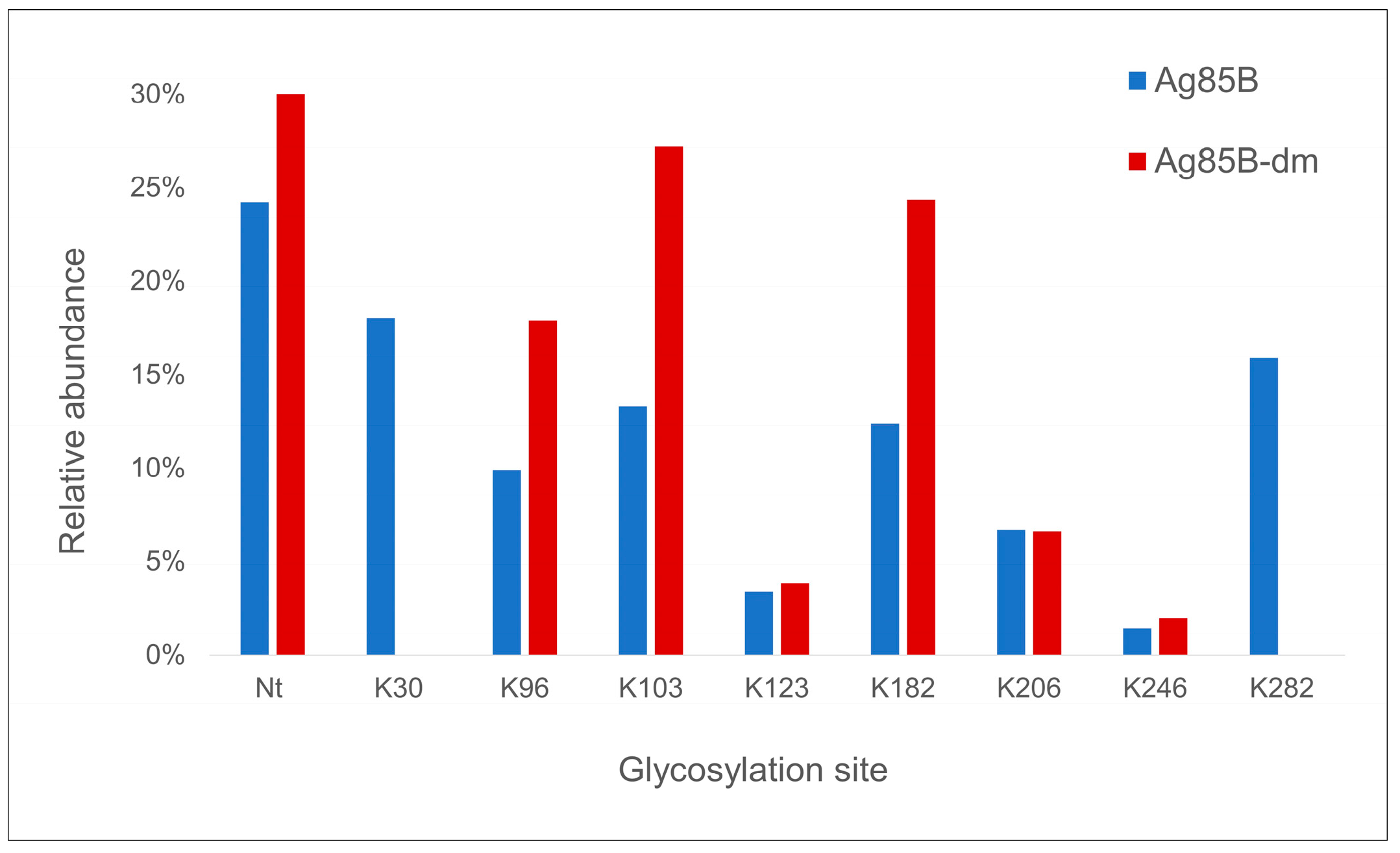
By glycopeptide mapping analysis, the N-terminal alanine residue was found to be the most reactive site, thus confirming the higher reactivity of the N-terminal amino group in these experimental conditions. This is a positive result in the case of recombinant Ag85B proteins, as the N-terminal region is not involved in their antigenicity [
17]. After the N-terminal amino group, the most reactive sites in Ag85B wt were K30 and K282, where two lysine residues which are missing in Ag85B-dm. The same glycan loading was achieved for both Ag85B variants mainly because of the increased reactivity of three lysine residues, namely K96, K103, and K182, in Ag85B-dm. This evidence suggests a competitive mechanism among the different reactive sites and explains the great influence of molar ratio on carbohydrate loading.
Based on the achieved results, conjugation via disuccinimidyl linkers appears to be a convenient strategy in the case of the optimized Ag85B-dm carrier; future research will consider this for its glycosylation with larger-sized antigenic oligosaccharides, and in the context of the rational development of a glycoconjugate vaccine.
5. Conclusions
In the present work, a common conjugation chemistry based on the use of a disuccinimidyl functional linker (namely DSG) was exploited for the synthesis of glycoconjugates. A detailed analytical characterization confirmed that the intrinsic tendency of the linker for a hydrolysis reaction limited the conjugation approach. We therefore addressed this issue by developing a novel, mostly selective, purification method based on HILIC chromatography. Once the purification protocol was set, we focused our attention on the optimization of glycosylation yield by using the model protein RNase A and the 3-aminopropyl mannose 19. By applying the selected conditions, excellent yields were obtained in the conjugation of RNase A with newly synthesized mono-, di-, and tri-saccharide derivatives. The same protocol was then applied to the antigenic proteins Ag85B wt and -dm. The glycosylation outcome was highly satisfactory (glycosylation yields > 99.5%), even in the case of mutated carrier.
In summary, the proposed purification method, together with a detailed structural characterization and a rational optimization of glycosylation conditions, can make this conjugation approach suitable for the synthesis of pure and high-loaded glycoconjugates, and should be considered for carbohydrate-based vaccine synthesis.

 ,
, 













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}