

Approaches to Assure Similarity between Pharmaceutical Heparins from Two Different Manufacturers

 ,
,
Abstract
:
1. Introduction
2. Material and Methods
2.1. Heparins and Standards
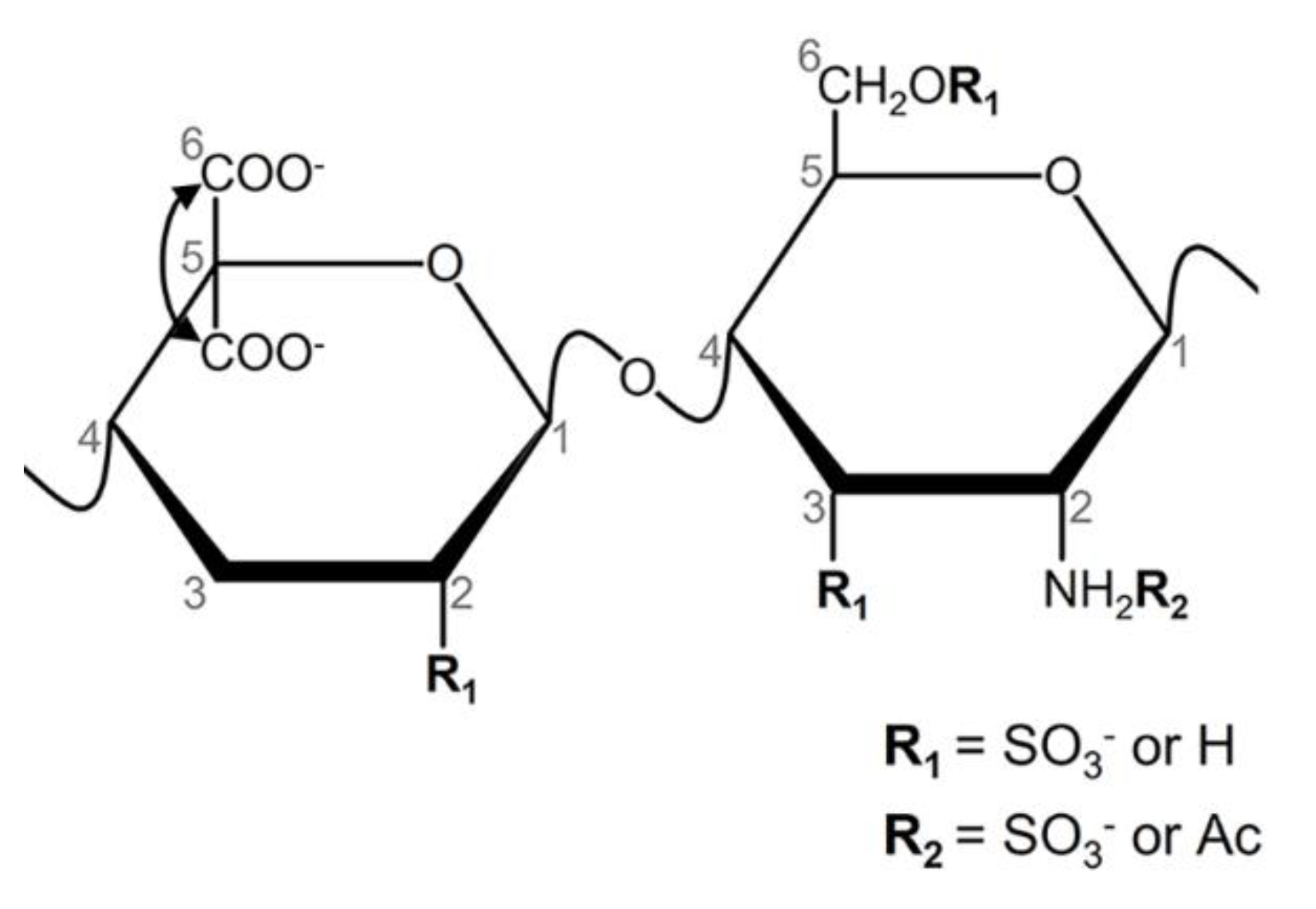
2.2. Biochemical Assays
2.3. Ion-Exchange Chromatography
2.4. Size-Exclusion Chromatography
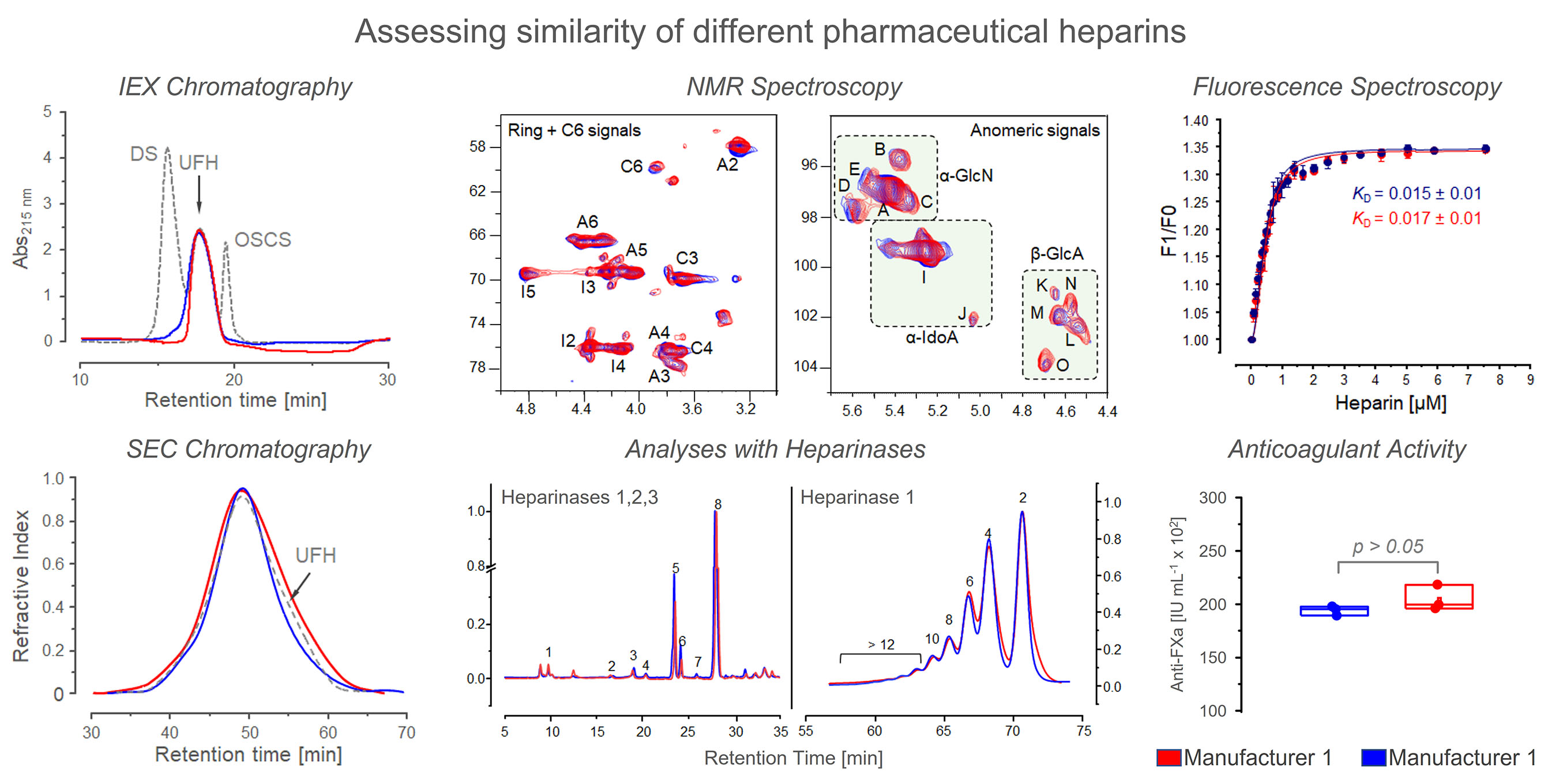
2.5. NMR Spectrometry
2.6. Disaccharide Analyses with SAX-HPLC Chromatography
2.7. Oligosaccharide Analyses with Size-Exclusion Chromatography
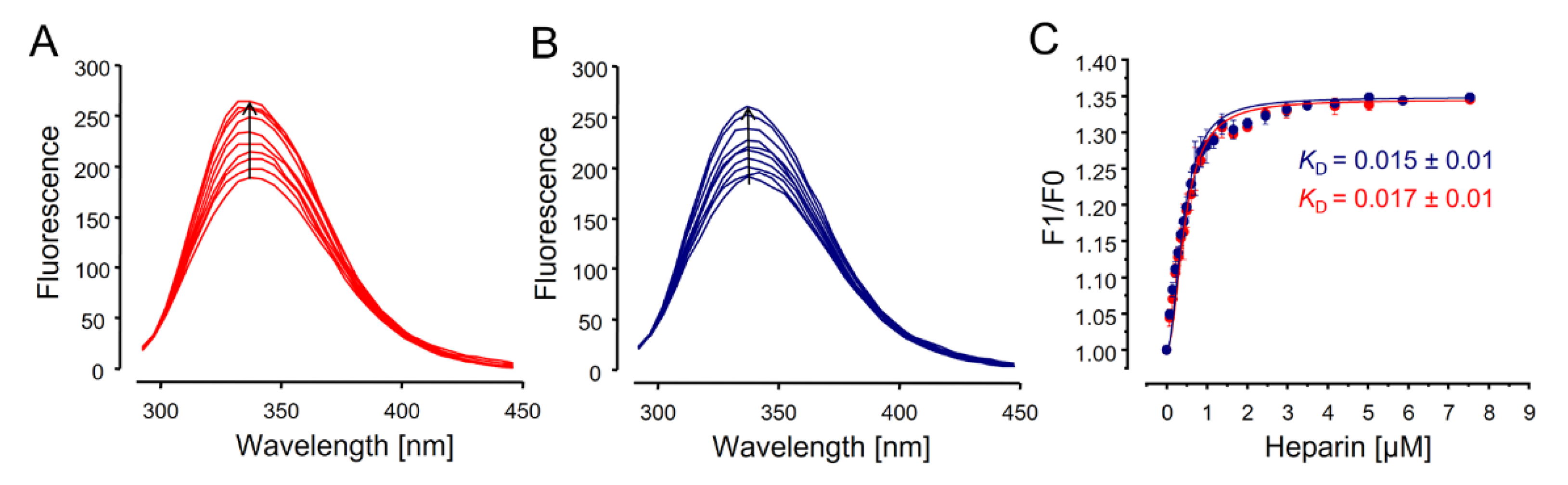
2.8. Fluorescence Spectroscopy to Determine Heparin–Antithrombin Interactions
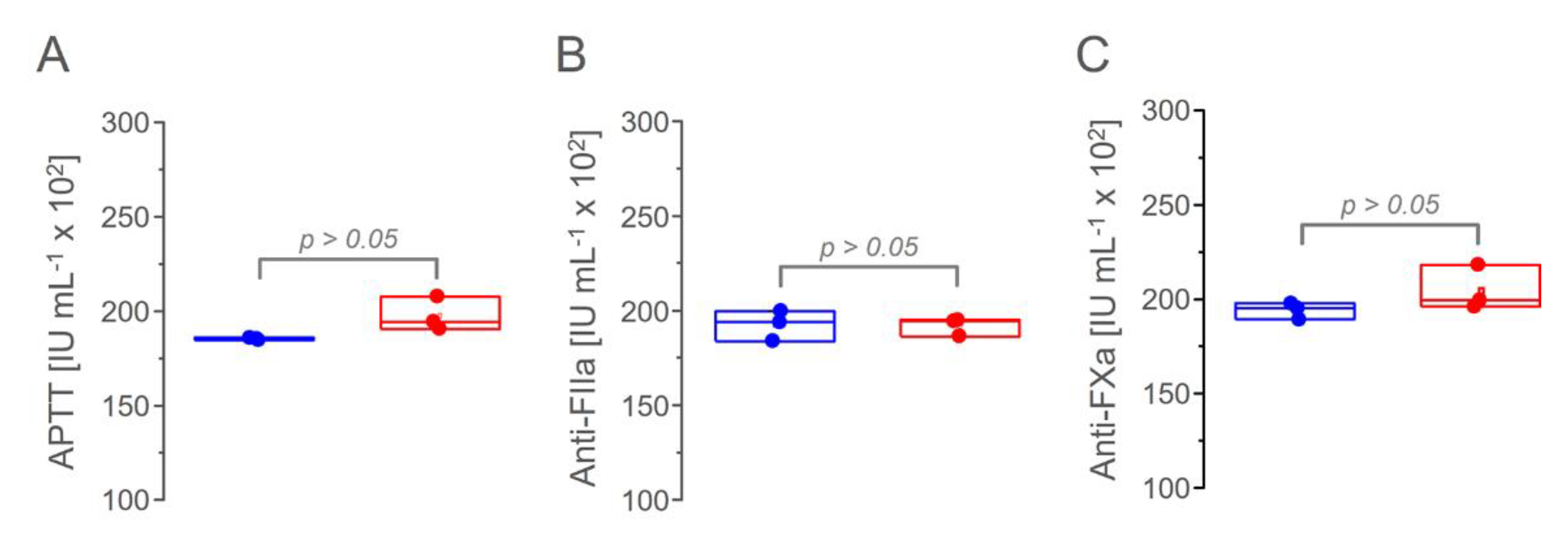
2.9. Activated Partial Thromboplastin Time Assay (APTT)
2.10. Anti-FXa and Anti-FIIa Activities
3. Results and Discussion
3.1. Biochemical Methods
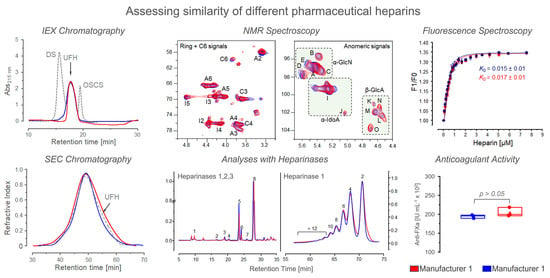
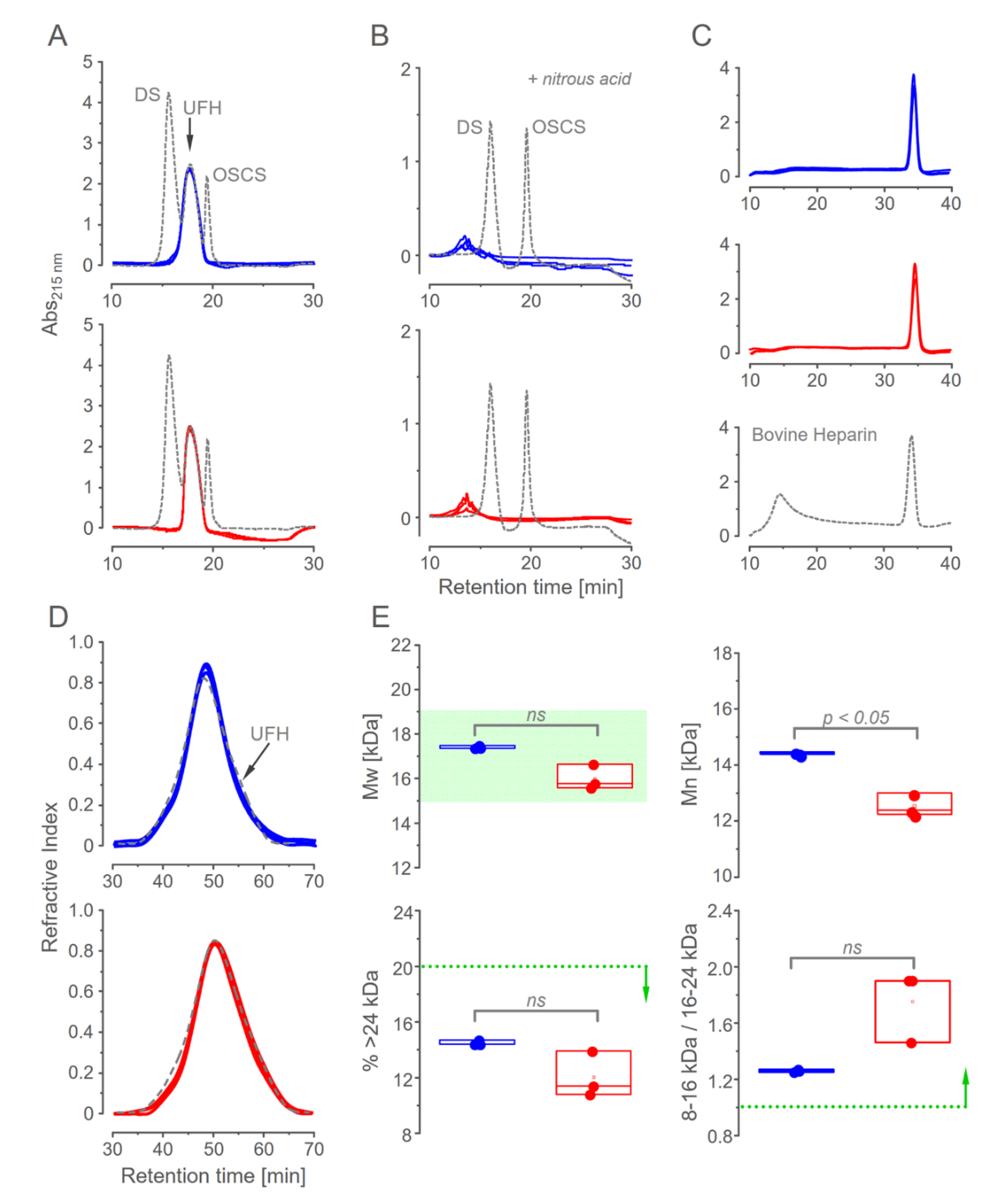
3.2. Chromatographic Analysis
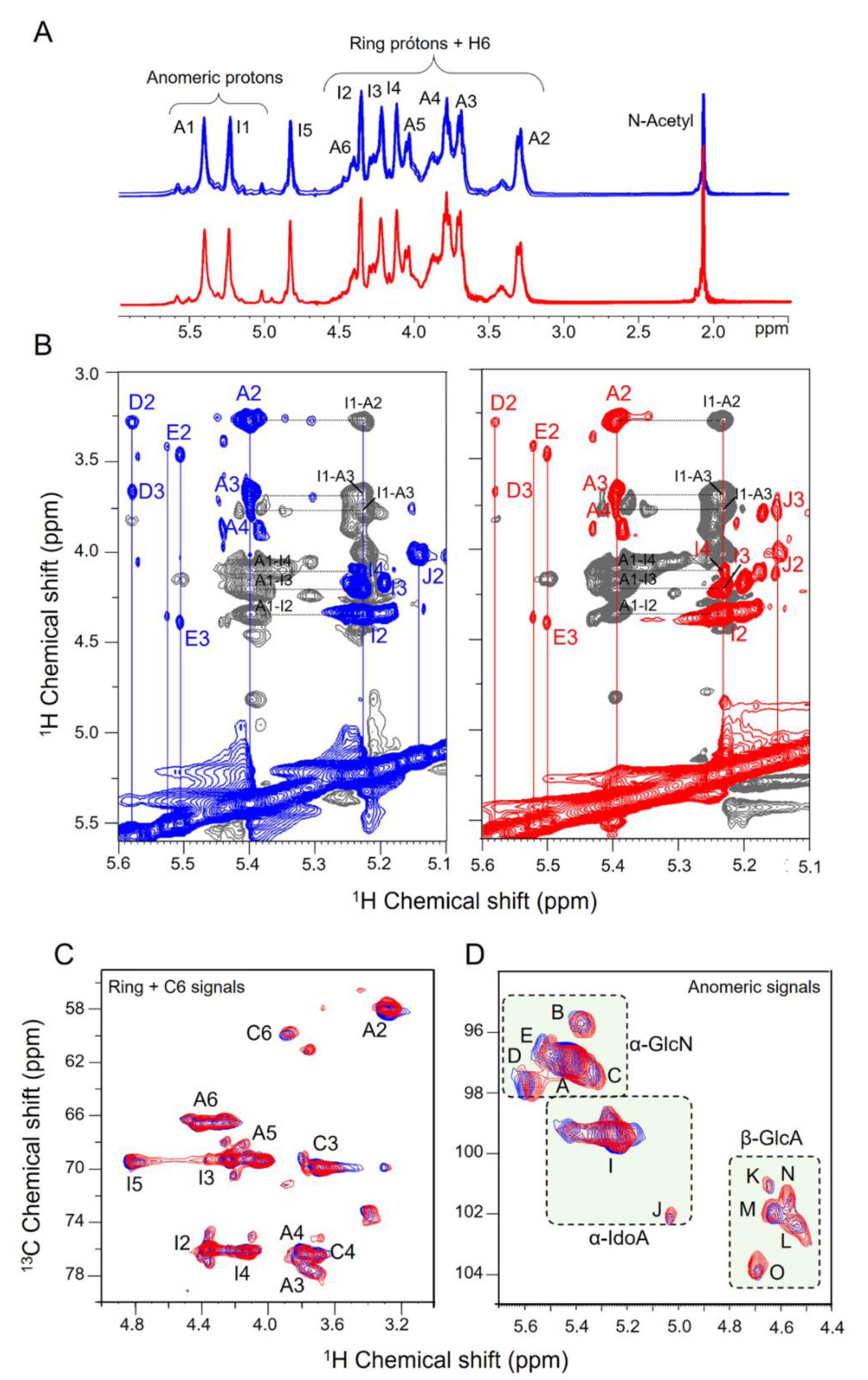
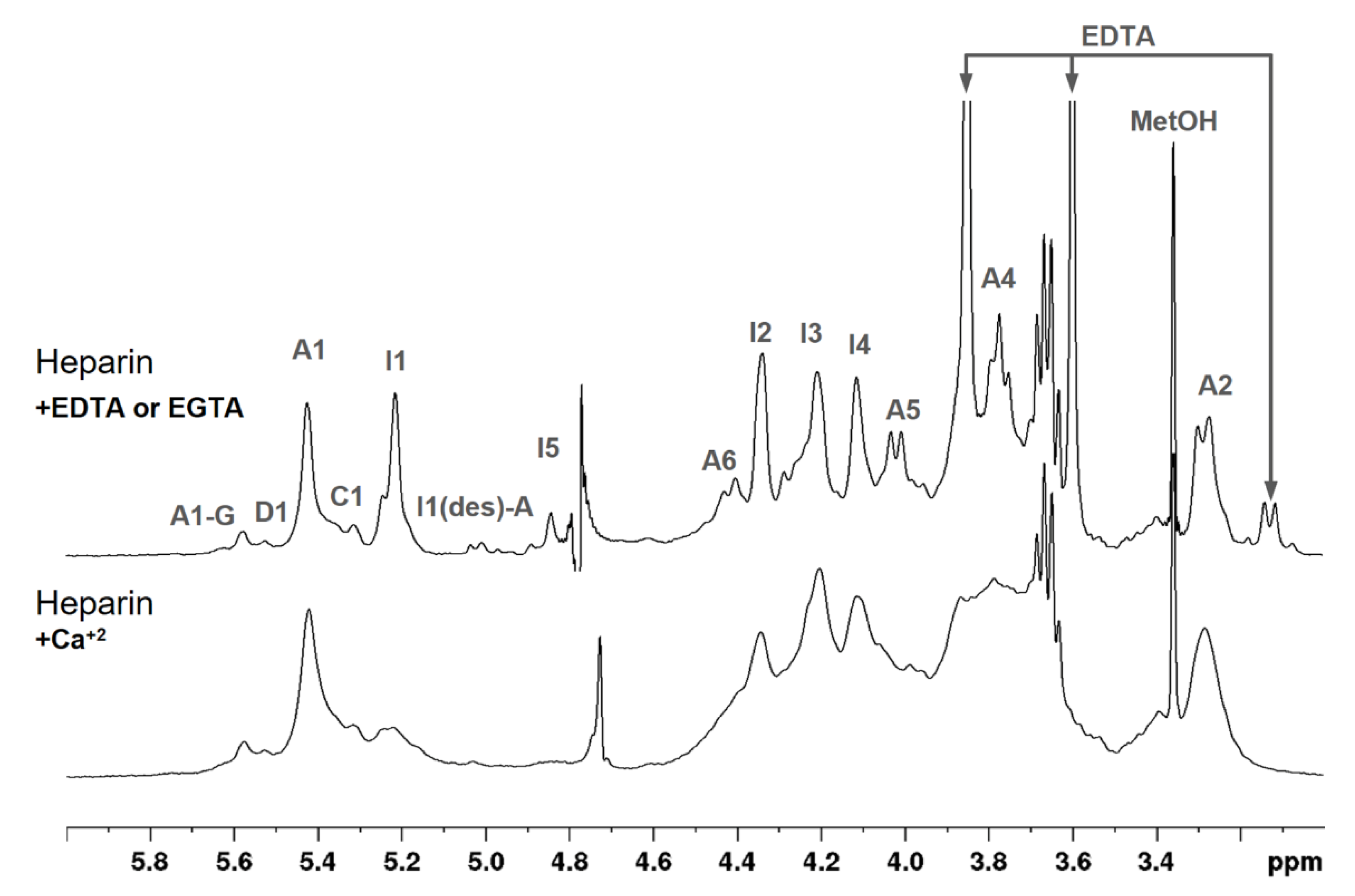
3.3. NMR Spectroscopy
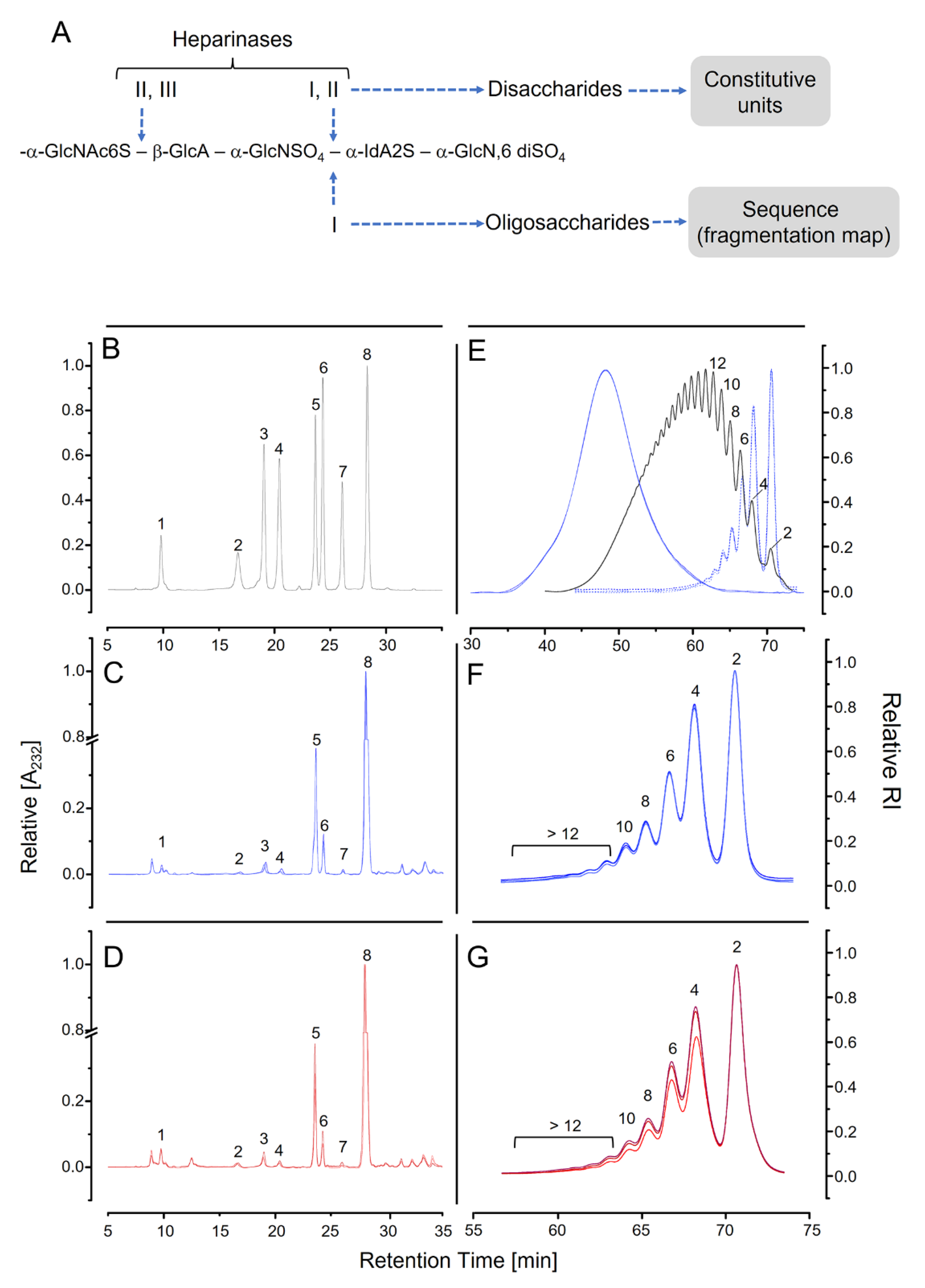
3.4. Analysis Based on Digestion with Heparinases
3.5. Fluorescence Spectroscopy
3.6. Anticoagulant Activity
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Casu, B.; Naggi, A.; Torri, G. Re-visiting the structure of heparin. Carbohydr. Res. 2015, 403, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Mulloy, B.; Hogwood, J.; Gray, E.; Lever, R.; Page, C.P. Pharmacology of heparin and related drugs. Pharmacol. Rev. 2016, 68, 76–141. [Google Scholar] [CrossRef] [PubMed]
- Torri, G.; Naggi, A. Heparin centenary—An ever-young life-saving drug. Int. J. Cardiol. 2016, 212 (Suppl. S1), S1–S4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunaratne, R.; Kumar, S.; Frederiksen, J.W.; Stayrook, S.; Lohrmann, J.L.; Perry, K.; Bompiani, K.M.; Chabata, C.V.; Thalji, N.K.; Ho, M.D.; et al. Combination of aptamer and drug for reversible anticoagulation in cardiopulmonary bypass. Nat. Biotechnol. 2018, 36, 606–613. [Google Scholar] [CrossRef]
- Hogwood, J.; Mulloy, B.; Lever, R.; Gray, E.; Page, C.P. Pharmacology of Heparin and Related Drugs: An Update. Pharmacol. Rev. 2023, 75, 328–379. [Google Scholar] [CrossRef]
- Beurskens, D.M.H.; Huckriede, J.P.; Schrijver, R.; Hemker, H.C.; Reutelingsperger, C.P.; Nicolaes, G.A.F. The Anticoagulant and Nonanticoagulant Properties of Heparin. Thromb. Haemost. 2020, 120, 1371–1383. [Google Scholar] [CrossRef]
- Spyropoulos, A.C.; Goldin, M.; Giannis, D.; Diab, W.; Wang, J.; Khanijo, S.; Mignatti, A.; Gianos, E.; Cohen, M.; Sharifova, G.; et al. Efficacy and Safety of Therapeutic-Dose Heparin vs Standard Prophylactic or Intermediate-Dose Heparins for Thromboprophylaxis in High-risk Hospitalized Patients With COVID-19: The HEP-COVID Randomized Clinical Trial. JAMA Intern. Med. 2021, 181, 1612–1620. [Google Scholar] [CrossRef]
- van der Meer, J.Y.; Kellenbach, E.; van der Bos, L.J. From Farm to Pharma: An Overview of Industrial Heparin Manufacturing Methods. Molecules 2017, 22, E1025. [Google Scholar] [CrossRef] [Green Version]
- Perlin, A.S.; Mazurek, M.; Jaques, L.B.; Kavanagh, L.W. A proton magnetic resonance spectral study of heparin. Carbohydr. Res. 1968, 7, 369–379. [Google Scholar] [CrossRef]
- Tovar, A.M.F.; Vairo, B.C.; Oliveira, S.M.C.G.; Glauser, B.F.; Santos, G.R.C.; Capillé, N.V.; Piquet, A.A.; Santana, P.S.; Micheli, K.V.A.; Pereira, M.S.; et al. Converting the Distinct Heparins Sourced from Bovine or Porcine Mucosa into a Single Anticoagulant Drug. Thromb. Haemost. 2019, 119, 618–632. [Google Scholar] [CrossRef]
- Vilanova, E.; Vairo, B.C.; Oliveira, S.M.C.G.; Glauser, B.F.; Capillé, N.V.; Santos, G.R.C.; Tovar, A.M.F.; Pereira, M.S.; Mourão, P.A.S. Heparins Sourced from Bovine and Porcine Mucosa Gain Exclusive Monographs in the Brazilian Pharmacopeia. Front. Med. 2019, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, S.N.M.C.G.; Tovar, A.M.F.; Bezerra, F.F.; Piquet, A.A.; Capillé, N.V.; Santos, P.S.; Vilanova, E.; Mourão, P.A.S. Anticoagulant Activity of Heparins from Different Animal Sources are Driven by a Synergistic Combination of Physical-chemical Factors. TH Open. 2022, 6, e309–e322. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Kurgan, L. Survey of Similarity-Based Prediction of Drug-Protein Interactions. Curr. Med. Chem. 2020, 27, 5856–5886. [Google Scholar] [CrossRef] [PubMed]
- Tovar, A.M.F.; Santos, G.R.C.; Capillé, N.V.; Piquet, A.A.; Glauser, B.F.; Pereira, M.S.; Vilanova, E.; Mourão, P.A.S. Structural and haemostatic features of pharmaceutical heparins from different animal sources: Challenges to define thresholds separating distinct drugs. Sci. Rep. 2016, 6, 35619. [Google Scholar] [CrossRef]
- Madhu, M.; Tseng, W.L. Recent developments in sensing of oversulfated chondroitin sulfate in heparin. A review. J. Food Drug. Anal. 2021, 29, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, M.; Zhang, Z.; Shriver, Z.; Naggi, A.; Masuko, S.; Langer, R.; Casu, B.; Linhardt, R.J.; Torri, G.; Sasisekharan, R. Orthogonal analytical approaches to detect potential contaminants in heparin. Proc. Natl. Acad. Sci. USA 2009, 106, 16956–16961. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Li, B.; Suwan, J.; Zhang, F.; Wang, Z.; Liu, H.; Mulloy, B.; Linhardt, R.J. Analysis of pharmaceutical heparins and potential contaminants using (1)H-NMR and PAGE. J. Pharm. Sci. 2009, 98, 4017–4026. [Google Scholar] [CrossRef]
- Li, B.; Suwan, J.; Martin, J.G.; Zhang, F.; Zhang, Z.; Hoppensteadt, D.; Clark, M.; Fareed, J.; Linhardt, R.J. Oversulfated chondroitin sulfate interaction with heparin-binding proteins: New insights into adverse reactions from contaminated heparins. Biochem. Pharmacol. 2009, 78, 292–300. [Google Scholar] [CrossRef] [Green Version]
- Goligher, E.C.; Bradbury, C.A.; McVerry, B.J.; Lawler, P.R.; Berger, J.S.; Gong, M.N.; Carrier, M.; Reynolds, H.R.; Kumar, A.; Turgeon, A.F.; et al. Therapeutic Anticoagulation with Heparin in Critically Ill Patients with COVID-19. N. Engl. J. Med. 2021, 385, 777–789. [Google Scholar]
- Vilanova, E.; Glauser, B.F.; Oliveira, S.M.; Tovar, A.M.; Mourão, P.A. Update on Brazilian biosimilar enoxaparins. Expert. Rev. Hematol. 2016, 9, 1015–1021. [Google Scholar] [CrossRef]
- FDA Response to Citizen Petition, Docket No. FDA-2003-P-0273. Washington DC: US Food and Drug Administration. 2010. Available online: http://www.fda.gov/downloads/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/UCM220083.pdf (accessed on 5 November 2022).
- Bowness, J.M. Application of the carbazole reaction to the estimation of glucuronic acid and glucose in some acidic polysaccharides and in urine. Biochem. J. 1957, 67, 295–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, J.M. The bicinchoninic acid (BCA) assay for protein quantitation. Methods Mol. Biol. 1994, 32, 5–8. [Google Scholar] [PubMed]
- Santos, G.R.; Piquet, A.A.; Glauser, B.F.; Tovar, A.M.; Pereira, M.S.; Vilanova, E.; Mourão, P.A. Systematic Analysis of Pharmaceutical Preparations of Chondroitin Sulfate Combined with Glucosamine. Pharmaceuticals 2017, 10, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olson, S.T.; Björk, I.; Shore, J.D. Kinetic characterization of heparin-catalyzed and uncatalyzed inhibition of blood coagulation proteinases by antithrombin. Methods Enzymol. 1993, 222, 525–559. [Google Scholar]
- U.S. Pharmacopoeia-National Formulary. Heparin Sodium Monograph: USP41-NF36; USP Convention: Rockville, MD, USA, 2018. [Google Scholar]
- European Pharmacopoeia. Heparin Sodium Monograph: EP, 9th ed.; EDQM Commission: Strasbourg, France, 2018. [Google Scholar]
- Brazilian Pharmacopoeia. Bovine Heparin Sodium Monograph: Brazilian Pharmacopeia, 5th ed.; 2nd Suppl.; ANVISA: Brasilia, Brazil, 2017. [Google Scholar]
- Matanović, M.R.; Grabnar, I.; Grabnar, P.A.; Roškar, R. Development and validation of a simple and sensitive size-exclusion chromatography method for quantitative determination of heparin in pharmaceuticals. Acta Pharm. 2015, 65, 43–52. [Google Scholar] [CrossRef] [Green Version]
- Kawai, Y.; Seno, N.; Anno, K. Chondroitin polysulfate of squid cartilage. J. Biochem. 1966, 60, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Devlin, A.J.; Mycroft-West, C.J.; Tunbull, J.E.; Lima, M.A.; Guerrini, M.; Yates, E.A.; Skidmore, M.A. Analysis of Heparin Samples by Attenuated Total Reflectance Fourier-transform Infrared Spectroscopy in the Solid State. ACS Cent. Sci. 2023, 3, 381–392. [Google Scholar] [CrossRef]
- Mourão, P.A.; Vilanova, E.; Soares, P.A. Unveiling the structure of sulfated fucose-rich polysaccharides via nuclear magnetic resonance spectroscopy. Curr. Opin. Struct. Biol. 2018, 50, 33–41. [Google Scholar] [CrossRef]
- Ouyang, Y.; Han, X.; Yu, Y.; Chen, J.; Fu, L.; Zhang, F.; Linhardt, R.J.; Fareed, J.; Hoppensteadt, D.; Jeske, W.; et al. Chemometric analysis of porcine, bovine and ovine heparins. J. Pharm. Biomed. Anal. 2019, 164, 345–352. [Google Scholar] [CrossRef]
- Colombo, E.; Mauri, L.; Marinozzi, M.; Rudd, T.R.; Yates, E.A.; Ballabio, D.; Guerrini, M. NMR spectroscopy and chemometric models to detect a specific non-porcine ruminant contaminant in pharmaceutical heparin. J. Pharm. Biomed. Anal. 2022, 214, 114724. [Google Scholar] [CrossRef]
- Bax, A.; Davis, D.G. MLEV-17-based two-dimensional homonuclear magnetization transfer spectroscopy. J. Magn. Reson. 1985, 65, 355–360. [Google Scholar] [CrossRef]
- Mauri, L.; Marinozzi, M.; Phatak, N.; Karfunkle, M.; St Ange, K.; Guerrini, M.; Keire, D.A.; Linhardt, R.J. 1D and 2D-HSQC NMR: Two Methods to Distinguish and Characterize Heparin From Different Animal and Tissue Sources. Front. Med. 2019, 6, 142. [Google Scholar] [CrossRef] [PubMed]
- Mauri, L.; Boccardi, G.; Torri, G.; Karfunkle, M.; Macchi, E.; Muzi, L.; Keire, D.; Guerrini, M. Qualification of HSQC methods for quantitative composition of heparin and low molecular weight heparins. J. Pharm. Biomed. Anal. 2017, 136, 92–105. [Google Scholar] [CrossRef]
- Mourier, P. Heparinase Digestion of 3-O-Sulfated Sequences: Selective Heparinase II Digestion for Separation and Identification of Binding Sequences Present in ATIII Affinity Fractions of Bovine Intestinal Heparins. Front. Med. 2022, 9, 841726. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, H.; Futatsumori, H.; Suzuki, E.; Kimata, K. A quantitative method to detect non-antithrombin-binding 3-O-sulfated units in heparan sulfate. J. Biol. Chem. 2021, 296, 100115. [Google Scholar] [CrossRef]
- Spelta, F.; Liverani, L.; Peluso, A.; Marinozzi, M.; Urso, E.; Guerrini, M.; Naggi, A. SAX-HPLC and HSQC NMR Spectroscopy: Orthogonal Methods for Characterizing Heparin Batches Composition. Front. Med. 2019, 6, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasisekharan, R.; Venkataraman, G.; Godavarti, R.; Ernst, S.; Cooney, C.L.; Langer, R. Heparinase I from Flavobacterium heparinum. Mapping and characterization of the heparin binding domain. J. Biol. Chem. 1996, 271, 3124–3131. [Google Scholar] [CrossRef] [Green Version]
- Gallus, A.S.; Coghlan, D.W. Heparin pentasaccharide. Curr. Opin. Hematol. 2002, 9, 422–429. [Google Scholar] [CrossRef]
- Einarsson, R.; Andersson, L.O. Binding of heparin to human antithrombin III as studied by measurements of tryptophan fluorescence. Biochim. Biophys. Acta 1977, 490, 104–111. [Google Scholar] [CrossRef]
- Shao, M.; Wang, H.F. Heparin-mediated fluorescence anisotropy assay of antithrombin based on polyethyleneimine capped Mn-doped ZnS quantum dots. Analyst 2013, 138, 4618–4623. [Google Scholar] [CrossRef]
- Burmistrova, N.A.; Diehl, B.W.K.; Soboleva, P.M.; Rubtsova, E.; Legin, E.A.; Legin, A.V.; Kirsanov, D.O.; Monakhova, Y.B. Quality Control of Heparin Injections: Comparison of Four Established Methods. Anal. Sci. 2020, 36, 1467–1472. [Google Scholar] [CrossRef] [PubMed]
- Keire, D.; Mulloy, B.; Chase, C.; Al-Hakim, A.; Cairatti, D.C.; Gray, E.; Hogwood, J.; Morris, T.; Mourão, P.A.S.; Soares, M.L.C.; et al. Diversifying the global heparin supply chain. Reintroduction of bovine heparin in the United States? Pharm Technol. 2015, 39, 28–35. [Google Scholar]
- Vilanova, E.; Tovar, A.M.F.; Mourão, P.A.S. Imminent risk of a global shortage of heparin caused by the African Swine Fever afflicting the Chinese pig herd. J. Thromb. Haemost. 2018, 17, 254–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Criterium | Compendial Assays | In-Depth Approaches |
|---|---|---|
| (1) Absence of contaminants | Biochemical assays IEX-HPLC 1D 1H-NMR | IEX-HPLC/nitrous acid deamination |
| (2) Equivalence of molecular mass | SEC-HPLC | - |
| (3) Animal source sameness | IEX-HPLC 1D 1H-NMR | Disaccharide analysis/IEX-HPLC/heparinases Fragmentation map/SEC-HPLC/heparinase I |
| (4) Exclude mixtures of heparins from different animal sources | 1D 1H-NMR | 2D 1H-13C HSQC NMR Disaccharide analysis/IEX-HPLC/heparinases |
| (5) Equivalence of the disaccharide composition | - | 2D 1H-13C HSQC NMR Disaccharide analysis/IEX-HPLC/heparinases |
| (6) Equivalence of minor components | - | 2D 1H-13C HSQC NMR 2D 1H-1H TOCSY NMR Disaccharide analysis/IEX-HPLC/heparinases Affinity heparin-AT/Fluorescence spectroscopy |
| (7) Equivalence in the sequence of residues | - | 2D 1H-13C HSQC NMR 2D 1H-1H TOCSY NMR Fragmentation map/SEC-HPLC/heparinase I |
| (8) Equivalence in biological assays | Anti-FXa activity assay Anti-FIIa activity assay | Fluorescence spectroscopy/Affinity heparin-AT |
| Heparin | Batch | Signal (Structure) 1 | |||
|---|---|---|---|---|---|
| A1 (α-GlcN,6-diS) | C1 (α-GlcNS) | I1 (α-IdoA2S) | C1 × 100/A1 2 | ||
| Manufaturer 1 | 1 | 1.00 | 0.09 | 0.83 | 9 |
| 2 | 1.00 | 0.10 | 0.82 | 10 | |
| 3 | 1.00 | 0.10 | 0.83 | 10 | |
| Mean ± SD 3 | 1 ± 0 | 0.09 ± 0.005 | 0.83 ± 0.005 | 10 ± 0.5 | |
| Manufacturer 2 | 4 | 1.00 | 0.11 | 0.80 | 11 |
| 5 | 1.00 | 0.10 | 0.81 | 10 | |
| 6 | 1.00 | 0.10 | 0.84 | 10 | |
| Mean ± DP 3 | 1 ± 0 | 0.10 ± 0.005 | 0.82 ± 0.02 | 10 ± 0.5 | |
| Unit 1 | Proportion % | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Manufacturer 1 | Manufacturer 2 | ||||||||
| 1 | 2 | 3 | ± dp | 4 | 5 | 6 | ± dp | ||
| α-GlcN: | |||||||||
| E | α-GlcN,3,6-triS-(?) | 5.2 | 5.1 | 5.4 | 5.2 ± 0.2 | 5.7 | 5.5 | 5.6 | 5.6 ± 0.1 |
| A-a | α-GlcN,6-diS-(α-IdoA2S) | 66.5 | 66.1 | 61.5 | 64.7 ± 2.8 | 60.4 | 61.7 | 62.2 | 61.4 ± 0.9 |
| A-b | α-GlcNAc-(β-GlcA) | 3.8 | 4.2 | 4.3 | 4.1 ± 0.3 | 6.1 | 6.1 | 7.5 | 6.6 ± 0.8 |
| B | α-GlcNS-(α-IdoA) | 6.9 | 4.9 | 7.1 | 6.3 ± 1.2 | 8.2 | 7.0 | 9.2 | 8.1 ± 1.1 |
| D | α-GlcNS-(β-GlcA) | 9.1 | 11.3 | 14.0 | 11.5 ± 2.4 | 10.6 | 12.6 | 7.5 | 10.2 ± 2.6 |
| C | α-GlcNS-(α-IdoA2S) | 8.6 | 8.4 | 7.8 | 8.3 ± 0.4 | 9.0 | 7.0 | 8.0 | 8.0 ± 1.0 |
| ∑ α-GlcN | 100 | 100 | 100 | 100.0 | 100 | 100 | 100 | 100.0 | |
| α-IdoA: | |||||||||
| I | α-IdoA2S | 79.4 | 77.9 | 81.0 | 79.4 ± 1.6 | 77.2 | 76.6 | 77.6 | 77.1 ± 0.5 |
| J | α-IdoA-(GlcN,6S) | 2.1 | 3.2 | 3.0 | 2.8 ± 0.6 | 1.6 | 2.1 | 1.6 | 1.8 ± 0.3 |
| - | α-IdoA | 1.5 | 0.9 | 1.1 | 1.2 ± 0.3 | 1.0 | 1.4 | 1.1 | 1.2 ± 0.2 |
| ∑ α-IdoA | 83.0 | 82.1 | 85.1 | 83.4 ± 1.5 | 79.8 | 80.0 | 80.3 | 80.0 ± 0.3 | |
| β-GlcA: | |||||||||
| K | β-GlcA-(α-GlcN,3,6-triS) | 3.0 | 2.4 | 2.6 | 2.7 ± 0.3 | 1.4 | 1.2 | 2.1 | 1.6 ± 0.5 |
| L | β-GlcA-(α-GlcNAc) | 6.9 | 8.0 | 4.7 | 6.5 ± 1.7 | 11.4 | 11.1 | 10.6 | 11.0 ± 0.4 |
| M | β-GlcA-(α-GlcNS) | 7.1 | 7.5 | 7.7 | 7.4 ± 0.3 | 7.5 | 7.7 | 7.0 | 7.4 ± 0.4 |
| ∑ β-GlcA | 17.0 | 17.9 | 14.9 | 16.6 ± 1.5 | 20.2 | 20.0 | 19.7 | 20.0 ± 0.3 | |
| α-GlcN | N-sulfatation 2 | 96.3 | 95.8 | 95.7 | 95.9 ± 0.3 | 93.9 | 93.9 | 92.5 | 93.4 ± 0.8 |
| 6-Sulfatation 3 | 87.4 | 86.2 | 84.1 | 85.9 ± 1.7 | 86.3 | 84.6 | 85 | 85.0 ± 0.9 | |
| 3-Sulfatation 2 | 5.2 | 5.1 | 5.4 | 5.2 ± 0.2 | 5.7 | 5.5 | 5.6 | 5.6 ± 0.1 | |
| N-Acetylation 2 | 3.8 | 4.2 | 4.3 | 4.1 ± 1.6 | 6.1 | 6.1 | 7.5 | 6.6 ± 0.5 | |
| α-IdoA | 2-Sulfatation 2 | 79.4 | 77.9 | 81.0 | 79.4 ± 0.3 | 77.2 | 76.6 | 77.6 | 77.1 ± 0.8 |
| Disaccharides 1 | Manufacturer 1 | Manufacturer 2 | ||||||
|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | Mean ± SD | 4 | 5 | 6 | Mean ± SD | |
| 1: ΔUA-GlcNAc | 1.7 | 1.1 | 1.0 | 1.2 ± 0.39 | 4.0 | 3.4 | 3.1 | 3.5 ± 0.45 |
| 2: ΔUA-GlcNS | 0.7 | 0.6 | 0.7 | 0.6 ± 0.07 | 1.7 | 1.4 | 1.3 | 1.5 ± 0.22 |
| 3: ΔUA-GlcNAc,6S | 2.5 | 2.4 | 2.6 | 2.5 ± 0.09 | 2.8 | 2.7 | 3.0 | 2.8 ± 0.17 |
| 4: ΔUA2S-GlcNAc | 1.3 | 0.8 | 1.1 | 1.0 ± 0.23 | 1.3 | 1.4 | 1.3 | 1.4 ± 0.06 |
| 5: ΔUA-GlcNS,6S | 18.5 | 18.7 | 18.8 | 18.7 ± 0.12 | 13.8 | 11.8 | 15.8 | 13.8 ± 2.00 |
| 6: ΔUA2S-GlcNS | 5.1 | 4.6 | 4.7 | 4.8 ± 0.29 | 3.7 | 3.4 | 4.6 | 3.9 ± 0.65 |
| 7: ΔUA2S-GlcNAc,6S | 0.9 | 0.7 | 0.7 | 0.8 ± 0.08 | 0.9 | 0.6 | 1.1 | 0.9 ± 0.27 |
| 8: ΔUA2S-GlcNS,6S | 69.3 | 71.1 | 70.5 | 70.5 ± 0.94 | 71.8 | 75.3 | 69.7 | 72.3 ± 2.82 |
| 2-sulfation | 76.5 | 77.2 | 77.0 | 76.9 ± 0.35 | 77.7 | 80.7 | 76.8 | 78.4 ± 2.05 |
| 6-sulfation | 91.2 | 92.9 | 92.6 | 92.3 ± 0.91 | 89.3 | 90.4 | 89.7 | 89.7 ± 0.53 |
| N-sulfation | 93.7 | 95.0 | 97.1 | 94.7 ± 0.70 | 90.9 | 91.9 | 91.5 | 91.4 ± 0.47 |
| N-acetylation | 6.3 | 5.0 | 5.3 | 5.6 ± 0.70 | 9.1 | 8.1 | 8.5 | 8.6 ± 0.47 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bezerra, F.F.; Oliveira, S.N.M.C.G.; Sales, R.A.; Piquet, A.A.; Capillé, N.V.; Vilanova, E.; Tovar, A.M.F.; Mourão, P.A.S. Approaches to Assure Similarity between Pharmaceutical Heparins from Two Different Manufacturers. Pharmaceutics 2023, 15, 1115. https://doi.org/10.3390/pharmaceutics15041115
Bezerra FF, Oliveira SNMCG, Sales RA, Piquet AA, Capillé NV, Vilanova E, Tovar AMF, Mourão PAS. Approaches to Assure Similarity between Pharmaceutical Heparins from Two Different Manufacturers. Pharmaceutics. 2023; 15(4):1115. https://doi.org/10.3390/pharmaceutics15041115
Chicago/Turabian StyleBezerra, Francisco Felipe, Stephan N.M.C.G. Oliveira, Rodrigo A. Sales, Adriana A. Piquet, Nina V. Capillé, Eduardo Vilanova, Ana M.F. Tovar, and Paulo A.S. Mourão. 2023. "Approaches to Assure Similarity between Pharmaceutical Heparins from Two Different Manufacturers" Pharmaceutics 15, no. 4: 1115. https://doi.org/10.3390/pharmaceutics15041115