
Experimental Elucidation of Templated Crystallization and Secondary Processing of Peptides

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
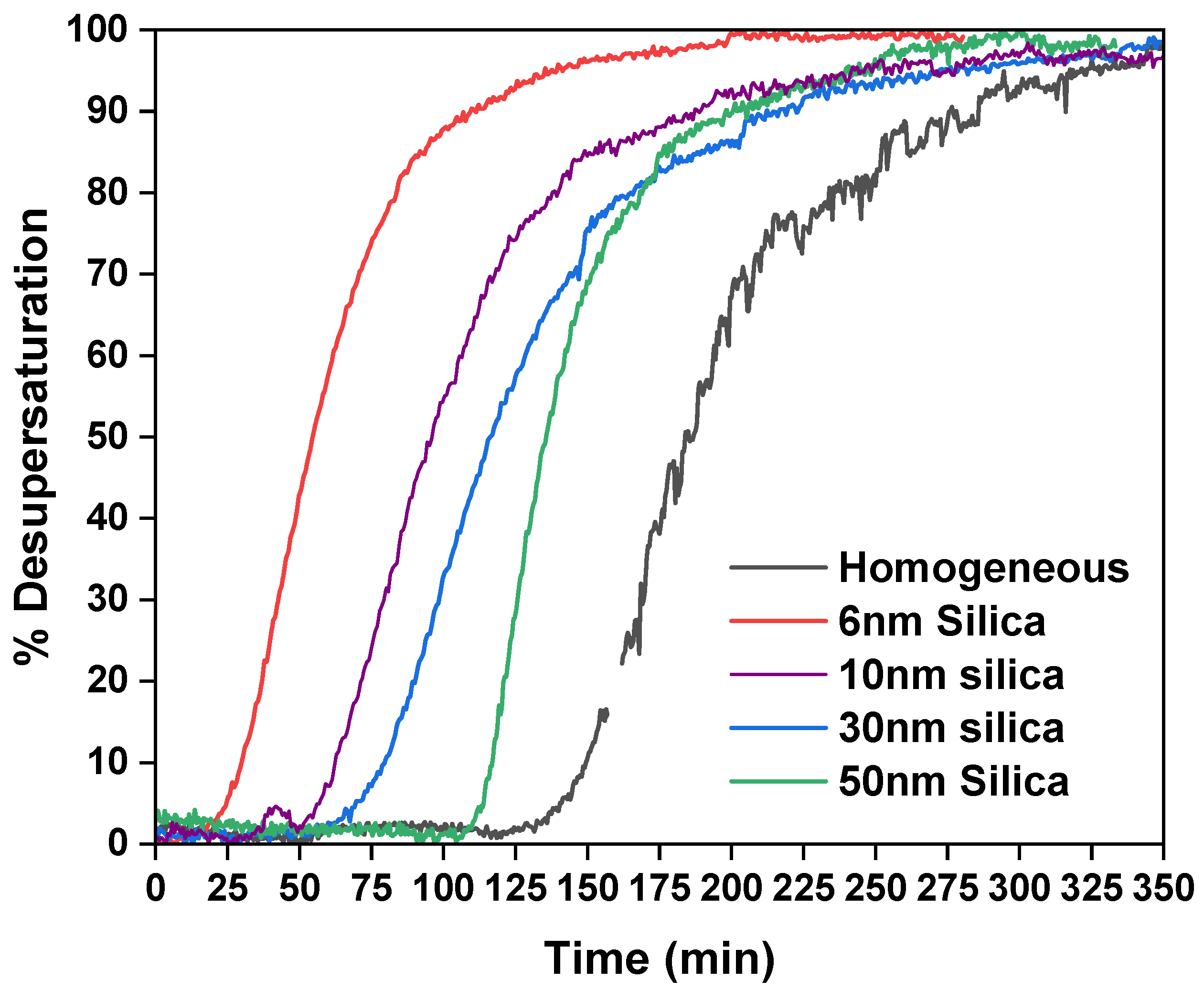
2.2. Isothermal Colling Crystallization Experiments
2.3. Dynamic Light Scattering (DLS)
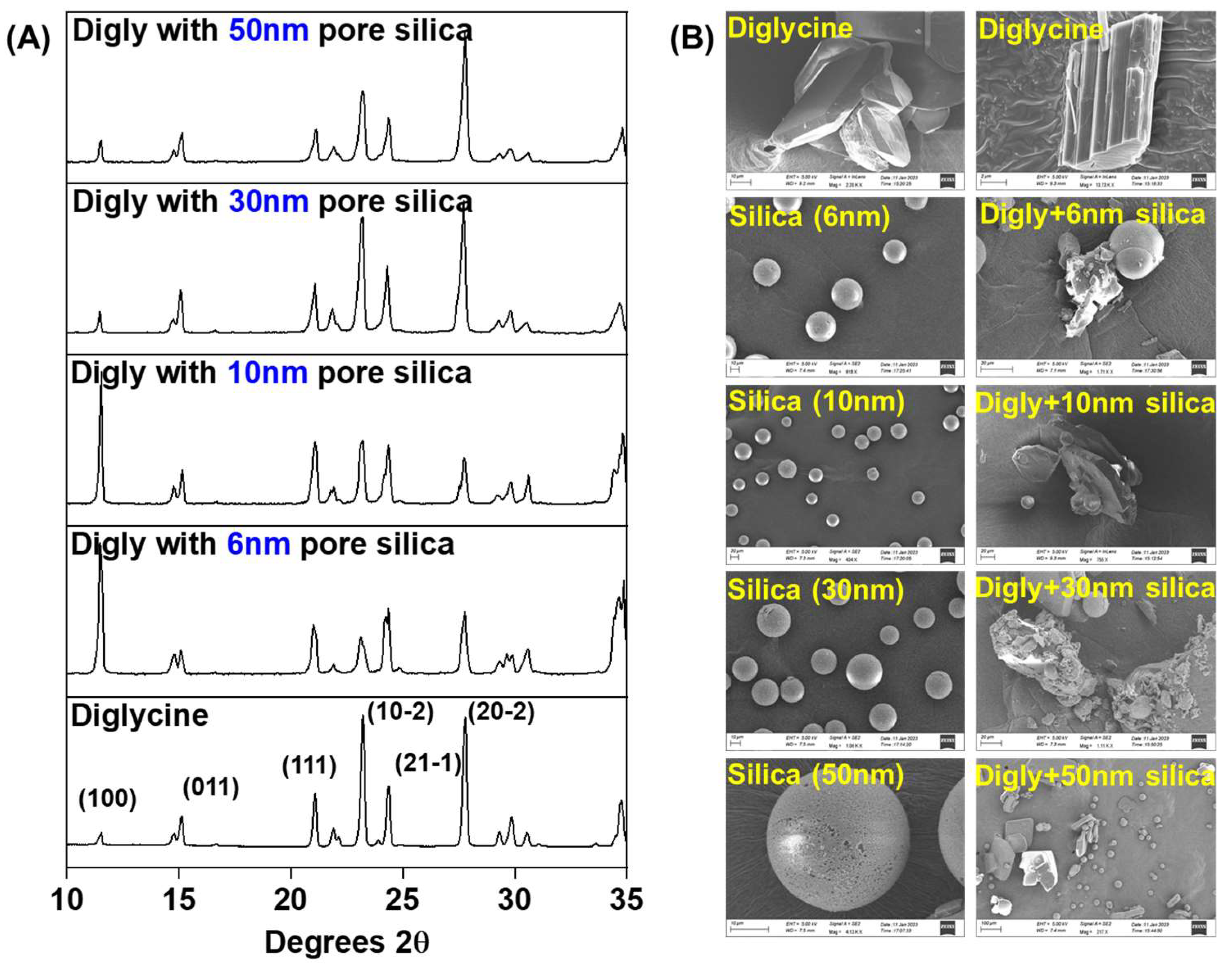
2.4. Solid State Characterization of Isolated Solids
2.4.1. Powder X-ray Diffraction (PXRD)
2.4.2. Scanning Electron Microscopy (SEM)
2.4.3. True Density Measurement
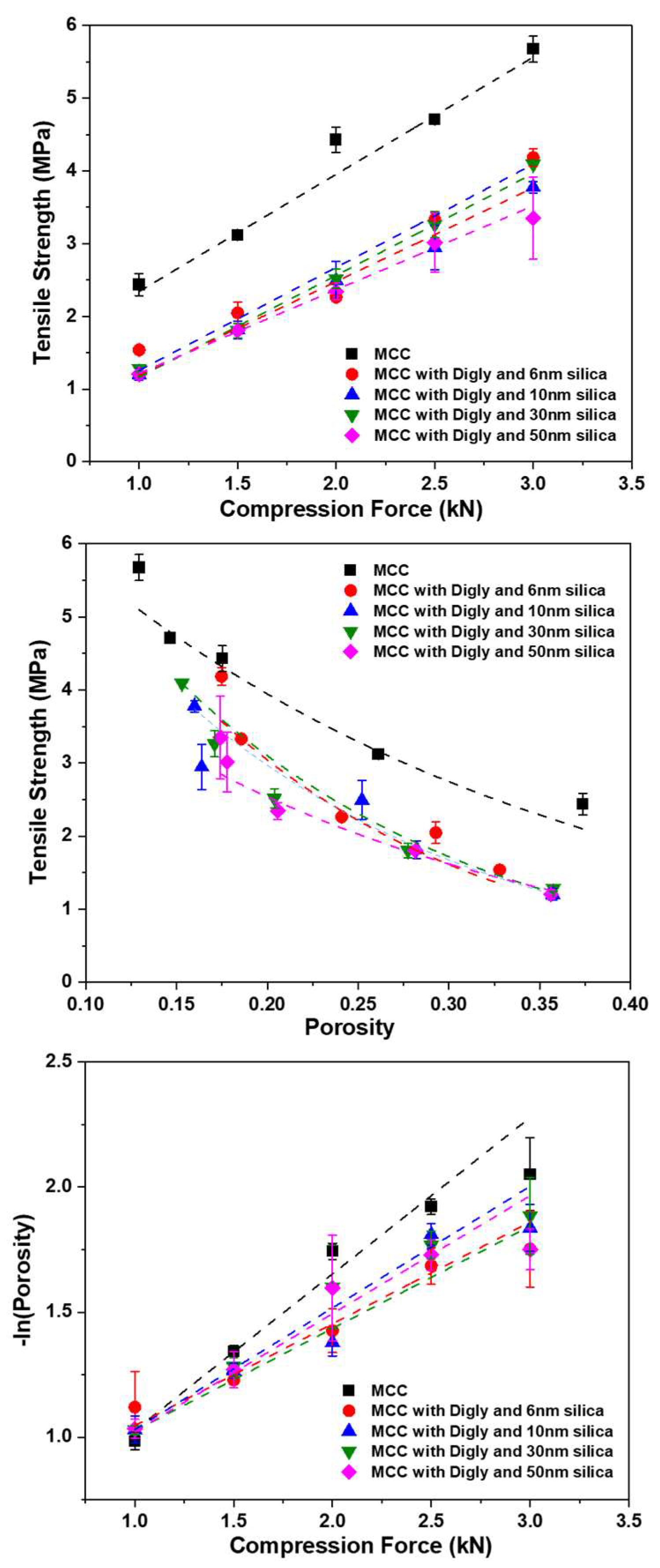
2.5. Tabletting of the Diglycine–Silica–MCC Composite
2.5.1. Tablet Porosity
2.5.2. Tabletability
2.5.3. Compactability
2.5.4. Compressibility
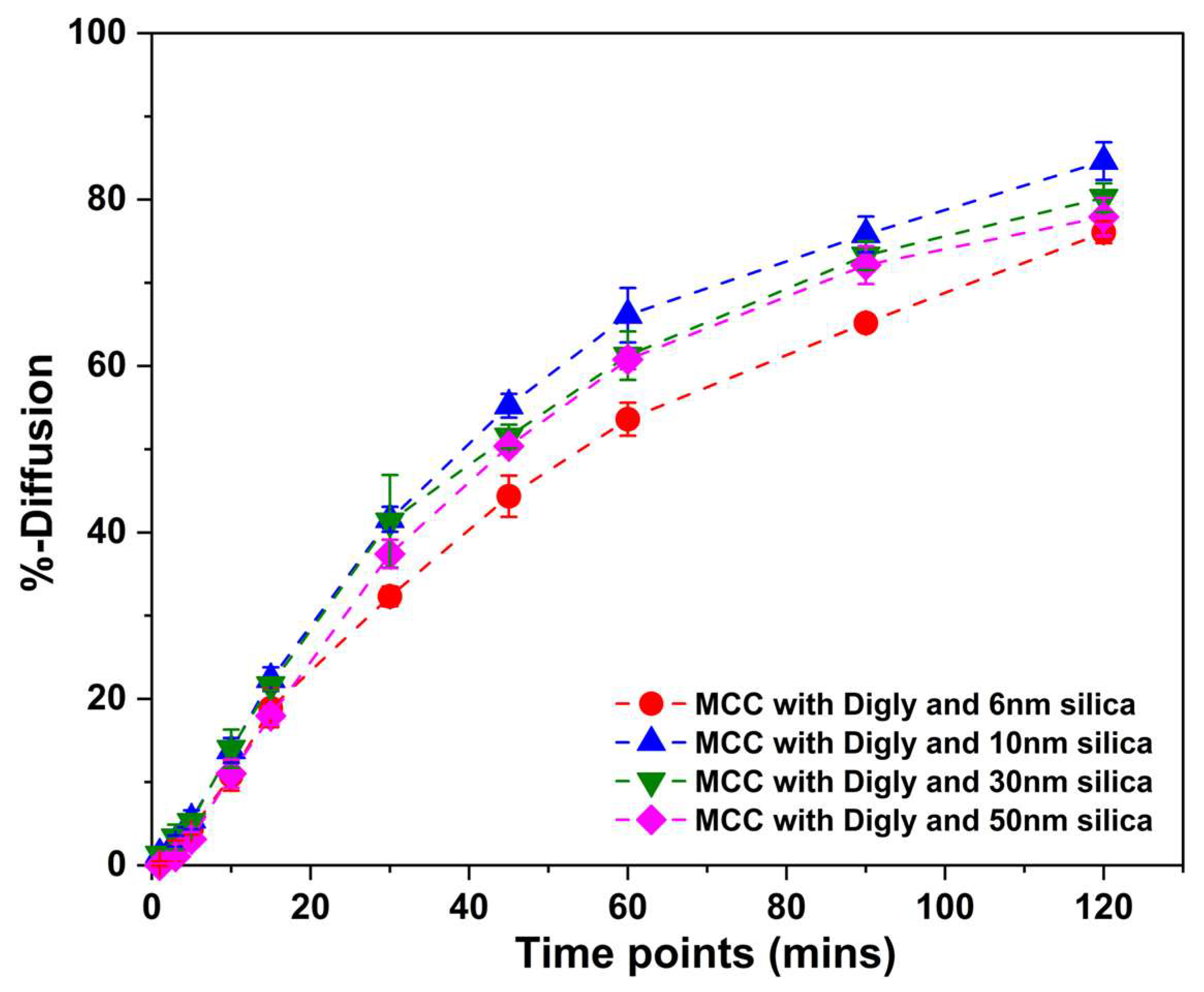
2.6. In Vitro Diffusion Studies
3. Results and Discussion
- paracellular, involving passing in between enterocyte cells, only allowing smaller peptides with a low molecular weight;
- transcellular, involving passage through the enterocyte cell with the help of other cells;
- ligand-assisted transport, allowing transportation through the intestinal mucosal membrane using a permeation enhancer.
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, L.; Wang, N.; Zhang, W.; Cheng, X.; Yan, Z.; Shao, G.; Wang, X.; Wang, R.; Fu, C. Therapeutic peptides: Current applications and future directions. Sig. Transduct. Target Ther. 2022, 7, 48. [Google Scholar] [CrossRef]
- Muttenthaler, M.; King, G.F.; Adams, D.J.; Alewood, P.F. Trends in peptide drug discovery. Nat. Rev. Drug Discov. 2021, 20, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Craik, D.J.; Fairlie, D.P.; Liras, S.; Price, D. The Future of Peptide-based Drugs. Chem. Biol. Drug Des. 2013, 81, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Ferrazzano, L.; Catani, M.; Cavazzini, A.; Martelli, G.; Corbisiero, D.; Cantelmi, P.; Fantoni, T.; Mattellone, A.; De Luca, C.; Felletti, S.; et al. Sustainability in peptide chemistry: Current synthesis and purification technologies and future challenges. Green Chem. 2022, 24, 975–1020. [Google Scholar] [CrossRef]
- Isidro-Llobet, A.; Kenworthy, M.N.; Mukherjee, S.; Kopach, M.E.; Wegner, K.; Gallou, F.; Smith, A.G.; Roschangar, F. Sustainability Challenges in Peptide Synthesis and Purification: From R&D to Production. J. Org. Chem. 2019, 84, 4615–4628. [Google Scholar] [CrossRef]
- Roque, A.C.A.; Pina, A.S.; Azevedo, A.M.; Aires-Barros, R.; Jungbauer, A.; Di Profio, G.; Heng, J.Y.Y.; Haigh, J.; Ottens, M. Anything but Conventional Chromatography Approaches in Bioseparation. Biotechnol. J. 2020, 15, 1900274. [Google Scholar] [CrossRef]
- Guo, M.; Jones, M.J.; Goh, R.; Verma, V.; Guinn, E.; Heng, J.Y.Y. The Effect of Chain Length and Conformation on the Nucleation of Glycine Homopeptides during the Crystallization Process. Cryst. Growth Des. 2023, 23, 1668–1675. [Google Scholar] [CrossRef]
- Guo, M.; Rosbottom, I.; Zhou, L.; Yong, C.W.; Zhou, L.; Yin, Q.; Todorov, I.T.; Errington, E.; Heng, J.Y.Y. Triglycine (GGG) Adopts a Polyproline II (pPII) Conformation in Its Hydrated Crystal Form: Revealing the Role of Water in Peptide Crystallization. J. Phys. Chem. Lett. 2021, 12, 8416–8422. [Google Scholar] [CrossRef]
- Link, F.J.; Heng, J.Y.Y. Enhancing the crystallisation of insulin using amino acids as soft-templates to control nucleation. CrystEngComm 2021, 23, 3951–3960. [Google Scholar] [CrossRef]
- Chen, W.; Park, S.J.; Kong, F.; Li, X.; Yang, H.; Heng, J.Y.Y. High Protein-Loading Silica Template for Heterogeneous Protein Crystallization. Cryst. Growth Des. 2020, 20, 866–873. [Google Scholar] [CrossRef]
- Li, X.; Heng, J.Y.Y. Protein crystallisation facilitated by silica particles to compensate for the adverse impact from protein impurities. CrystEngComm 2021, 23, 8386–8391. [Google Scholar] [CrossRef]
- Li, X.; Heng, J.Y.Y. The critical role of agitation in moving from preliminary screening results to reproducible batch protein crystallisation. Chem. Eng. Res. Des. 2021, 173, 81–88. [Google Scholar] [CrossRef]
- Verma, V.; Mitchell, H.; Guo, M.; Hodnett, B.K.; Heng, J.Y.Y. Studying the impact of the pre-exponential factor on templated nucleation. Faraday Discuss. 2022, 235, 199–218. [Google Scholar] [CrossRef]
- Verma, V.; Mitchell, H.; Errington, E.; Guo, M.; Heng, J.Y.Y. Templated Crystallization of Glycine Homopeptides: Experimental and Computational Developments. Chem. Eng. Technol. 2023. [Google Scholar] [CrossRef]
- Haddadzadegan, S.; Dorkoosh, F.; Bernkop-Schnürch, A. Oral delivery of therapeutic peptides and proteins: Technology landscape of lipid-based nanocarriers. Adv. Drug Deliv. Rev. 2022, 182, 114097. [Google Scholar] [CrossRef]
- Jenkins, K., II. Needle phobia: A psychological perspective. Br. J. Anaesth. 2014, 113, 4–6. [Google Scholar] [CrossRef]
- Karsdal, M.A.; Byrjalsen, I.; Riis, B.J.; Christiansen, C. Optimizing bioavailability of oral administration of small peptides through pharmacokinetic and pharmacodynamic parameters: The effect of water and timing of meal intake on oral delivery of Salmon Calcitonin. BMC Clin. Pharmacol. 2008, 8, 5. [Google Scholar] [CrossRef]
- Klepach, A.; Tran, H.; Ahmad Mohammed, F.; ElSayed, M.E.H. Characterization and impact of peptide physicochemical properties on oral and subcutaneous delivery. Adv. Drug. Deliv. Rev. 2022, 186, 114322. [Google Scholar] [CrossRef]
- Choonara, B.F.; Choonara, Y.E.; Kumar, P.; Bijukumar, D.; du Toit, L.C.; Pillay, V. A review of advanced oral drug delivery technologies facilitating the protection and absorption of protein and peptide molecules. Biotechnol. Adv. 2014, 32, 1269–1282. [Google Scholar] [CrossRef]
- Maher, S.; Brayden, D.J. Formulation strategies to improve the efficacy of intestinal permeation enhancers. Adv. Drug Deliv. Rev. 2021, 177, 113925. [Google Scholar] [CrossRef]
- Maher, S.; Ryan, B.; Duffy, A.; Brayden, D.J. Formulation strategies to improve oral peptide delivery. Pharm. Pat. Anal. 2014, 3, 313–336. [Google Scholar] [CrossRef]
- Zhu, Q.; Chen, Z.; Paul, P.K.; Lu, Y.; Wu, W.; Qi, J. Oral delivery of proteins and peptides: Challenges, status quo and future perspectives. Acta Pharm. Sin. B 2021, 11, 2416–2448. [Google Scholar] [CrossRef]
- Anselmo, A.C.; Gokarn, Y.; Mitragotri, S. Non-invasive delivery strategies for biologics. Nat. Rev. Drug Discov. 2019, 18, 19–40. [Google Scholar] [CrossRef]
- Harrison, G.A. Insulin in Alcoholic Solution by the Mouth. Br. Med. J. 1923, 2, 1204. [Google Scholar] [CrossRef]
- Basu, S.K.; Govardhan, C.P.; Jung, C.W.; Margolin, A.L. Protein crystals for the delivery of biopharmaceuticals. Expert Opin. Biol. Ther. 2004, 4, 301–317. [Google Scholar] [CrossRef]
- Aguirre, T.A.S.; Teijeiro-Osorio, D.; Rosa, M.; Coulter, I.S.; Alonso, M.J.; Brayden, D.J. Current status of selected oral peptide technologies in advanced preclinical development and in clinical trials. Adv. Drug Deliv. Rev. 2016, 106, 223–241. [Google Scholar] [CrossRef]
- Drucker, D.J. Advances in oral peptide therapeutics. Nat. Rev. Drug Discov. 2020, 19, 277–289. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Brayden, D.J.; Hill, T.A.; Fairlie, D.P.; Maher, S.; Mrsny, R.J. Systemic delivery of peptides by the oral route: Formulation and medicinal chemistry approaches. Adv. Drug Deliv. Rev. 2020, 157, 2–36. [Google Scholar] [CrossRef]
- Verma, V.; Zeglinski, J.; Hudson, S.; Davern, P.; Hodnett, B.K. Dependence of Heterogeneous Nucleation on Hydrogen Bonding Lifetime and Complementarity. Cryst. Growth Des. 2018, 18, 7158–7172. [Google Scholar] [CrossRef]
- Guo, M.; Chang, Z.H.; Liang, E.; Mitchell, H.; Zhou, L.; Yin, Q.; Guinn, E.J.; Heng, J.Y.Y. The effect of chain length and side chains on the solubility of peptides in water from 278.15 K to 313.15 K: A case study in glycine homopeptides and dipeptides. J. Mol. Liq. 2022, 352, 118681. [Google Scholar] [CrossRef]
- Newton, J.M.; Rowley, G.; Fell, J.T.; Peacock, D.G.; Ridgway, K. Computer analysis of the relation between tablet strength and compaction pressure. J. Pharm. Pharmacol. 1971, 23, 195S–201S. [Google Scholar] [CrossRef] [PubMed]
- Ryshkewitch, E. Compression Strength of Porous Sintered Alumina and Zirconia. J. Am. Ceram. Soc. 1953, 36, 65–68. [Google Scholar] [CrossRef]
- Steendam, R.; Lerk, C.F. Poly(dl-lactic acid) as a direct compression excipient in controlled release tablets: Part I. Compaction behaviour and release characteristics of poly(dl-lactic acid) matrix tablets. Int. J. Pharm. 1998, 175, 33–46. [Google Scholar] [CrossRef]
- de Andrade, D.F.; Zuglianello, C.; Pohlmann, A.R.; Guterres, S.S.; Beck, R.C.R. Assessing the In Vitro Drug Release from Lipid-Core Nanocapsules: A New Strategy Combining Dialysis Sac and a Continuous-Flow System. AAPS PharmSciTech 2015, 16, 1409–1417. [Google Scholar] [CrossRef]
- Shah, U.V.; Williams, D.R.; Heng, J.Y.Y. Selective Crystallization of Proteins Using Engineered Nanonucleants. Cryst. Growth Des. 2012, 12, 1362–1369. [Google Scholar] [CrossRef]
- Yang, H.; Belviso, B.D.; Li, X.; Chen, W.; Mastropietro, T.F.; Di Profio, G.; Caliandro, R.; Heng, J.Y.Y. Optimization of Vapor Diffusion Conditions for Anti-CD20 Crystallization and Scale-Up to Meso Batch. Crystals 2019, 9, 230. [Google Scholar] [CrossRef]
- Gerard, C.J.J.; Briuglia, M.L.; Rajoub, N.; Mastropietro, T.F.; Chen, W.; Heng, J.Y.Y.; Di Profio, G.; ter Horst, J.H. Template-Assisted Crystallization Behavior in Stirred Solutions of the Monoclonal Antibody Anti-CD20: Probability Distributions of Induction Times. Cryst. Growth Des. 2022, 22, 3637–3645. [Google Scholar] [CrossRef]
- Michrafy, A.; Michrafy, M.; Kadiri, M.S.; Dodds, J.A. Predictions of tensile strength of binary tablets using linear and power law mixing rules. Int. J. Pharm. 2007, 333, 118–126. [Google Scholar] [CrossRef]
- Wu, C.-Y.; Best, S.M.; Bentham, A.C.; Hancock, B.C.; Bonfield, W. A simple predictive model for the tensile strength of binary tablets. Eur. J. Pharm. Sci. 2005, 25, 331–336. [Google Scholar] [CrossRef]
- Pudasaini, N.; Upadhyay, P.P.; Parker, C.R.; Hagen, S.U.; Bond, A.D.; Rantanen, J. Downstream Processability of Crystal Habit-Modified Active Pharmaceutical Ingredient. Org. Process Res. Dev. 2017, 21, 571–577. [Google Scholar] [CrossRef]
- Chen, H.; Aburub, A.; Sun, C.C. Direct Compression Tablet Containing 99% Active Ingredient&-A Tale of Spherical Crystallization. J. Pharm. Sci. 2019, 108, 1396–1400. [Google Scholar] [CrossRef] [PubMed]
- Barralet, J.E.; Gaunt, T.; Wright, A.J.; Gibson, I.R.; Knowles, J.C. Effect of porosity reduction by compaction on compressive strength and microstructure of calcium phosphate cement. J. Biomed. Mater. Res. 2002, 63, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Shah, U.V.; Olusanmi, D.; Narang, A.S.; Hussain, M.A.; Gamble, J.F.; Tobyn, M.J.; Heng, J.Y.Y. Effect of crystal habits on the surface energy and cohesion of crystalline powders. Int. J. Pharm. 2014, 472, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Föger, F.; Kopf, A.; Loretz, B.; Albrecht, K.; Bernkop-Schnürch, A. Correlation of in vitro and in vivo models for the oral absorption of peptide drugs. Amino Acids 2008, 35, 233–241. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pore Diameter | Induction Time (min) |
|---|---|
| Homogeneous | 145 ± 7 |
| 6 nm pore | 29 ± 5 |
| 10 nm pore | 43 ± 17 |
| 30 nm pore | 72 ± 3 |
| 50 nm pore | 115 ± 7 |
| Sample | Tabletability | Compactability | Compressibility | ||
|---|---|---|---|---|---|
| Cp | σt0 | −b | k | Py (kN) | |
| MCC | 1.61 | 7.98 | 3.30 | 0.54 | 1.84 |
| Digly-6 nm pore size Silica-MCC | 1.33 | 10.40 | 5.82 | 0.34 | 2.90 |
| Digly-10 nm pore size Silica-MCC | 1.26 | 7.95 | 5.16 | 0.43 | 2.32 |
| Digly-30 nm pore size Silica-MCC | 1.41 | 8.34 | 5.38 | 0.44 | 2.27 |
| Digly-50 nm pore size Silica-MCC | 1.10 | 7.60 | 5.19 | 0.38 | 2.64 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Verma, V.; Bade, I.; Karde, V.; Heng, J.Y.Y. Experimental Elucidation of Templated Crystallization and Secondary Processing of Peptides. Pharmaceutics 2023, 15, 1288. https://doi.org/10.3390/pharmaceutics15041288
Verma V, Bade I, Karde V, Heng JYY. Experimental Elucidation of Templated Crystallization and Secondary Processing of Peptides. Pharmaceutics. 2023; 15(4):1288. https://doi.org/10.3390/pharmaceutics15041288
Chicago/Turabian StyleVerma, Vivek, Isha Bade, Vikram Karde, and Jerry Y. Y. Heng. 2023. "Experimental Elucidation of Templated Crystallization and Secondary Processing of Peptides" Pharmaceutics 15, no. 4: 1288. https://doi.org/10.3390/pharmaceutics15041288