
pH-Responsive and Mucoadhesive Nanoparticles for Enhanced Oral Insulin Delivery: The Effect of Hyaluronic Acid with Different Molecular Weights

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Preparation of NPs
2.3. Physicochemical Properties of NPs
2.3.1. FT-IR
2.3.2. Particle Sizes and Zeta Potentials of NPs
2.3.3. Encapsulation Efficiency (EE) and Drug Loading (DL)
2.3.4. Transmission Electron Microscopy (TEM)
2.3.5. Storage Stability
2.4. Stability of NPs in Simulated Gastrointestinal Fluids
2.5. In Vitro Drug Release
2.5.1. In Vitro INS Release
2.5.2. Kinetics of Drug Release
2.5.3. Stability of INS in NPs
2.6. Mucin Adhesion and Ex Vivo Intestinal Permeation Study
2.6.1. Mucin Adhesion
2.6.2. Ex Vivo Intestinal Permeation Experiment
2.6.3. Confocal Laser Scanning Microscope (CLSM)
2.7. Caco-2 Cells
2.7.1. CCK 8
2.7.2. Live–Dead Cell Staining
2.7.3. Transwell
2.7.4. Cellular Uptake of NPs
2.8. In Vivo Hypoglycemic Effect
2.8.1. Establishment of the Diabetic Rat Model
2.8.2. Pharmacokinetics and Pharmacodynamics
2.9. In Vivo Toxicity
2.9.1. Systemic Toxicity
2.9.2. Hemolysis Test
2.9.3. Hematoxylin and Eosin Staining (H&E)
2.10. Statistical Analysis
3. Results
3.1. Formulation Screening and Process Optimization
3.2. Characterization of NPs
3.2.1. FT-IR
3.2.2. Morphology, Particle Sizes, and Zeta Potentials
3.3. EE and DL
3.4. Stability
3.5. Anti-Protease Degradation Ability
3.6. In Vitro Release
3.6.1. pH-Responsive Release of NPs
3.6.2. Structural Stability of INS
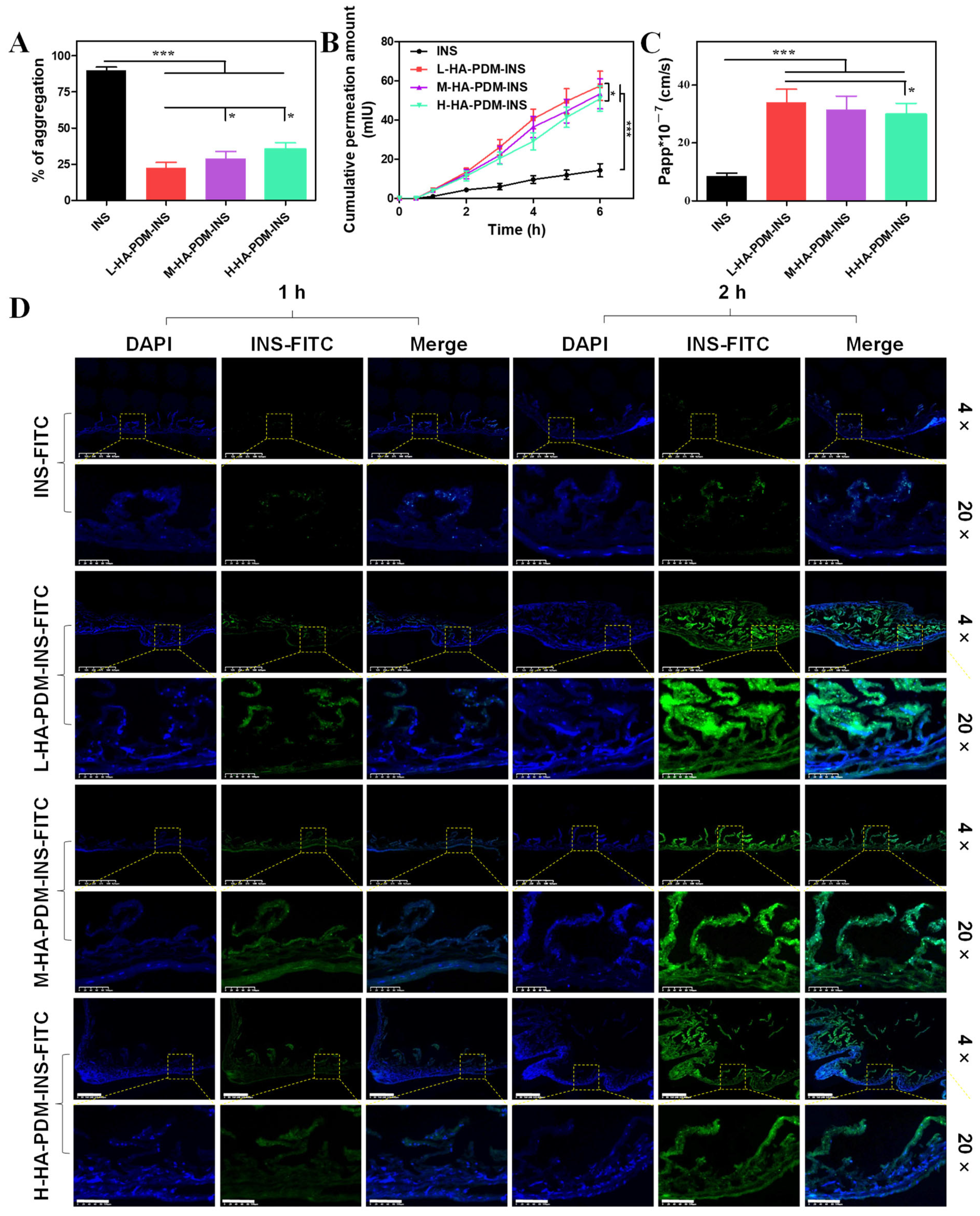
3.7. In Vitro Intestinal Penetration Study
3.8. Caco-2 Study
3.8.1. Biocompatibility
3.8.2. Transwell Experiments
3.8.3. Cellular Uptake
3.9. In Vivo Study
3.9.1. Pharmacodynamics
3.9.2. Pharmacokinetic
3.10. Safety Evaluation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Jeyagaran, A.; En-Lu, C.; Zbinden, A.; Birkenfeld, A.L.; Brucker, S.Y.; Layland, S.L. Type 1 Diabetes and Engineering Enhanced Islet Transplantation. Adv. Drug Deliv. Rev. 2022, 189, 114481. [Google Scholar] [CrossRef]
- Mishra, V.; Nayak, P.; Sharma, M.; Albutti, A.; Alwashmi, A.S.S. Emerging Treatment Strategies for Diabetes Mellitus and Associated Complications: An Update. Pharmaceutics 2021, 13, 1568. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Wang, C.; Ren, S.; Pan, J.; Wang, Y.; Shen, Y.; Zeng, Z.; Cui, H.; Zhao, X. Versatile Oral Insulin Delivery Nanosystems: From Materials to Nanostructures. Int. J. Mol. Sci. 2022, 23, 3362. [Google Scholar] [CrossRef] [PubMed]
- Domingo-Lopez, D.A.; Lattanzi, G.; Schreiber, L.H.J.; Wallace, E.J.; Wylie, R.; O’Sullivan, J.; Dolan, E.B.; Duffy, G.P. Medical Devices, Smart Drug Delivery, Wearables and Technology for the Treatment of Diabetes Mellitus. Adv. Drug Deliv. Rev. 2022, 185, 114280. [Google Scholar] [CrossRef]
- Puri, V.; Kaur, V.P.; Singh, A.; Singh, C. Recent Advances on Drug Delivery Applications of Mucopenetrative/Mucoadhesive Particles: A Review. J. Drug Deliv. Sci. Technol. 2022, 75, 103712. [Google Scholar] [CrossRef]
- Wong, C.Y.; Al-Salami, H.; Dass, C.R. Potential of Insulin Nanoparticle Formulations for Oral Delivery and Diabetes Treatment. J. Control. Release 2017, 264, 247–275. [Google Scholar] [CrossRef] [PubMed]
- Fonte, P.; Araújo, F.; Silva, C.; Pereira, C.; Reis, S.; Santos, H.A.; Sarmento, B. Polymer-Based Nanoparticles for Oral Insulin Delivery: Revisited Approaches. Biotechnol. Adv. 2015, 33, 1342–1354. [Google Scholar] [CrossRef] [PubMed]
- Jørgensen, J.R.; Thamdrup, L.H.E.; Kamguyan, K.; Nielsen, L.H.; Nielsen, H.M.; Boisen, A.; Rades, T.; Müllertz, A. Design of a Self-Unfolding Delivery Concept for Oral Administration of Macromolecules. J. Control. Release 2021, 329, 948–954. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.Y.; Al-Salami, H.; Dass, C.R. Recent Advancements in Oral Administration of Insulin-Loaded Liposomal Drug Delivery Systems for Diabetes Mellitus. Int. J. Pharm. 2018, 549, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Ibsen, K.; Brown, T.; Chen, R.; Agatemor, C.; Mitragotri, S. Ionic Liquids for Oral Insulin Delivery. Proc. Natl. Acad. Sci. USA 2018, 115, 7296–7301. [Google Scholar] [CrossRef] [Green Version]
- Kristensen, M.; Diedrichsen, R.G.; Vetri, V.; Foderà, V.; Nielsen, H.M. Increased Carrier Peptide Stability through Ph Adjustment Improves Insulin and Pth(1-34) Delivery in Vitro and in Vivo Rather than by Enforced Carrier Peptide-Cargo Complexation. Pharmaceutics 2020, 12, 993. [Google Scholar] [CrossRef] [PubMed]
- Fernandes Patta, A.C.M.; Mathews, P.D.; Madrid, R.R.M.; Rigoni, V.L.S.; Silva, E.R.; Mertins, O. Polyionic Complexes of Chitosan-N-Arginine with Alginate as PH Responsive and Mucoadhesive Particles for Oral Drug Delivery Applications. Int. J. Biol. Macromol. 2020, 148, 550–564. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, C.A.B.; Carriero, M.M.; Leomil, F.S.C.; Moro de Sousa, R.L.; de Miranda, A.; Mertins, O.; Mathews, P.D. Complexation of a Polypeptide-Polyelectrolytes Bioparticle as a Biomaterial of Antibacterial Activity. Pharmaceutics 2022, 14, 2746. [Google Scholar] [CrossRef] [PubMed]
- Bami, M.S.; Raeisi Estabragh, M.A.; Khazaeli, P.; Ohadi, M.; Dehghannoudeh, G. PH-Responsive Drug Delivery Systems as Intelligent Carriers for Targeted Drug Therapy: Brief History, Properties, Synthesis, Mechanism and Application. J. Drug Deliv. Sci. Technol. 2022, 70, 102987. [Google Scholar] [CrossRef]
- Wang, Z.; Deng, X.; Ding, J.; Zhou, W.; Zheng, X.; Tang, G. Mechanisms of Drug Release in PH-Sensitive Micelles for Tumour Targeted Drug Delivery System: A Review. Int. J. Pharm. 2018, 535, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Tan, P.; Fu, S.; Tian, X.; Zhang, H.; Ma, X.; Gu, Z.; Luo, K. Preparation and Application of PH-Responsive Drug Delivery Systems. J. Control. Release 2022, 348, 206–238. [Google Scholar] [CrossRef]
- Wang, L.; Dai, W.; Yang, M.; Wei, X.; Ma, K.; Song, B.; Jia, P.; Gong, Y.; Yang, J.; Zhao, J. Cell Membrane Mimetic Copolymer Coated Polydopamine Nanoparticles for Combined PH-Sensitive Drug Release and near-Infrared Photothermal Therapeutic. Colloids Surf. B Biointerfaces 2019, 176, 1–8. [Google Scholar] [CrossRef]
- Ghobashy, M.M.; Elbarbary, A.M.; Hegazy, D.E.; Maziad, N.A. Radiation Synthesis of PH-Sensitive 2-(Dimethylamino)Ethyl Methacrylate/ Polyethylene Oxide/ZnS Nanocomposite Hydrogel Membrane for Wound Dressing Application. J. Drug Deliv. Sci. Technol. 2022, 73, 103399. [Google Scholar] [CrossRef]
- Foss, A.C.; Goto, T.; Morishita, M.; Peppas, N.A. Development of Acrylic-Based Copolymers for Oral Insulin Delivery. Eur. J. Pharm. Biopharm. 2004, 57, 163–169. [Google Scholar] [CrossRef]
- Raza, A.; Rasheed, T.; Nabeel, F.; Hayat, U.; Bilal, M.; Iqbal, H.M.N. Endogenous and Exogenous Stimuli-Responsive Drug Delivery Systems for Programmed Site-Specific Release. Molecules 2019, 24, 1117. [Google Scholar] [CrossRef] [Green Version]
- Panahi, Y.; Gharekhani, A.; Hamishehkar, H.; Zakeri-Milani, P.; Gharekhani, H. Stomach-Specific Drug Delivery of Clarithromycin Using a Semi Interpenetrating Polymeric Network Hydrogel Made of Montmorillonite and Chitosan: Synthesis, Characterization and in Vitro Drug Release Study. Adv. Pharm. Bull. 2019, 9, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Panic, V.V.; Spasojevic, P.M.; Radoman, T.S.; Dzunuzovic, E.S.; Popovic, I.G.; Velickovic, S.J. Methacrylic Acid Based Polymer Networks with a High Content of Unfunctionalized Nanosilica: Particle Distribution, Swelling, and Rheological Properties. J. Phys. Chem. C 2015, 119, 610–622. [Google Scholar] [CrossRef]
- Brown, H.R. A Model of the Fracture of Double Network Gels. Macromolecules 2007, 40, 3815–3818. [Google Scholar] [CrossRef]
- Li, S.; Chen, Z.; Wang, J.; Yan, L.; Chen, T.; Zeng, Q. Fabrication and Characterization of a Novel Semi-Interpenetrating Network Hydrogel Based on Sodium Carboxymethyl Cellulose and Poly(Methacrylic Acid) for Oral Insulin Delivery. J. Biomater. Appl. 2020, 35, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Nayak, A.K.; Hasnain, M.S.; Aminabhavi, T.M. Drug Delivery Using Interpenetrating Polymeric Networks of Natural Polymers: A Recent Update. J. Drug Deliv. Sci. Technol. 2021, 66, 102915. [Google Scholar] [CrossRef]
- Kumar, R.; Islam, T.; Nurunnabi, M. Mucoadhesive Carriers for Oral Drug Delivery. J. Control. Release 2022, 351, 504–559. [Google Scholar] [CrossRef] [PubMed]
- Li, M.G.; Lu, W.L.; Wang, J.C.; Zhang, X.; Wang, X.Q.; Zheng, A.P.; Zhang, Q. Distribution, Transition, Adhesion and Release of Insulin Loaded Nanoparticles in the Gut of Rats. Int. J. Pharm. 2007, 329, 182–191. [Google Scholar] [CrossRef]
- Chen, J.; Huang, Y.; Ma, X.; Lei, Y. Functional Self-Healing Materials and Their Potential Applications in Biomedical Engineering. Adv. Compos. Hybrid Mater. 2018, 1, 94–113. [Google Scholar] [CrossRef]
- Kim, J.; Park, K.; Hahn, S.K. Effect of Hyaluronic Acid Molecular Weight on the Morphology of Quantum Dot-Hyaluronic Acid Conjugates. Int. J. Biol. Macromol. 2008, 42, 41–45. [Google Scholar] [CrossRef]
- Ibrahim, Y.H.E.Y.; Regdon, G.; Hamedelniel, E.I.; Sovány, T. Review of Recently Used Techniques and Materials to Improve the Efficiency of Orally Administered Proteins/Peptides. DARU J. Pharm. Sci. 2020, 28, 403–416. [Google Scholar] [CrossRef] [Green Version]
- Evanko, S.P.; Angello, J.C.; Wight, T.N. Formation of Hyaluronan- and Versican-Rich Pericellular Matrix Is Required for Proliferation and Migration of Vascular Smooth Muscle Cells. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1004–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segura, T.; Anderson, B.C.; Chung, P.H.; Webber, R.E.; Shull, K.R.; Shea, L.D. Crosslinked Hyaluronic Acid Hydrogels: A Strategy to Functionalize and Pattern. Biomaterials 2005, 26, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Chiesa, E.; Greco, A.; Riva, F.; Dorati, R.; Conti, B.; Modena, T.; Genta, I. CD44-Targeted Carriers: The Role of Molecular Weight of Hyaluronic Acid in the Uptake of Hyaluronic Acid-Based Nanoparticles. Pharmaceuticals 2022, 15, 103. [Google Scholar] [CrossRef]
- Liu, D.; Jiang, G.; Yu, W.; Li, L.; Tong, Z.; Kong, X.; Yao, J. Oral Delivery of Insulin Using CaCO3-Based Composite Nanocarriers with Hyaluronic Acid Coatings. Mater. Lett. 2017, 188, 263–266. [Google Scholar] [CrossRef]
- Li, L.; Jiang, G.; Yu, W.; Liu, D.; Chen, H.; Liu, Y.; Huang, Q.; Tong, Z.; Yao, J.; Kong, X. A Composite Hydrogel System Containing Glucose-Responsive Nanocarriers for Oral Delivery of Insulin. Mater. Sci. Eng. C 2016, 69, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Petkov, C.I.; Flecknell, P.; Murphy, K.; Basso, M.A.; Mitchell, A.S.; Hartig, R.; Thompson-Iritani, S. Unified Ethical Principles and an Animal Research ‘Helsinki’ Declaration as Foundations for International Collaboration. Curr. Res. Neurobiol. 2022, 3, 100060. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Xia, D.; Zhu, Q.; Li, X.; He, S.; Zhu, C.; Guo, S.; Hovgaard, L.; Yang, M.; Gan, Y. Functional Nanoparticles Exploit the Bile Acid Pathway to Overcome Multiple Barriers of the Intestinal Epithelium for Oral Insulin Delivery. Biomaterials 2018, 151, 13–23. [Google Scholar] [CrossRef]
- Mauro, N.; Fiorica, C.; Varvarà, P.; Di Prima, G.; Giammona, G. A Facile Way to Build up Branched High Functional Polyaminoacids with Tunable Physicochemical and Biological Properties. Eur. Polym. J. 2016, 77, 124–138. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Y.; Yu, M.; Wang, A.; Qiu, Y.; Fan, W.; Hovgaard, L.; Yang, M.; Li, Y.; Wang, R.; et al. The Upregulated Intestinal Folate Transporters Direct the Uptake of Ligand-Modified Nanoparticles for Enhanced Oral Insulin Delivery. Acta Pharm. Sin. B 2022, 12, 1460–1472. [Google Scholar] [CrossRef]
- Wu, H.; Nan, J.; Yang, L.; Park, H.J.; Li, J. Insulin-Loaded Liposomes Packaged in Alginate Hydrogels Promote the Oral Bioavailability of Insulin. J. Control Release 2023, 353, 51–62. [Google Scholar] [CrossRef]
- Kaur, I.; Nallamothu, B.; Kuche, K.; Katiyar, S.S.; Chaudhari, D.; Jain, S. Exploring Protein Stabilized Multiple Emulsion with Permeation Enhancer for Oral Delivery of Insulin. Int. J. Biol. Macromol. 2021, 167, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Gao, Z.; Liu, L.; Li, M.; Zuo, A.; Guo, J. Preparation, in Vitro and in Vivo Evaluation of Chitosan-Sodium Alginate-Ethyl Cellulose Polyelectrolyte Film as a Novel Buccal Mucosal Delivery Vehicle. Eur. J. Pharm. Sci. 2022, 168, 106085. [Google Scholar] [CrossRef] [PubMed]
- Qamar, Z.; Ashhar, M.U.; Annu; Qizilibash, F.F.; Sahoo, P.K.; Ali, A.; Ali, J.; Baboota, S. Lipid Nanocarrier of Selegiline Augmented Anti-Parkinson’s Effect via P-Gp Modulation Using Quercetin. Int. J. Pharm. 2021, 609, 121131. [Google Scholar] [CrossRef]
- Jørgensen, J.R.; Yu, F.; Venkatasubramanian, R.; Nielsen, L.H.; Nielsen, H.M.; Boisen, A.; Rades, T.; Müllertz, A. In Vitro, Ex Vivo and in Vivo Evaluation of Microcontainers for Oral Delivery of Insulin. Pharmaceutics 2020, 12, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Y.; Jin, J.; Duan, H.; Liu, C.; Chen, L.; Huang, W.; Gao, Z.; Jin, M. Targeted Therapeutic Effects of Oral Inulin-Modified Double-Layered Nanoparticles Containing Chemotherapeutics on Orthotopic Colon Cancer. Biomaterials 2022, 283, 121440. [Google Scholar] [CrossRef] [PubMed]
- Meng, S.; Wang, S.; Piao, M.G. Prescription Optimization of Gastroretentive Furosemide Hollow-Bioadhesive Microspheres via Box-Behnken Design: In Vitro Characterization and in Vivo Evaluation. J. Drug Deliv. Sci. Technol. 2022, 70, 103235. [Google Scholar] [CrossRef]
- Mauro, N.; Drago, S.E.; Cavallaro, G.; Giammona, G. Near-Infrared, Light-Triggered, On-Demand Anti-Inflammatories and Antibiotics Release by Graphene Oxide/Elecrospun PCL Patch for Wound Healing. C 2019, 5, 63. [Google Scholar] [CrossRef] [Green Version]
- Mauro, N.; Utzeri, M.A.; Drago, S.E.; Nicosia, A.; Costa, S.; Cavallaro, G.; Giammona, G. Hyaluronic Acid Dressing of Hydrophobic Carbon Nanodots: A Self-Assembling Strategy of Hybrid Nanocomposites with Theranostic Potential. Carbohydr. Polym. 2021, 267, 118213. [Google Scholar] [CrossRef]
- He, Z.; Santos, J.L.; Tian, H.; Huang, H.; Hu, Y.; Liu, L.; Leong, K.W.; Chen, Y.; Mao, H.Q. Scalable Fabrication of Size-Controlled Chitosan Nanoparticles for Oral Delivery of Insulin. Biomaterials 2017, 130, 28–41. [Google Scholar] [CrossRef]
- Andreani, T.; Miziara, L.; Lorenzón, E.N.; De Souza, A.L.R.; Kiill, C.P.; Fangueiro, J.F.; Garcia, M.L.; Gremião, P.D.; Silva, A.M.; Souto, E.B. Effect of Mucoadhesive Polymers on the in Vitro Performance of Insulin-Loaded Silica Nanoparticles: Interactions with Mucin and Biomembrane Models. Eur. J. Pharm. Biopharm. 2015, 93, 118–126. [Google Scholar] [CrossRef]
- Chickering, D.E.; Mathiowitz, E. Bioadhesive Microspheres: I. A Novel Electrobalance-Based Method to Study Adhesive Interactions between Individual Microspheres and Intestinal Mucosa. J. Control Release 1995, 34, 251–262. [Google Scholar] [CrossRef]
- Buckley, C.; Murphy, E.J.; Montgomery, T.R.; Major, I. Hyaluronic Acid: A Review of the Drug Delivery Capabilities of This Naturally Occurring Polysaccharide. Polymers 2022, 14, 3442. [Google Scholar] [CrossRef] [PubMed]
- Pornpitchanarong, C.; Rojanarata, T.; Opanasopit, P.; Ngawhirunpat, T.; Patrojanasophon, P. Catechol-Modified Chitosan/Hyaluronic Acid Nanoparticles as a New Avenue for Local Delivery of Doxorubicin to Oral Cancer Cells. Colloids Surf. B Biointerfaces 2020, 196, 111279. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gu, Z.; Li, W.; Guo, L.; Wang, L.; Guo, L.; Ma, S.; Han, B.; Chang, J. PH-Sensitive O-Carboxymethyl Chitosan/Sodium Alginate Nanohydrogel for Enhanced Oral Delivery of Insulin. Int. J. Biol. Macromol. 2022, 223, 433–445. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Jiang, L.; Meng, S.; Liu, C.; Wang, H.; Gao, Z.; Guo, J. Hollow Mesoporous Silica Nanoparticles-Loaded Ion-Crosslinked Bilayer Films with Excellent Mechanical Properties and High Bioavailability for Buccal Delivery. Int. J. Pharm. 2022, 624, 122056. [Google Scholar] [CrossRef]
- Sonaje, K.; Lin, K.J.; Wey, S.P.; Lin, C.K.; Yeh, T.H.; Nguyen, H.N.; Hsu, C.W.; Yen, T.C.; Juang, J.H.; Sung, H.W. Biodistribution, Pharmacodynamics and Pharmacokinetics of Insulin Analogues in a Rat Model: Oral Delivery Using PH-Responsive Nanoparticles vs. Subcutaneous Injection. Biomaterials 2010, 31, 6849–6858. [Google Scholar] [CrossRef]
- Lima, G.G.; Rocha Neto, J.B.M.; de Carvalho, H.F.; Beppu, M.M. Control of Surface Properties of Hyaluronan/Chitosan Multilayered Coatings for Tumor Cell Capture. Polysaccharides 2021, 2, 387–399. [Google Scholar] [CrossRef]
- Didaskalou, P.; Kafetzi, M.; Pispas, S. Cationic Polyelectrolytes Containing Perfluorinated Groups: Synthesis and Self-Assembly Properties in Aqueous Solutions. Macromol 2022, 2, 194–210. [Google Scholar] [CrossRef]
- Mohammadi, M.; Salami-Kalajahi, M.; Roghani-Mamaqani, H.; Golshan, M. Effect of Molecular Weight and Polymer Concentration on the Triple Temperature/PH/Ionic Strength-Sensitive Behavior of Poly(2-(Dimethylamino)Ethyl Methacrylate). Int. J. Polym. Mater. Polym. Biomater. 2017, 66, 455–461. [Google Scholar] [CrossRef]
- Du, J.; Fu, F.; Shi, X.; Yin, Z. Controlled Release of a Model Protein Drug Ovalbumin from Thiolated Hyaluronic Acid Matrix. J. Drug Deliv. Sci. Technol. 2015, 30, 74–81. [Google Scholar] [CrossRef]
- He, C.; Yin, L.; Tang, C.; Yin, C. Size-Dependent Absorption Mechanism of Polymeric Nanoparticles for Oral Delivery of Protein Drugs. Biomaterials 2012, 33, 8569–8578. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xiong, G.M.; Ali, Y.; Boehm, B.O.; Huang, Y.Y.; Venkatraman, S. Layer-by-Layer Coated Nanoliposomes for Oral Delivery of Insulin. Nanoscale 2021, 13, 776–789. [Google Scholar] [CrossRef] [PubMed]
- Varanko, A.; Saha, S.; Chilkoti, A. Recent Trends in Protein and Peptide-Based Biomaterials for Advanced Drug Delivery. Adv. Drug Deliv. Rev. 2020, 156, 133–187. [Google Scholar] [CrossRef] [PubMed]
- Andretto, V.; Rosso, A.; Briançon, S.; Lollo, G. Nanocomposite Systems for Precise Oral Delivery of Drugs and Biologics. Drug Deliv. Transl. Res. 2021, 11, 445–470. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HA–PDM (L, M, and H) | |
|---|---|
| DM (μL) | 100 |
| 300 | |
| 500 | |
| KPS (mL) | 0.5 |
| 1.5 | |
| 2.5 | |
| Stirring speed (rpm) | 1040 |
| 1300 | |
| 1560 | |
| Temperature (°C) | 50 |
| 60 | |
| 70 |
| Value | Size | PDI | Zeta | ||
|---|---|---|---|---|---|
| L-HA–PDM | DM (μL) | 100 | 445.99 ± 16.94 | 0.51 ± 0.16 | −36.98 ± 2.41 |
| 300 | 272.53 ± 8.32 | 0.32 ± 0.04 | −31.64 ± 3.09 | ||
| 500 | 276.06 ± 10.42 | 0.29 ± 0.04 | −32.08 ± 1.98 | ||
| KPS (mL) | 0.5 | 340.11 ± 13.16 | 0.46 ± 0.06 | −37.29 ± 2.19 | |
| 1.5 | 255.68 ± 12.94 | 0.26 ± 0.05 | −31.58 ± 1.97 | ||
| 2.5 | 522.62 ± 14.39 | 0.33 ± 0.13 | −23.42 ± 2.05 | ||
| Stirring speed (rpm) | 1040 | 577.09 ± 15.92 | 0.34 ± 0.13 | −32.27 ± 1.97 | |
| 1300 | 324.18 ± 11.68 | 0.22 ± 0.08 | −31.64 ± 1.67 | ||
| 1560 | 451.67 ± 9.97 | 0.21 ± 0.07 | −34.92 ± 2.21 | ||
| Temperature (°C) | 50 | 250.48 ±17.97 | 0.17 ± 0.07 | −42.02 ± 4.02 | |
| 60 | 286.22 ± 18.66 | 0.15 ± 0.11 | −40.68 ± 4.19 | ||
| 70 | 445.99 ± 16.94 | 0.51 ± 0.16 | −36.98 ± 2.41 | ||
| M-HA-PDM | DM (μL) | 100 | 534.02 ±25.64 | 0. 62 ± 0.18 | −33.96 ± 2.89 |
| 300 | 762.46 ± 19.64 | 0.54 ± 0.13 | −36.39 ± 2.06 | ||
| 500 | 453.04 ± 15.62 | 0.32 ± 0.09 | −34.88 ± 2.14 | ||
| KPS (mL) | 0.5 | 728.25 ± 17.69 | 0.33 ± 0.11 | −36.92 ± 2.01 | |
| 1.5 | 443.11 ± 15.21 | 0.28 ± 0.09 | −38.57 ± 1.97 | ||
| 2.5 | 412.67 ± 12.71 | 0.23 ± 0.08 | −35.04 ± 2.15 | ||
| Stirring speed (rpm) | 1040 | 292.07 ± 11.29 | 0.24 ± 0.05 | −33.84 ± 2.67 | |
| 1300 | 264.82 ± 11.06 | 0.16 ± 0.08 | −34.02 ± 2.81 | ||
| 1560 | 437.43 ± 10.11 | 0.21 ± 0.07 | −38.59 ± 2.09 | ||
| Temperature (°C) | 50 | 452.39 ± 13.42 | 0.19 ± 0.08 | −27.99 ± 1.87 | |
| 60 | 262.59 ± 10.97 | 0.15 ± 0.04 | −36.52 ± 3.14 | ||
| 70 | 269.55 ± 10.67 | 0.16 ± 0.04 | −26.98 ± 1.62 | ||
| H-HA–PDM | DM (μL) | 100 | 452.37 ± 19.64 | 0.67 ± 0.21 | −28.64 ± 2.61 |
| 300 | 304.62 ±16.35 | 0.33 ± 0.16 | −32.05 ± 2.01 | ||
| 500 | 287.88 ± 12.62 | 0.47 ± 0.11 | −28.64 ± 1.99 | ||
| KPS (mL) | 0.5 | 557.41 ± 16.98 | 0.38 ± 0.10 | −38.65 ± 2.23 | |
| 1.5 | 400.21 ± 14.62 | 0.32 ± 0.08 | −41.02 ± 1.97 | ||
| 2.5 | 239.11 ± 10.98 | 0.24 ± 0.05 | −37.88 ± 2.09 | ||
| Stirring speed (rpm) | 1040 | 306.13 ± 15.09 | 0.22 ± 0.09 | −30.94 ± 2.64 | |
| 1300 | 332.82 ± 12.01 | 0.18 ± 0.05 | −26.48 ± 1.96 | ||
| 1560 | 277.98 ± 10.93 | 0.14 ± 0.04 | −29.54 ± 1.89 | ||
| Temperature (°C) | 50 | 592.59 ± 18.19 | 0.16 ± 0.07 | −34.62 ± 3.01 | |
| 60 | 395.92 ± 10.67 | 0.15 ± 0.08 | −31.82 ± 1.67 | ||
| 70 | 275.24 ± 8.00 | 0.14 ± 0.05 | −25.61 ± 1.98 |
| DM (μL) | KPS (mL) | Stirring Speed (rpm) | Temperature (°C) | |
|---|---|---|---|---|
| L-HA–PDM | 300 | 1.5 | 1300 | 50 |
| M-HA–PDM | 500 | 2.5 | 1300 | 60 |
| H-HA–PDM | 300 | 2.5 | 1560 | 70 |
| Zero Order | First Order | Higuchi | Ritger–Pappas | |||
|---|---|---|---|---|---|---|
| Mathematical Equation | Qt = k0t + Q0 | Log Qt = LogQ0 − k0t/2.303 | Qt = kHt1/2 | Mt/M∞ = ktn | ||
| R2 | R2 | R2 | R2 | n | ||
| pH 1.2 | L-HA–PDM–INS | 0.8336 | 0.9983 | 0.9766 | 0.9766 | 0.4945 |
| M-HA–PDM–INS | 0.8660 | 0.9983 | 0.9822 | 0.9823 | 0.5314 | |
| H-HA–PDM–INS | 0.8411 | 0.9968 | 0.9772 | 0.9768 | 0.5049 | |
| pH 6.8 | L-HA–PDM–INS | 0.8164 | 0.9890 | 0.9500 | 0.9492 | 0.5219 |
| M-HA–PDM–INS | 0.9074 | 0.9781 | 0.9303 | 0.9540 | 0.7017 | |
| H-HA–PDM–INS | 0.9468 | 0.9843 | 0.9261 | 0.9709 | 0.7783 | |
| pH 7.4 | L-HA–PDM–INS | 0.8320 | 0.9950 | 0.9636 | 0.9630 | 0.5199 |
| M-HA–PDM–INS | 0.9243 | 0.9959 | 0.9672 | 0.9825 | 0.6504 | |
| H-HA–PDM–INS | 0.9351 | 0.9942 | 0.9593 | 0.9818 | 0.6864 | |
| S.C | Oral | L-HA–PDM–INS | M-HA–PDM–INS | H-HA–PDM–INS | |
|---|---|---|---|---|---|
| Dose (IU/kg) | 5 | 40 | 40 | 40 | 40 |
| AUC0–∞ (μIU/mL·h) | 180.31 ± 32.31 | 177.07 ± 83.24 | 189.83 ± 72.16 | 210.90 ± 113.06 | |
| MRT0–∞ (h) | 1.63 ± 0.07 | - | 3.96 ± 0.50 | 4.88 ± 0.82 | 6.17 ± 0.39 |
| VRT0–∞ (h2) | 2.13 ± 0.01 | - | 7.22 ± 5.89 | 10.82 ± 5.61 | 21.88 ± 9.12 |
| t1/2z (h) | 1.27 ± 0.09 | - | 1.88 ± 1.06 | 2.36 ± 0.33 | 3.35 ± 1.11 |
| Tmax (h) | 1 | - | 3 | 4 | 4 |
| CLz/F (L/h/kg) | 28.39 ± 4.30 | - | 183.89 ± 91.37 | 166.22 ± 64.64 | 143.90 ± 79.58 |
| Cmax (μIU/mL) | 104.67 ± 10.61 | - | 38.01 ± 11.31 | 31.98 ± 8.49 | 27.03 ± 9.89 |
| AAC | 330.1 | 17.3 | 293.2 | 324.8 | 377.1 |
| PA (%) | 100 | 0.66 | 11.10 | 12.30 | 14.28 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Meng, S.; Zhou, X.; Gao, Z.; Piao, M.G. pH-Responsive and Mucoadhesive Nanoparticles for Enhanced Oral Insulin Delivery: The Effect of Hyaluronic Acid with Different Molecular Weights. Pharmaceutics 2023, 15, 820. https://doi.org/10.3390/pharmaceutics15030820
Wang S, Meng S, Zhou X, Gao Z, Piao MG. pH-Responsive and Mucoadhesive Nanoparticles for Enhanced Oral Insulin Delivery: The Effect of Hyaluronic Acid with Different Molecular Weights. Pharmaceutics. 2023; 15(3):820. https://doi.org/10.3390/pharmaceutics15030820
Chicago/Turabian StyleWang, Shuangqing, Saige Meng, Xinlei Zhou, Zhonggao Gao, and Ming Guan Piao. 2023. "pH-Responsive and Mucoadhesive Nanoparticles for Enhanced Oral Insulin Delivery: The Effect of Hyaluronic Acid with Different Molecular Weights" Pharmaceutics 15, no. 3: 820. https://doi.org/10.3390/pharmaceutics15030820