
CD163 Monoclonal Antibody Modified Polymer Prodrug Nanoparticles for Targeting Tumor-Associated Macrophages (TAMs) to Enhance Anti-Tumor Effects

 , ,
, ,
Abstract
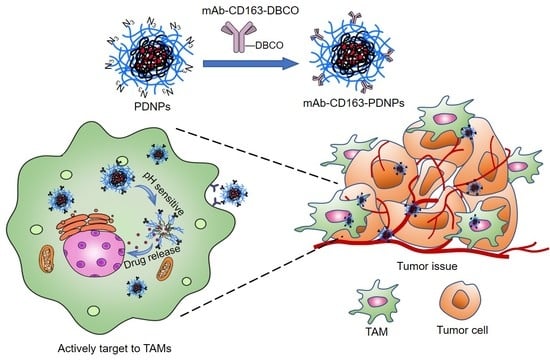
:
1. Introduction
2. Materials and Methods
2.1. Materials and Characerization
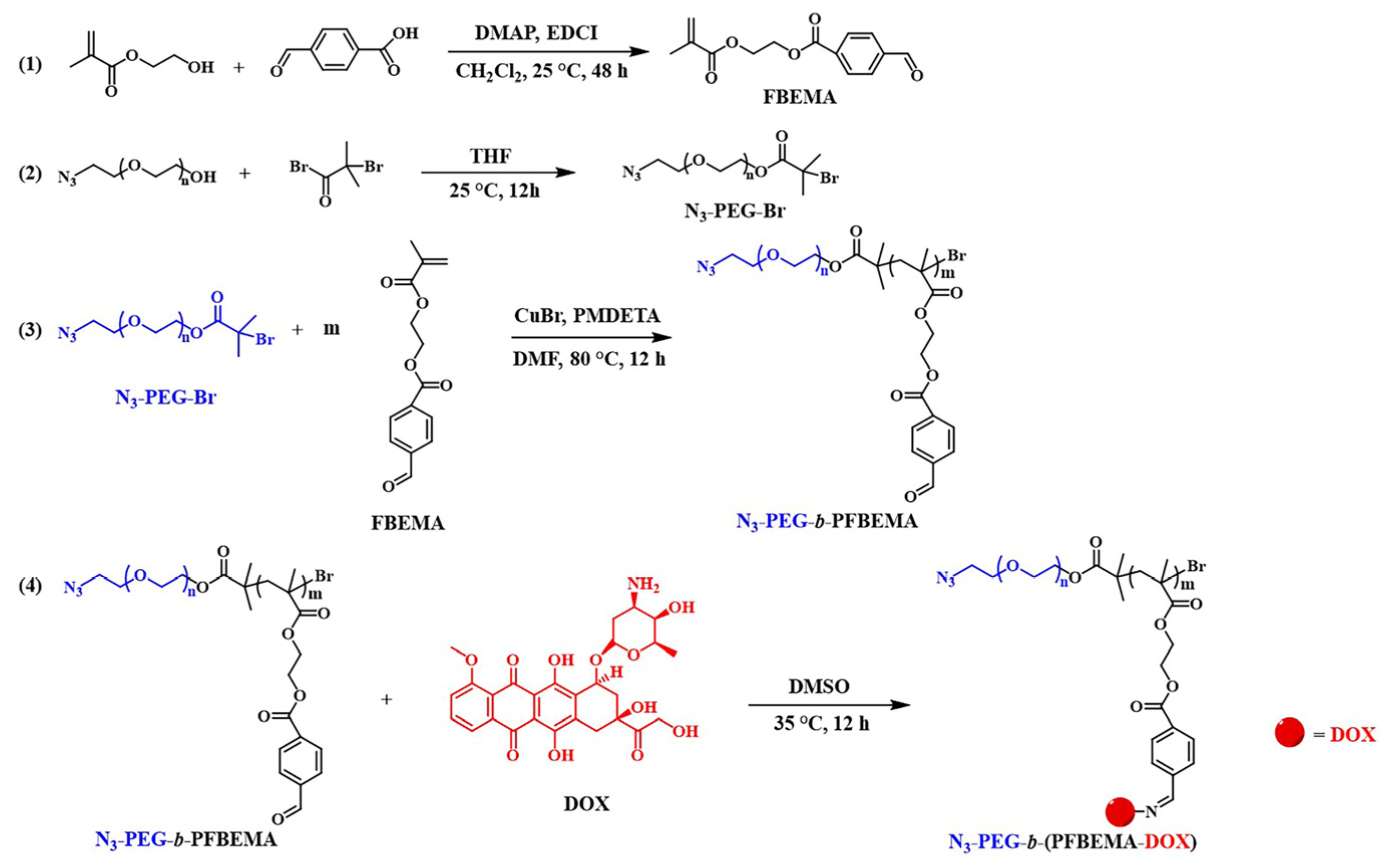
2.2. Synthesis of FBEMA Monomer
2.3. Synthesis of PEG Macromolecular ATRP Initiator
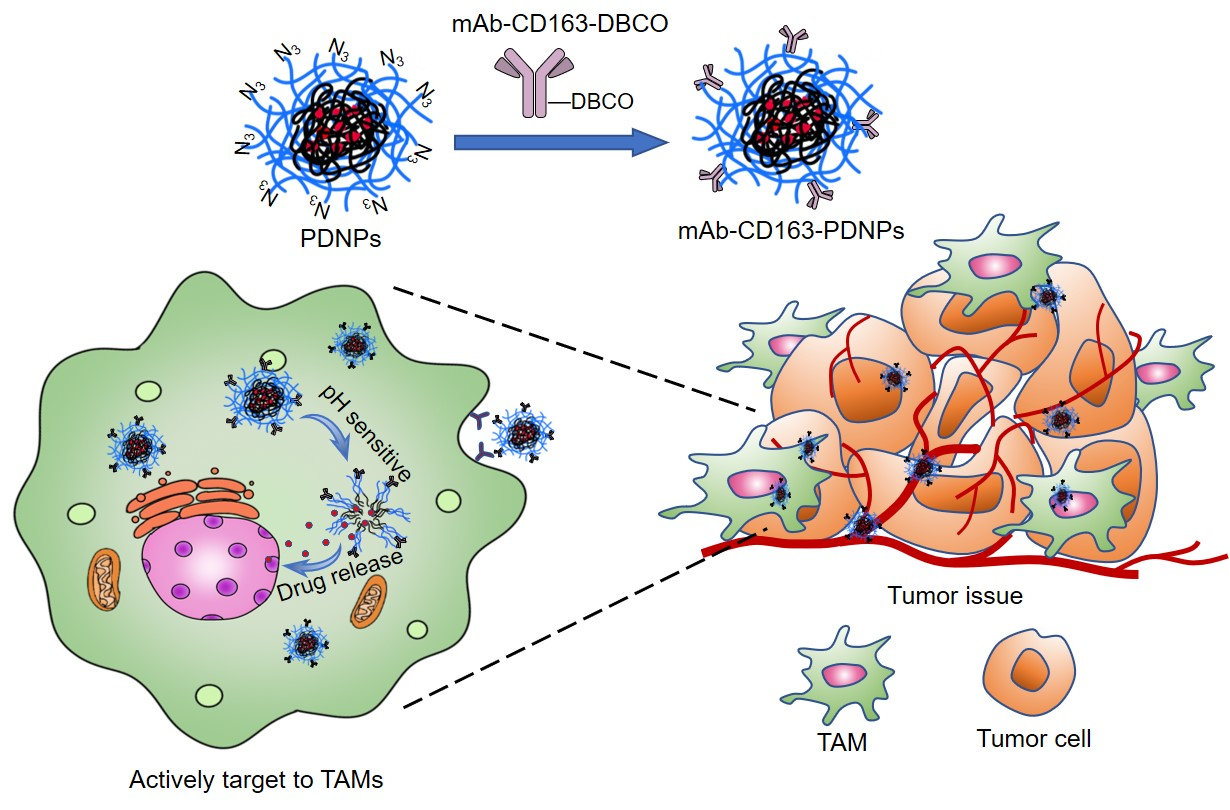
2.4. Synthesis of N3-PEG-b-PFBEMA Amphiphilic Polymer
2.5. Synthesis of N3-PEG-b-(PFBEMA-DOX) Polymer Prodrug
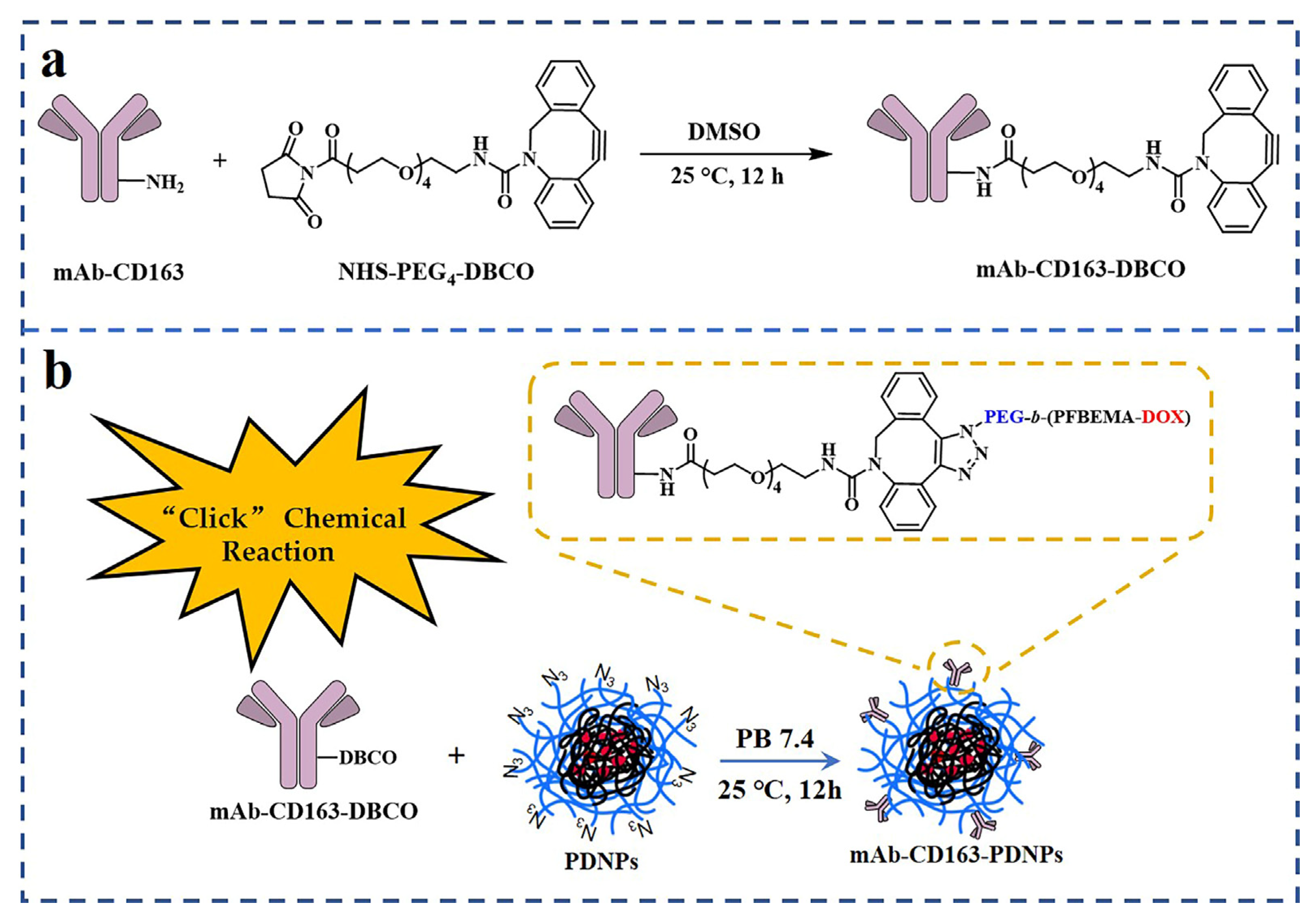
2.6. Synthesis of mAb-CD163-DBCO
2.7. Preparation and Characterization of mAb-CD163-PEG-b-(PFBEMA-DOX) Prodrug Nanoparticles
2.8. Self-Assembly Behavior
2.9. Stability and In Vitro Drug Release
2.10. In Vitro Hemolysis Activity
2.11. Cell Culture
2.12. In Vitro Cytotoxicity
2.13. Cellular Uptake
2.14. Animal Models
2.15. In Vivo Antitumor Efficacy
2.16. In Vivo Biodistribution and Fluorescence Imaging
2.17. Histopathology and Immunohistochemical Analysis
2.18. Statistical Analysis
3. Results and Discussion
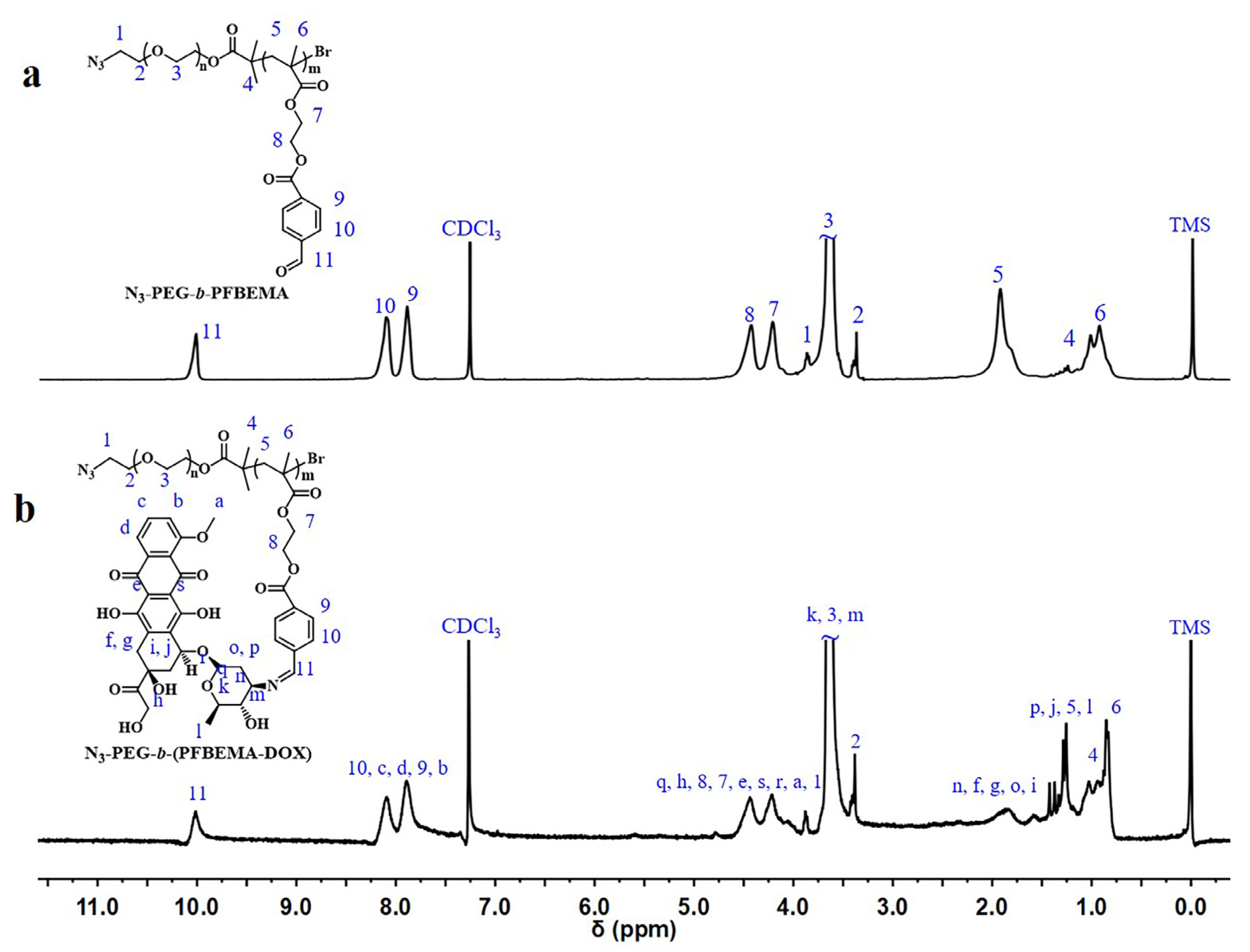
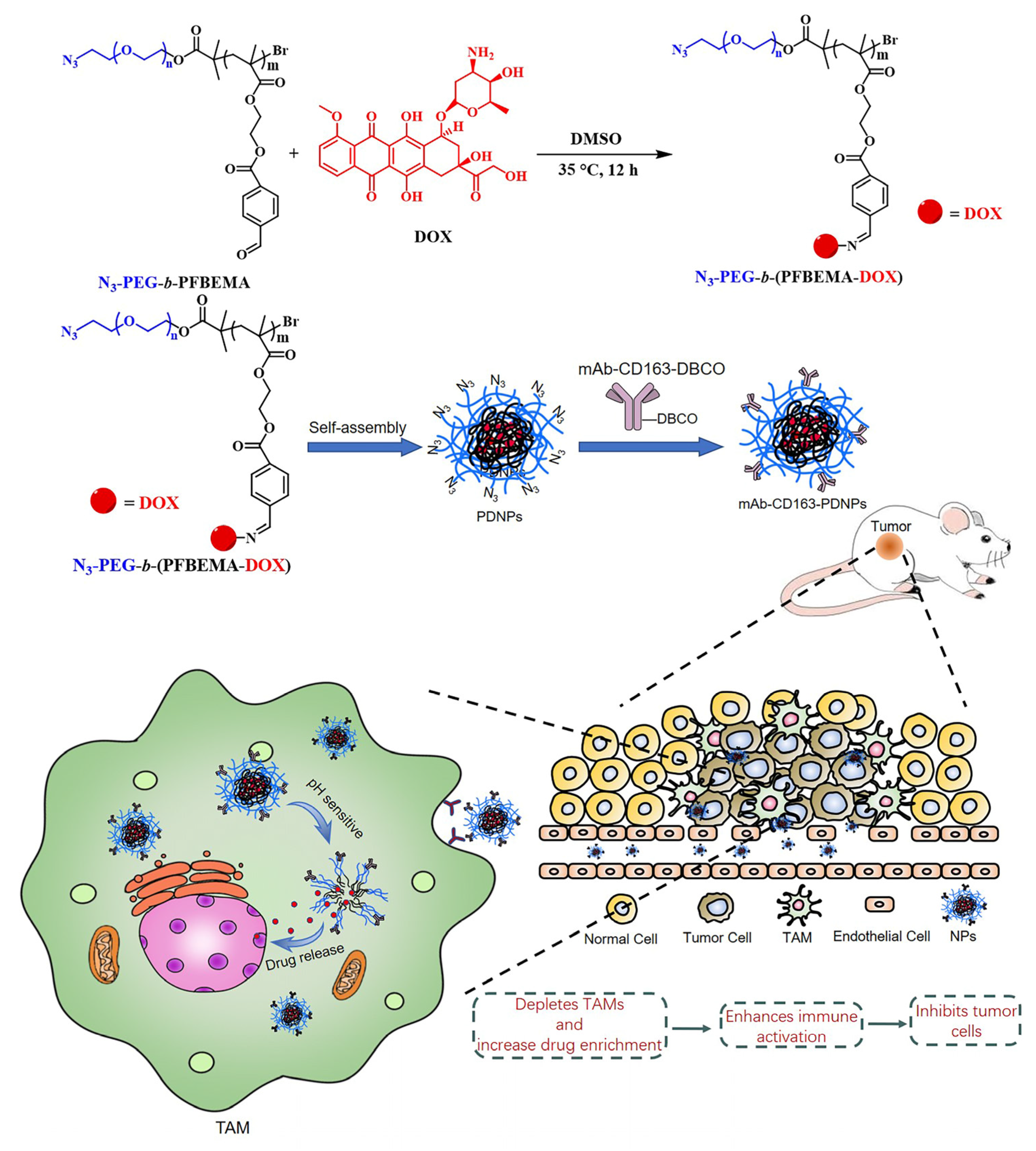
3.1. Synthesis and Characteristics of N3-PEG-b-(PFBEMA-DOX)
3.2. Modification of CD163 Monoclonal Antibodies
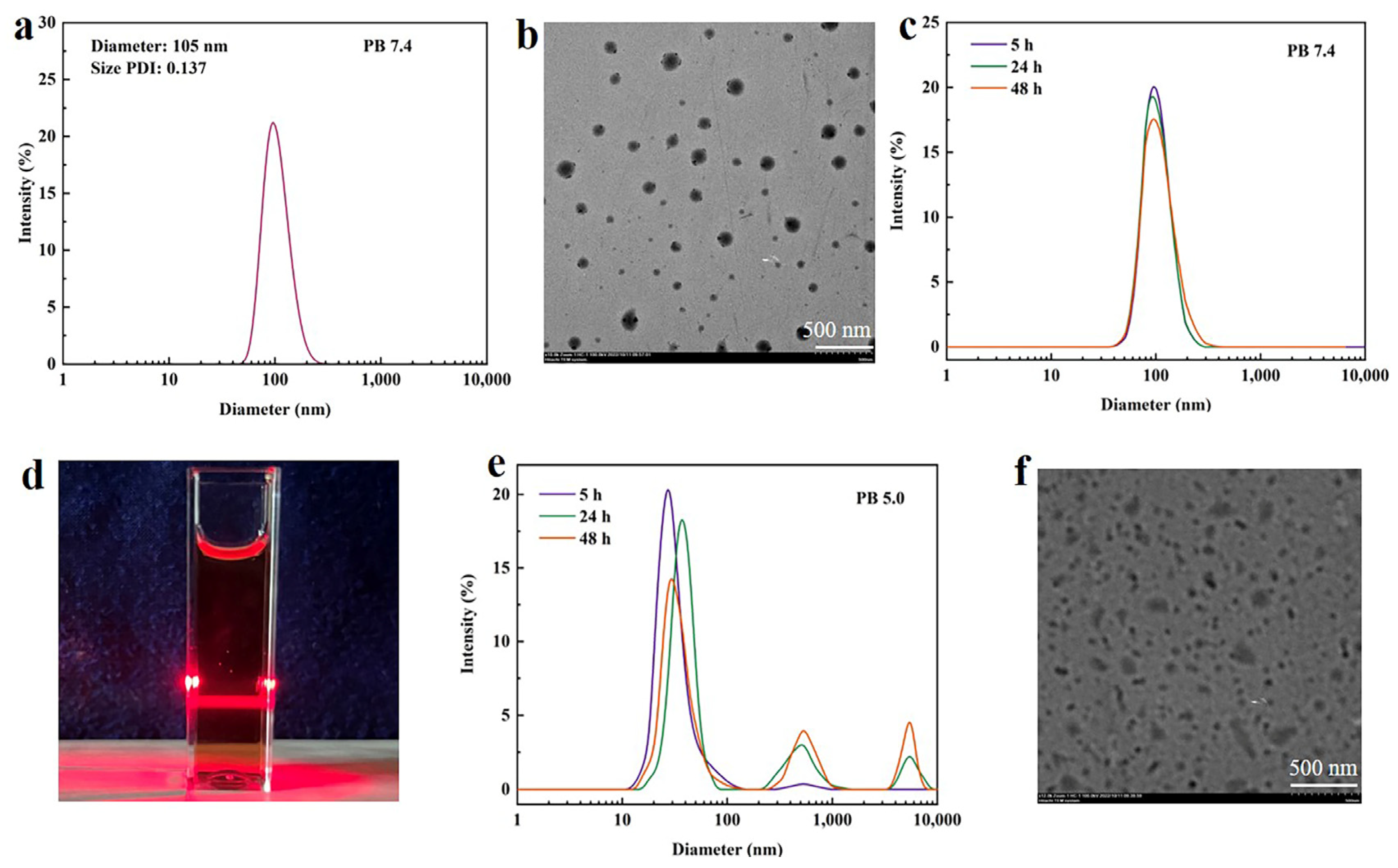
3.3. Stability of Nanoparticles
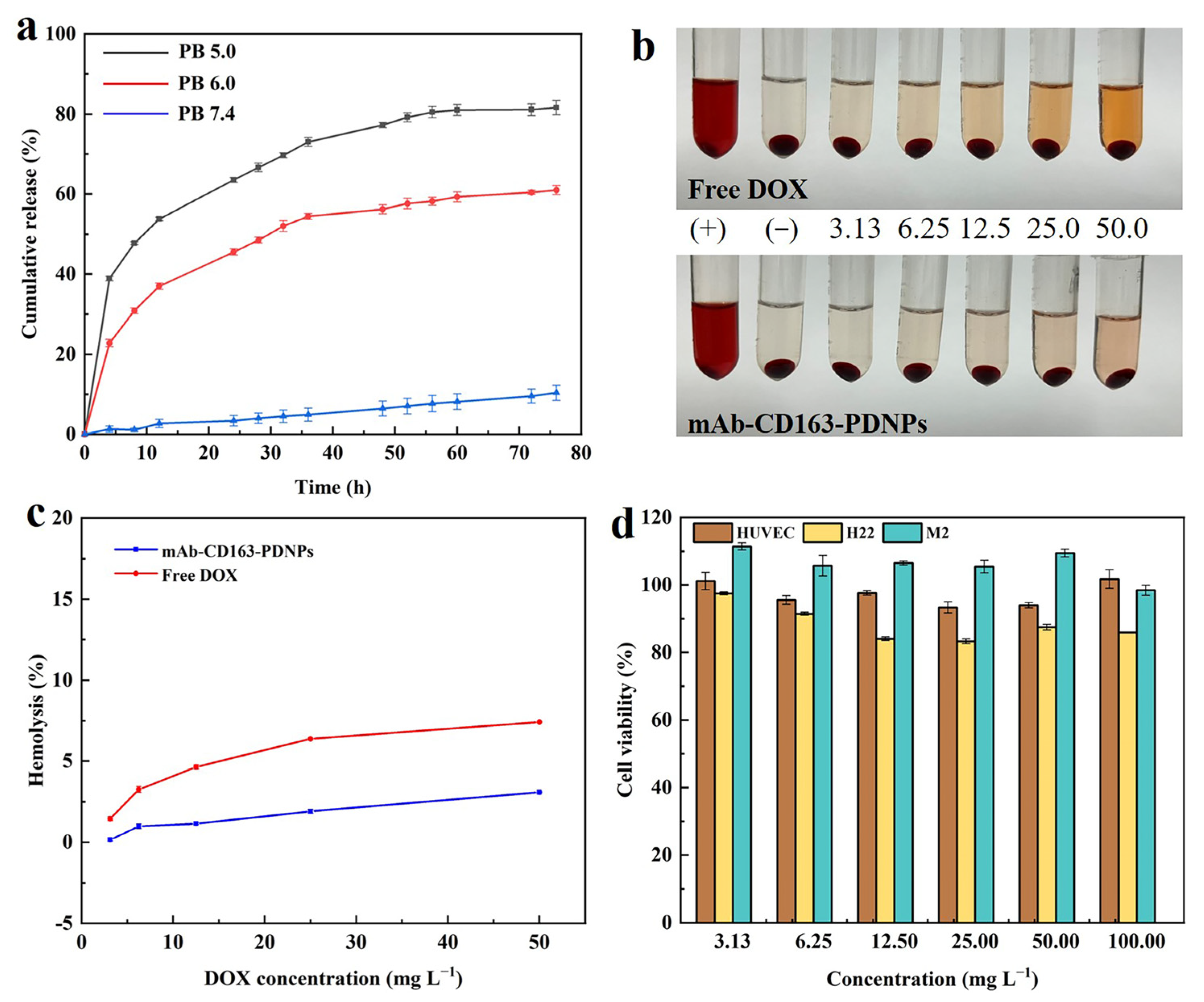
3.4. In Vitro Drug Release
3.5. Hemocompatibility
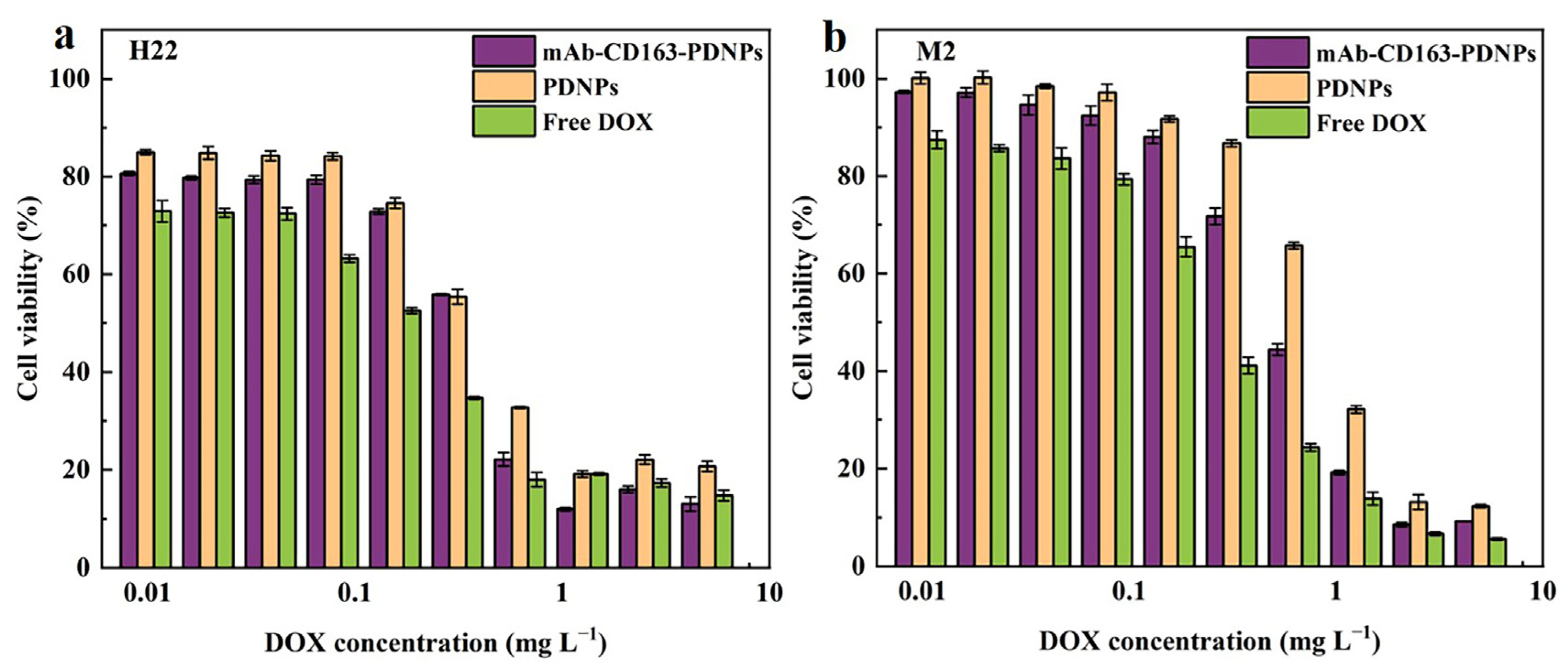
3.6. In Vitro Cytotoxicity
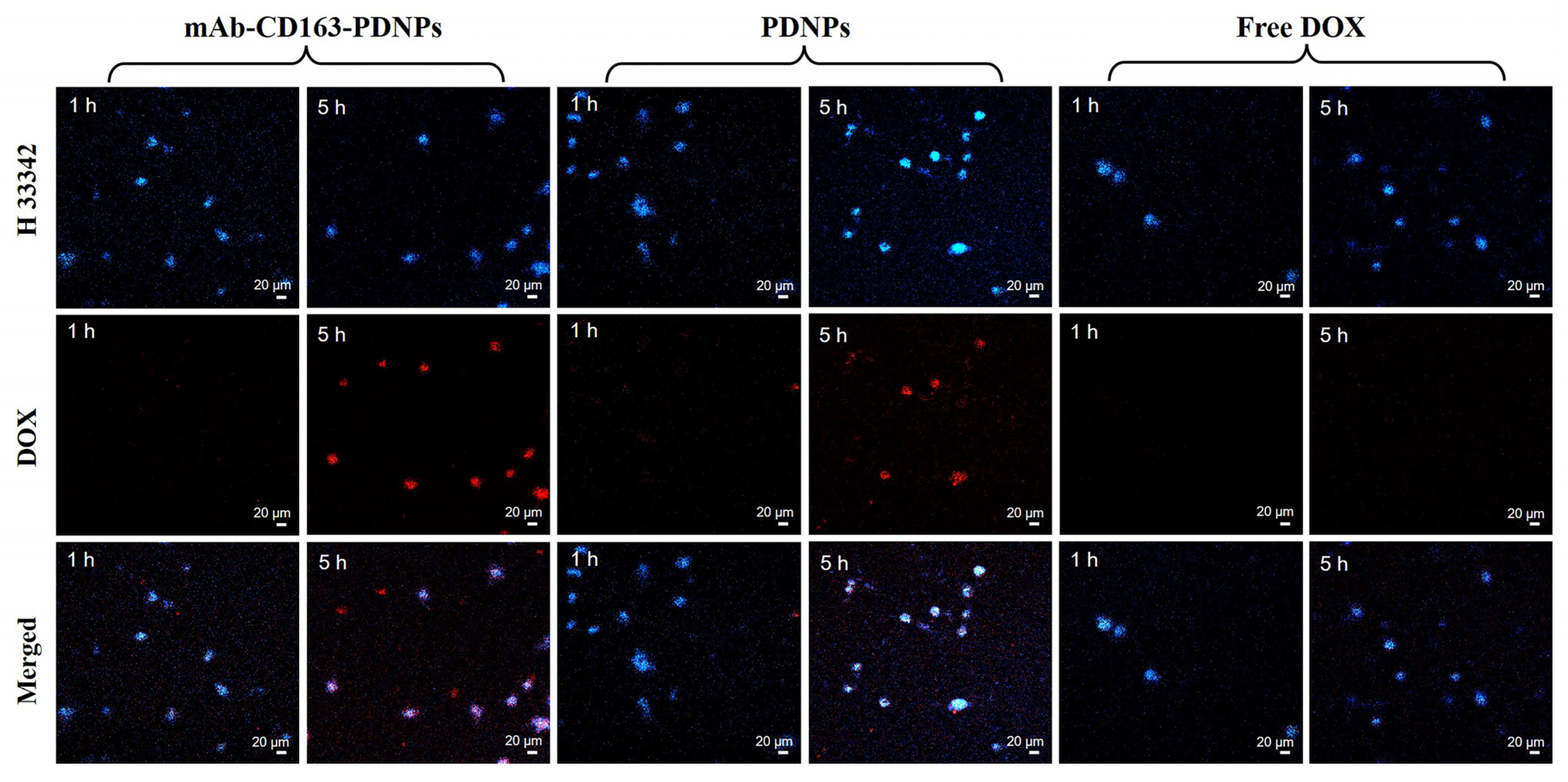
3.7. Cell Uptake
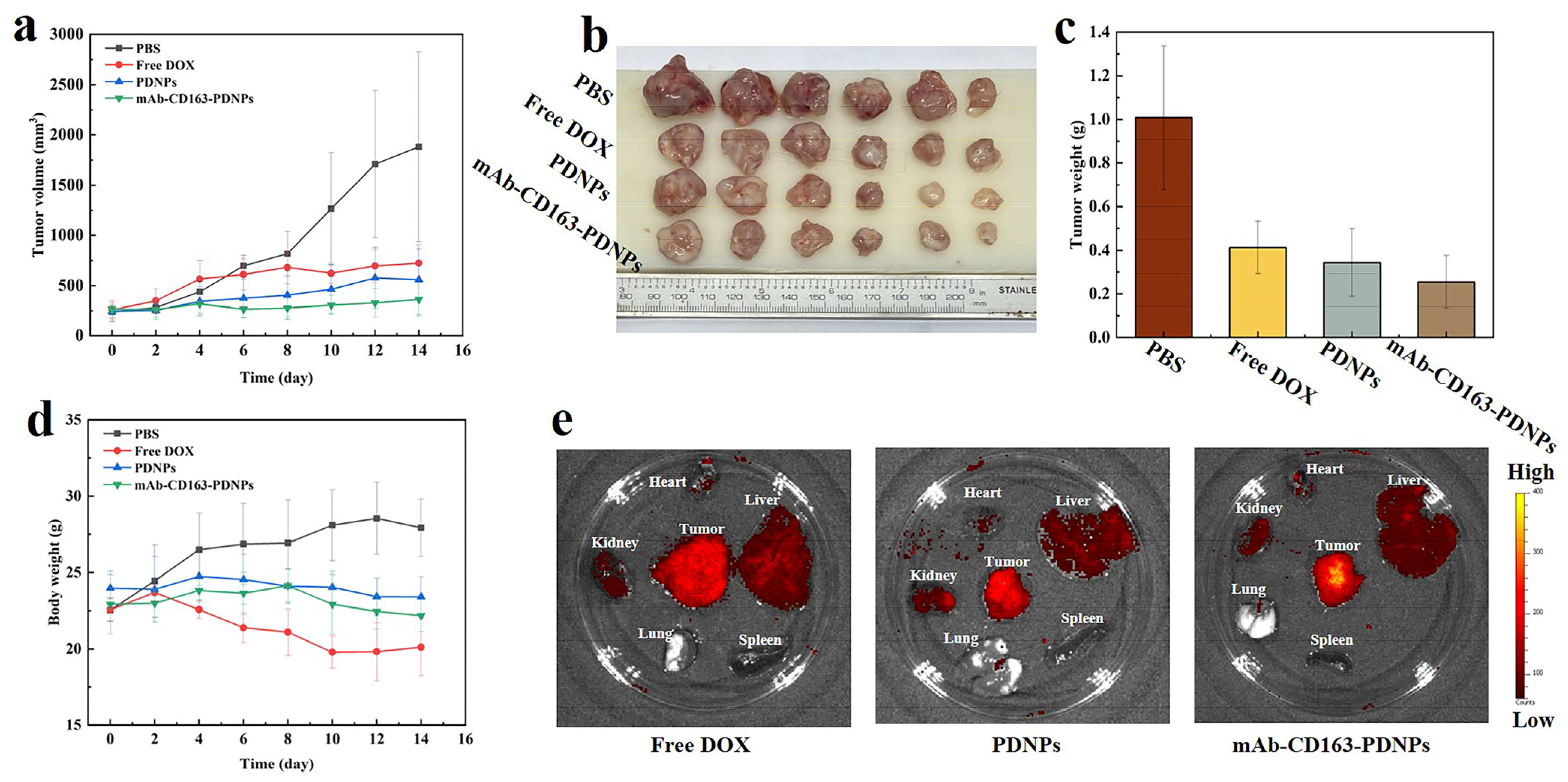
3.8. In Vivo Antitumor Effect
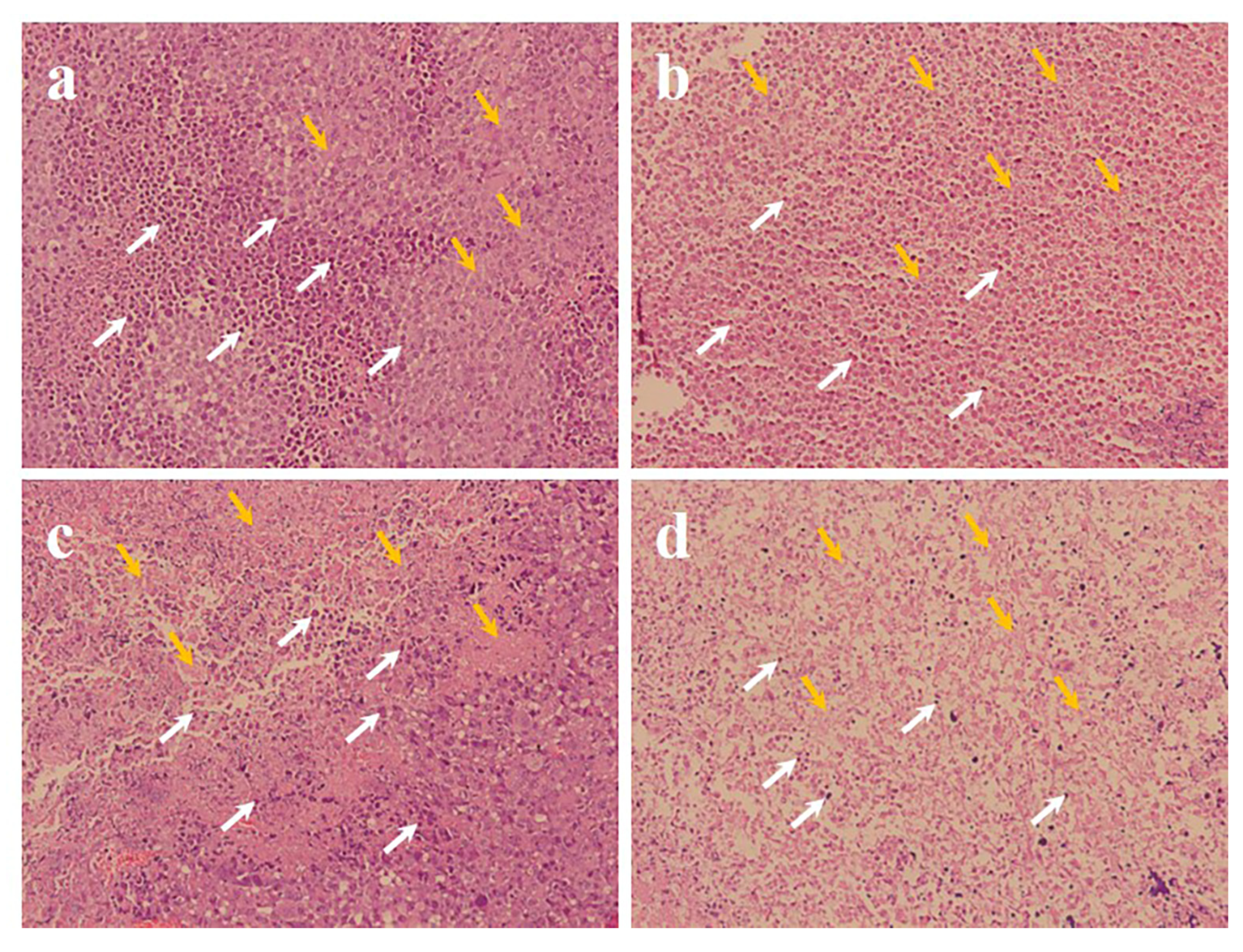
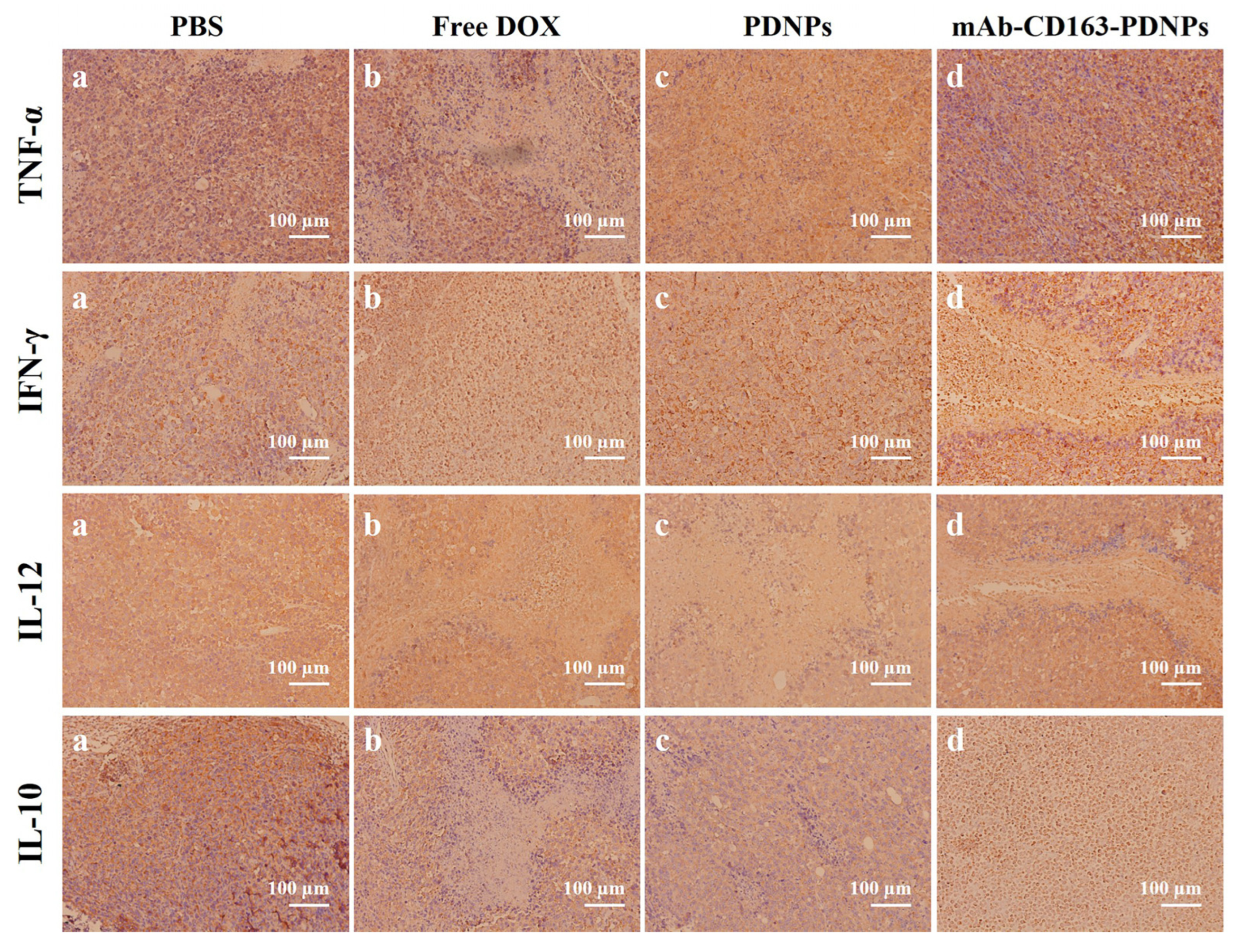
3.9. Histopathology and Immunohistochemical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, Y.Y.; Zhang, Z.M. The History and Advances in Cancer Immunotherapy: Understanding the Characteristics of Tumor-Infiltrating Immune Cells and Their Therapeutic Implications. Cell. Mol. Immunol. 2020, 17, 807–821. [Google Scholar] [CrossRef] [PubMed]
- Shields, C.W.; Wang, L.L.W.; Evans, M.A.; Mitragotri, S. Materials for Immunotherapy. Adv. Mater. 2020, 32, 1901633. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Guo, F.K. Recent Updates on Cancer Immunotherapy. Precis. Clin. Med. 2018, 1, 65–74. [Google Scholar] [CrossRef]
- Tan, S.Z.; Li, D.P.; Zhu, X. Cancer Immunotherapy: Pros, Cons and Beyond. Biomed. Pharmacother. 2020, 124, 109821. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.R.; Xu, W.G.; Liu, J.H.; Li, D.; Li, G.; Ding, J.X.; Chen, X.S. Polypeptide Nanoformulation-Induced Immunogenic Cell Death and Remission of Immunosuppression for Enhanced Chemoimmunotherapy. Sci. Bull. 2021, 66, 362–373. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Song, W.T.; Xu, Y.D.; Si, X.H.; Lv, S.X.; Zhang, Y.; Tang, Z.H.; Chen, X.S. Rationally Designed Polymer Conjugate for Tumor-Specific Amplification of Oxidative Stress and Boosting Antitumor Immunity. Nano Lett. 2020, 20, 2514–2521. [Google Scholar] [CrossRef]
- Wang, Y.B.; Xu, C.N.; Meng, M.; Lin, L.; Hu, Y.Y.; Hao, K.; Sheng, S.; Zhang, S.J.; Wu, J.Y.; Liu, F.; et al. Precise Regulation of Inflammation and Immunosuppressive Microenvironment for Amplified Photothermal/Immunotherapy Against Tumour Recurrence and Metastasis. Nano Today 2021, 40, 101266. [Google Scholar] [CrossRef]
- Jiang, M.; Li, W.; Zhu, C.; Li, X.; Zhang, J.; Luo, Z.; Qin, B.; Du, Y.; Luo, L.; You, J. Perdurable PD-1 Blockage Awakes Anti-Tumor Immunity Suppressed by Precise Chemotherapy. J. Control. Release 2021, 329, 1023–1036. [Google Scholar] [CrossRef]
- Socinski, M.A.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F.; et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N. Engl. J. Med. 2018, 378, 2288–2301. [Google Scholar] [CrossRef]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T Cell Immunotherapy for Human Cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.Z.; Wang, Q.X.; Liu, Q.; Zheng, Y.D.; Zheng, C.X.; Yi, K.K.; Zhao, Y.; Gu, Y.; Wang, Y.; Wang, C.; et al. Dual-Locking Nanoparticles Disrupt the PD-1/PD-L1 Pathway for Efficient Cancer Immunotherapy. Adv. Mater. 2019, 31, 1905751. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.-Z.; Pollard, J.W. Macrophage Diversity Enhances Tumor Progression and Metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, J.; Hagemann, T. Tumour-Associated Macrophages and Cancer. Curr. Opin. Pharmacol. 2013, 13, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental Regulation of Tumor Progression and Metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Miao, W.M.; Wu, J.S.; Tong, Q.S.; Huang, Y.C.; Du, J.Z. Polymeric Materials for the Regulation of Tumor Immune Microenvironment. Acta Polym. Sin. 2020, 51, 548–558. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-Related Inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Anfray, C.; Ummarino, A.; Andón, F.T.; Allavena, P. Current Strategies to Target Tumor-Associated-Macrophages to Improve Anti-Tumor Immune Responses. Cells 2020, 9, 46. [Google Scholar] [CrossRef] [Green Version]
- Ruan, S.B.; Huang, Y.Y.; He, M.; Gao, H.L. Advanced Biomaterials for Cell-Specific Modulation and Restore of Cancer Immunotherapy. Adv. Sci. 2022, 9, 2200027. [Google Scholar] [CrossRef]
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-Associated Macrophages as Treatment Targets in Oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Chanmee, T.; Ontong, P.; Konno, K.; Itano, N. Tumor-Associated Macrophages as Major Players in the Tumor Microenvironment. Cancers 2014, 6, 1670–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balkwill, F.; Charles, K.A.; Mantovani, A. Smoldering and Polarized Inflammation in the Initiation and Promotion of Malignant Disease. Cancer Cell 2005, 7, 211–217. [Google Scholar] [CrossRef] [Green Version]
- Allavena, P.; Sica, A.; Solinas, G.; Porta, C.; Mantovani, A. The Inflammatory Micro-Environment in Tumor Progression: The Role of Tumor-Associated Macrophages. Crit. Rev. Oncol. /Hematol. 2008, 66, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional Polarization of Tumour-Associated Macrophages by Tumour-Derived Lactic Acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohn, T.; Rapp, S.; Luther, N.; Klein, M.; Bruehl, T.-J.; Kojima, N.; Lopez, P.A.; Hahlbrock, J.; Muth, S.; Endo, S.; et al. Tumor Immunoevasion via Acidosis-Dependent Induction of Regulatory Tumor-Associated Macrophages. Nat. Immunol. 2018, 19, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Qiao, S.L.; Ma, Y.; Wang, Y.; Lin, Y.X.; An, H.W.; Li, L.L.; Wang, H. General Approach of Stimuli-Induced Aggregation for Monitoring Tumor Therapy. ACS Nano 2017, 11, 7301–7311. [Google Scholar] [CrossRef]
- Yang, Z.W.; Zhang, M.N.; Peng, R.Y.; Liu, J.L.; Wang, F.; Li, Y.Z.; Zhao, Q.; Liu, J. The Prognostic and Clinicopathological Value of Tumor-Associated Macrophages in Patients with Colorectal Cancer: A Systematic Review and Meta-Analysis. Int. J. Color. Dis. 2020, 35, 1651–1661. [Google Scholar] [CrossRef]
- Zhao, X.X.; Qu, J.K.; Sun, Y.C.; Wang, J.Z.; Liu, X.; Wang, F.D.; Zhang, H.; Wang, W.; Ma, X.C.; Gao, X.Y.; et al. Prognostic Significance of Tumor-Associated Macrophages in Breast Cancer: A Meta-Analysis of the Literature. Oncotarget 2017, 8, 30576–30586. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.L.; Yu, A.H.; Ye, L.; Zhai, G.X. Research Progress in Tumor Targeted Immunotherapy. Expert Opin. Drug Deliv. 2021, 18, 1067–1090. [Google Scholar] [CrossRef]
- He, Y.Y.; de Araújo Júnior, R.F.; Cruz, L.J.; Eich, C. Functionalized Nanoparticles Targeting Tumor-Associated Macrophages as Cancer Therapy. Pharmaceutics 2021, 13, 1670. [Google Scholar] [CrossRef]
- Singh, Y.; Pawar, V.K.; Meher, J.G.; Raval, K.; Kumar, A.; Shrivastava, R.; Bhadauria, S.; Chourasia, M.K. Targeting Tumor Associated Macrophages (TAMs) via Nanocarriers. J. Control. Release 2017, 254, 92–106. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.X.; Song, S.Y.; Wu, P.Y.; Lyu, B.; Qin, M.M.; Sun, Y.N.; Sun, A.N.; Mu, L.M.; Xu, F.; Zhang, L.; et al. Tumor Associated Macrophages and TAMs-Based Anti-Tumor Nanomedicines. Adv. Healthc. Mater. 2021, 10, 2100590. [Google Scholar] [CrossRef] [PubMed]
- Ngambenjawong, C.; Gustafson, H.H.; Pun, S.H. Progress in Tumor-Associated Macrophage (TAM)-Targeted Therapeutics. Adv. Drug Deliv. Rev. 2017, 114, 206–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Zhang, Y. Tumor-Associated Macrophages: From Basic Research to Clinical Application. J. Hematol. Oncol. 2017, 10, 58. [Google Scholar] [CrossRef] [Green Version]
- Tian, L.L.; Yi, X.; Dong, Z.L.; Xu, J.; Liang, C.; Chao, Y.; Wang, Y.X.; Yang, K.; Liu, Z. Calcium Bisphosphonate Nanoparticles with Chelator-Free Radiolabeling to Deplete Tumor-Associated Macrophages for Enhanced Cancer Radioisotope Therapy. ACS Nano 2018, 12, 11541–11551. [Google Scholar] [CrossRef]
- Miller, M.A.; Zheng, Y.R.; Suresh, G.W.; Pfirschke, C.; Zope, H.; Engblom, C.; Kohler, R.H.; Iwamoto, Y.; Yang, K.S.; Askevold, B.; et al. Tumour-Associated Macrophages Act as A Slow-Release Reservoir of Nano-Therapeutic Pt(IV) Pro-Drug. Nat. Commun. 2015, 6, 8692. [Google Scholar] [CrossRef] [Green Version]
- D’Angelo, N.A.; Noronha, M.A.; Câmara, M.C.C.; Kurnik, I.S.; Feng, C.; Araujo, V.H.S.; Santos, J.H.P.M.; Feitosa, V.; Molino, J.V.D.; Rangel-Yagui, C.O.; et al. Doxorubicin Nanoformulations on Therapy Against Cancer: An Overview from the Last 10 Years. Biomater. Adv. 2022, 133, 112623. [Google Scholar] [CrossRef]
- Cagel, M.; Grotz, E.; Bernabeu, E.; Moretton, M.A.; Chiappetta, D.A. Doxorubicin: Nanotechnological Overviews from Bench to Bedside. Drug Discov. Today 2017, 22, 270–281. [Google Scholar] [CrossRef]
- Pugazhendhi, A.; Edison, T.N.J.I.; Velmurugan, B.K.; Jacob, J.A.; Karuppusamy, I. Toxicity of Doxorubicin (Dox) to Different Experimental Organ Systems. Life Sci. 2018, 200, 26–30. [Google Scholar] [CrossRef]
- Slingerland, M.; Guchelaar, H.J.; Gelderblom, H. Liposomal Drug Formulations in Cancer Therapy: 15 Years along the Road. Drug Discov. Today 2012, 17, 160–166. [Google Scholar] [CrossRef]
- van der Meel, R.; Sulheim, E.; Shi, Y.; Kiessling, F.; Mulder, W.J.M.; Lammers, T. Smart Cancer Nanomedicine. Nat. Nanotechnol. 2019, 14, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.K.; Tian, H.R.; Zhang, M.Z.; He, J.L.; Ni, P.H. Monoclonal Antibody-Conjugated Polyphosphoester-hyd-DOX Prodrug Nanoparticles for Targeted Chemotherapy of Liver Cancer Cells. Chin. J. Polym. Sci. 2021, 39, 1392–1402. [Google Scholar] [CrossRef]
- Li, S.D.; Huang, L. Stealth Nanoparticles: High Density but Sheddable PEG is a Key for Tumor Targeting. J. Control. Release 2010, 145, 178–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, B.; Biswas, S. Polymeric Micelles in Cancer Therapy: State of the Art. J. Control. Release 2021, 332, 127–147. [Google Scholar] [CrossRef]
- Jin, Q.; Deng, Y.Y.; Chen, X.H.; Ji, J. Rational Design of Cancer Nanomedicine for Simultaneous Stealth Surface and Enhanced Cellular Uptake. ACS Nano 2019, 13, 954–977. [Google Scholar] [CrossRef]
- Berger, M.; Degey, M.; Leblond Chain, J.; Maquoi, E.; Evrard, B.; Lechanteur, A.; Piel, G. Effect of PEG Anchor and Serum on Lipid Nanoparticles: Development of a Nanoparticles Tracking Method. Pharmaceutics 2023, 15, 597. [Google Scholar] [CrossRef]
- Foster, J.C.; Matson, J.B. Functionalization of Methacrylate Polymers with Thiooximes: A Robust Postpolymerization Modification Reaction and a Method for the Preparation of H2S-Releasing Polymers. Macromolecules 2014, 47, 5089–5095. [Google Scholar] [CrossRef]
- Fuchs, A.-K.; Syrovets, T.; Haas, K.A.; Loos, C.; Musyanovych, A.; Mailänder, V.; Landfester, K.; Simmet, T. Carboxyl- and Amino-Functionalized Polystyrene Nanoparticles Differentially Affect the Polarization Profile of M1 and M2 Macrophage Subsets. Biomaterials 2016, 85, 78–87. [Google Scholar] [CrossRef]
- Feng, X.R.; Li, D.; Han, J.D.; Zhuang, X.L.; Ding, J.X. Schiff Base Bond-Linked Polysaccharide-Doxorubicin Conjugate for Upregulated Cancer Therapy. Mater. Sci. Eng. C 2017, 76, 1121–1128. [Google Scholar] [CrossRef]
- Song, Y.; Li, D.; Zhang, M.Z.; He, J.L.; Ni, P.H. Facile Preparation of pH-Responsive PEGylated Prodrugs for Activated Intracellular Drug Delivery. Chin. Chem. Lett. 2019, 30, 2027–2031. [Google Scholar] [CrossRef]
- Choimet, M.; Hyoung-Mi, K.; Jae-Min, O.; Tourrette, A.; Drouet, C. Nanomedicine: Interaction of Biomimetic Apatite Colloidal Nanoparticles with Human Blood Components. Colloids Surf. B 2016, 145, 87–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zielinski, C.; Mele, F.; Aschenbrenner, D.; Jarrossay, D.; Ronchi, F.; Gattorno, F.; Monticelli, S.; Lanzavecchia, A.; Sallusto, F. Pathogen-Induced Human TH17 Cells Produce IFN-γ or IL-10 and are Regulated by IL-1β. Nature 2012, 484, 514–518. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | N3-PEG-Br: PFBEMA (Feed Ratio m1:m2) | Polymer: Drug (Feed Ratio m3:m4) | Drug Loading (wt%) |
|---|---|---|---|
| N3-PEG-b-(PFBEMA-DOX)-1 | 1.0:0.6 | 100:20 | 2.4 |
| N3-PEG-b-(PFBEMA-DOX)-2 | 1.0:0.8 | 100:20 | 6.2 |
| N3-PEG-b-(PFBEMA-DOX)-3 | 1.0:1.0 | 100:20 | 6.8 |
| N3-PEG-b-(PFBEMA-DOX)-4 | 1.0:1.2 | 100:20 | 11.9 |
| Samples | CD163 mAb Added (μg·mg−1) | CD163 mAb Modified (μg·mg−1) | Modified Ratio (%) |
|---|---|---|---|
| mAb-CD163-PDNPs-1 | 2.56 | 2.27 | 88.6 |
| mAb-CD163-PDNPs-2 | 1.28 | 1.15 | 90.1 |
| mAb-CD163-PDNPs-3 | 0.64 | 0.59 | 92.9 |
| mAb-CD163-PDNPs-4 | 0.32 | 0.30 | 93.7 |
| IC50 | H22 (mg·L−1) | M2 (mg·L−1) |
|---|---|---|
| Free DOX | 0.166 | 0.235 |
| PDNPs | 0.367 | 0.860 |
| mAb-CD163-PDNPs | 0.340 | 0.541 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Z.; Li, H.; Zhang, W.; Zhang, M.; He, J.; Yu, Z.; Sun, X.; Ni, P. CD163 Monoclonal Antibody Modified Polymer Prodrug Nanoparticles for Targeting Tumor-Associated Macrophages (TAMs) to Enhance Anti-Tumor Effects. Pharmaceutics 2023, 15, 1241. https://doi.org/10.3390/pharmaceutics15041241
Yang Z, Li H, Zhang W, Zhang M, He J, Yu Z, Sun X, Ni P. CD163 Monoclonal Antibody Modified Polymer Prodrug Nanoparticles for Targeting Tumor-Associated Macrophages (TAMs) to Enhance Anti-Tumor Effects. Pharmaceutics. 2023; 15(4):1241. https://doi.org/10.3390/pharmaceutics15041241
Chicago/Turabian StyleYang, Zun, Haijiao Li, Wenrui Zhang, Mingzu Zhang, Jinlin He, Zepeng Yu, Xingwei Sun, and Peihong Ni. 2023. "CD163 Monoclonal Antibody Modified Polymer Prodrug Nanoparticles for Targeting Tumor-Associated Macrophages (TAMs) to Enhance Anti-Tumor Effects" Pharmaceutics 15, no. 4: 1241. https://doi.org/10.3390/pharmaceutics15041241