
Current Status of Oligonucleotide-Based Protein Degraders

Abstract
: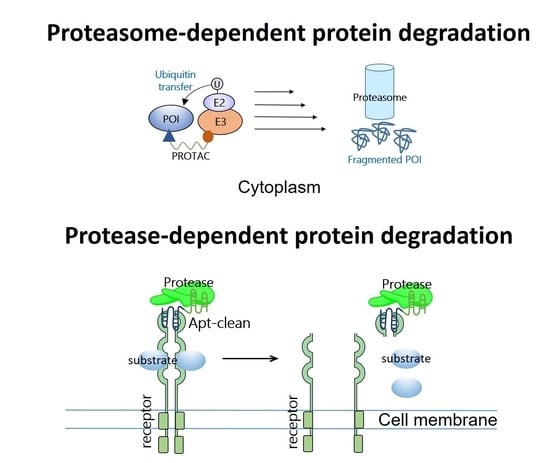
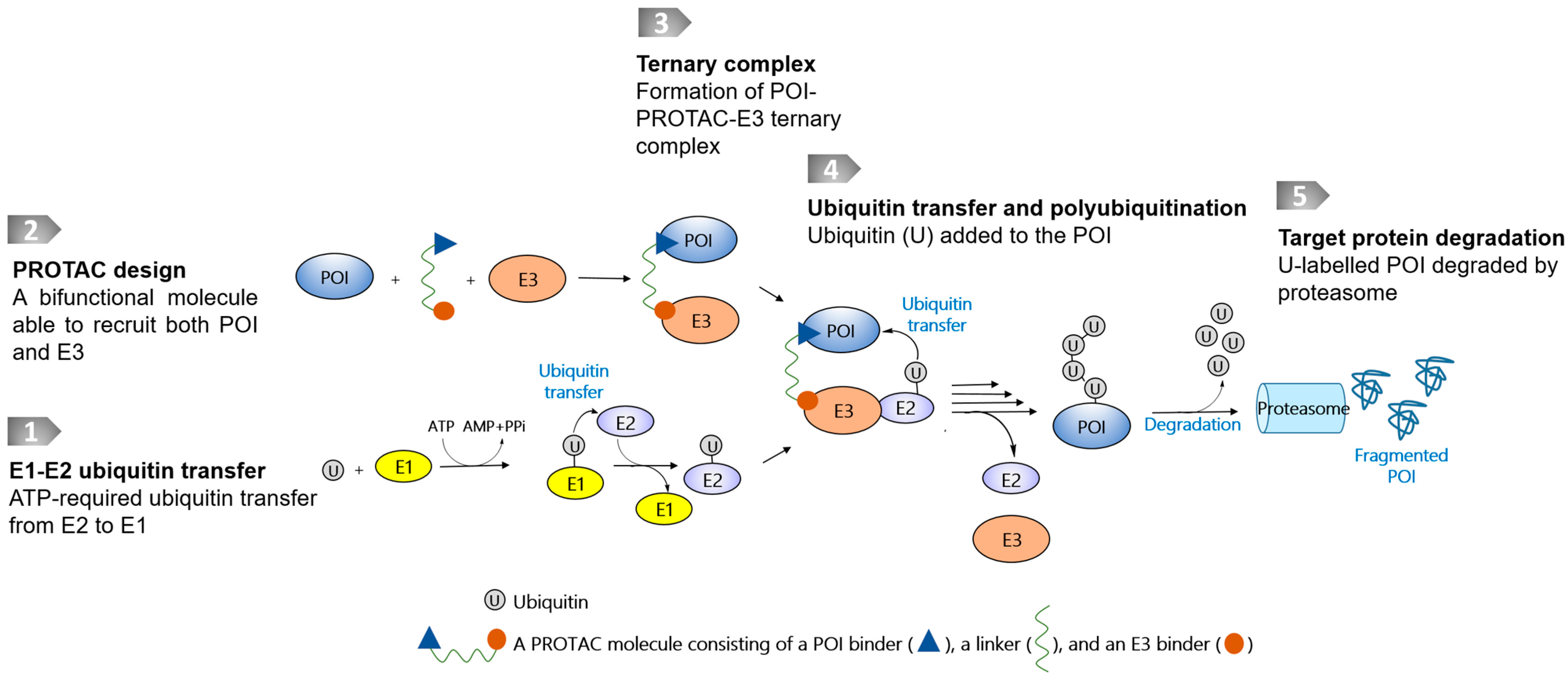
1. Introduction
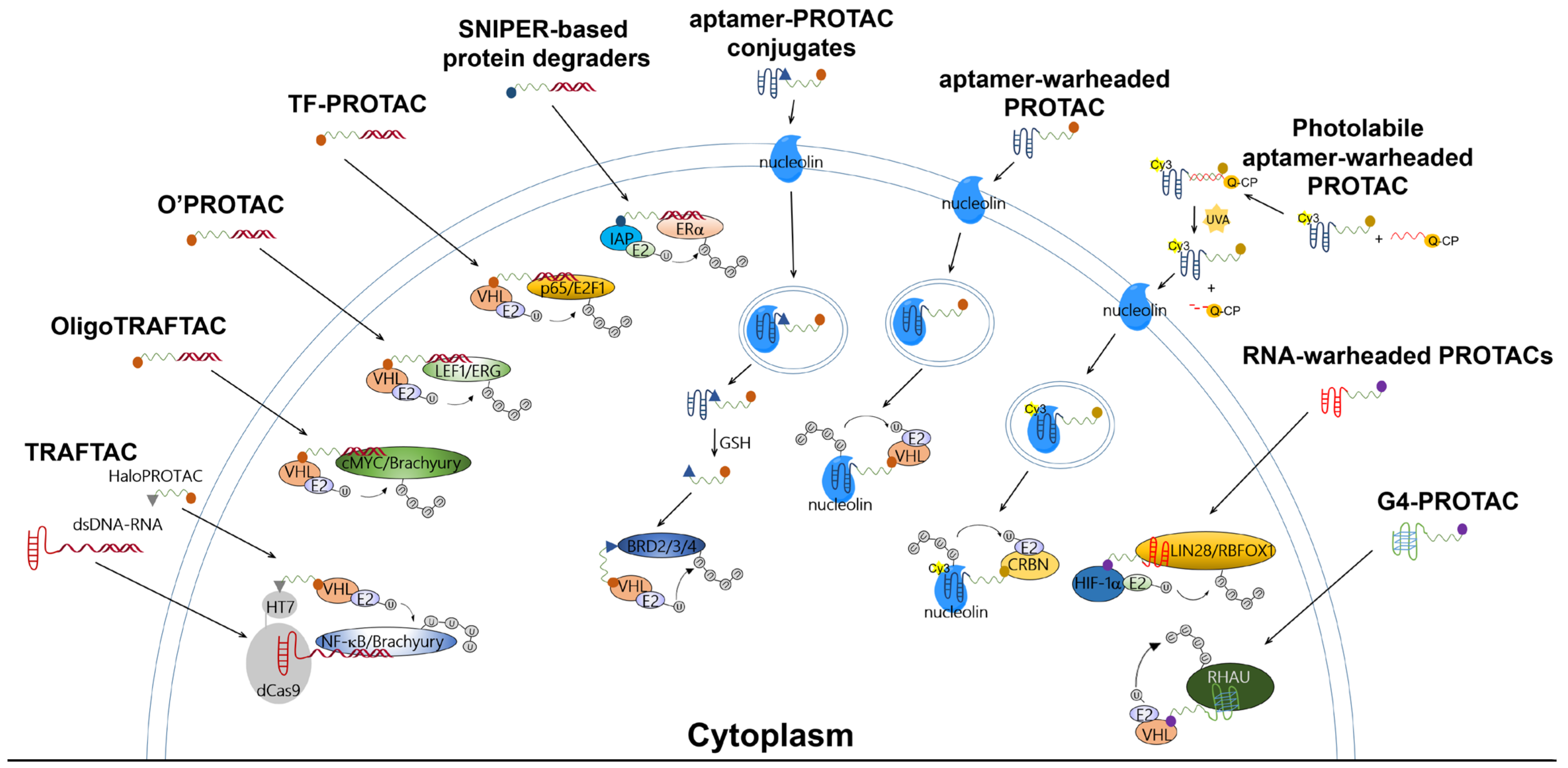
2. PROATC-Based Protein Degraders Warheaded with Single-Stranded or Double-Stranded Decoys: TRAFTAC, OligoTRAFTAC, O’PROTAC, TF-PROTAC, and SNIPER
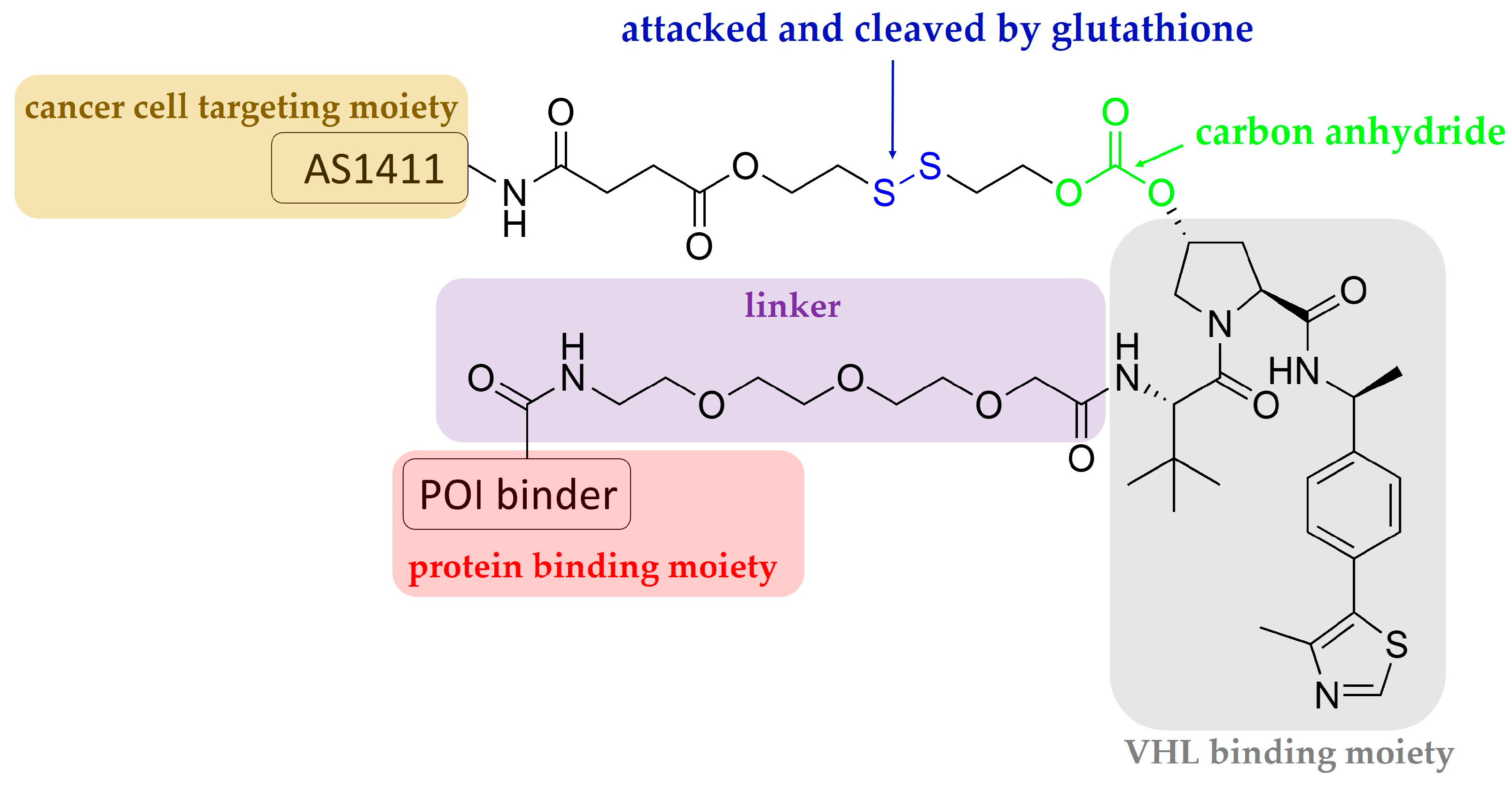
3. Aptamer-PROTAC Conjugates and Aptamer-Warheaded PROTAC
4. RNA-PROTACs: RNA-Warheaded PROTACs for Targeting RNA-Binding Proteins
5. G4-PROTAC: G-Quadruplex-Warheaded Protein Degraders
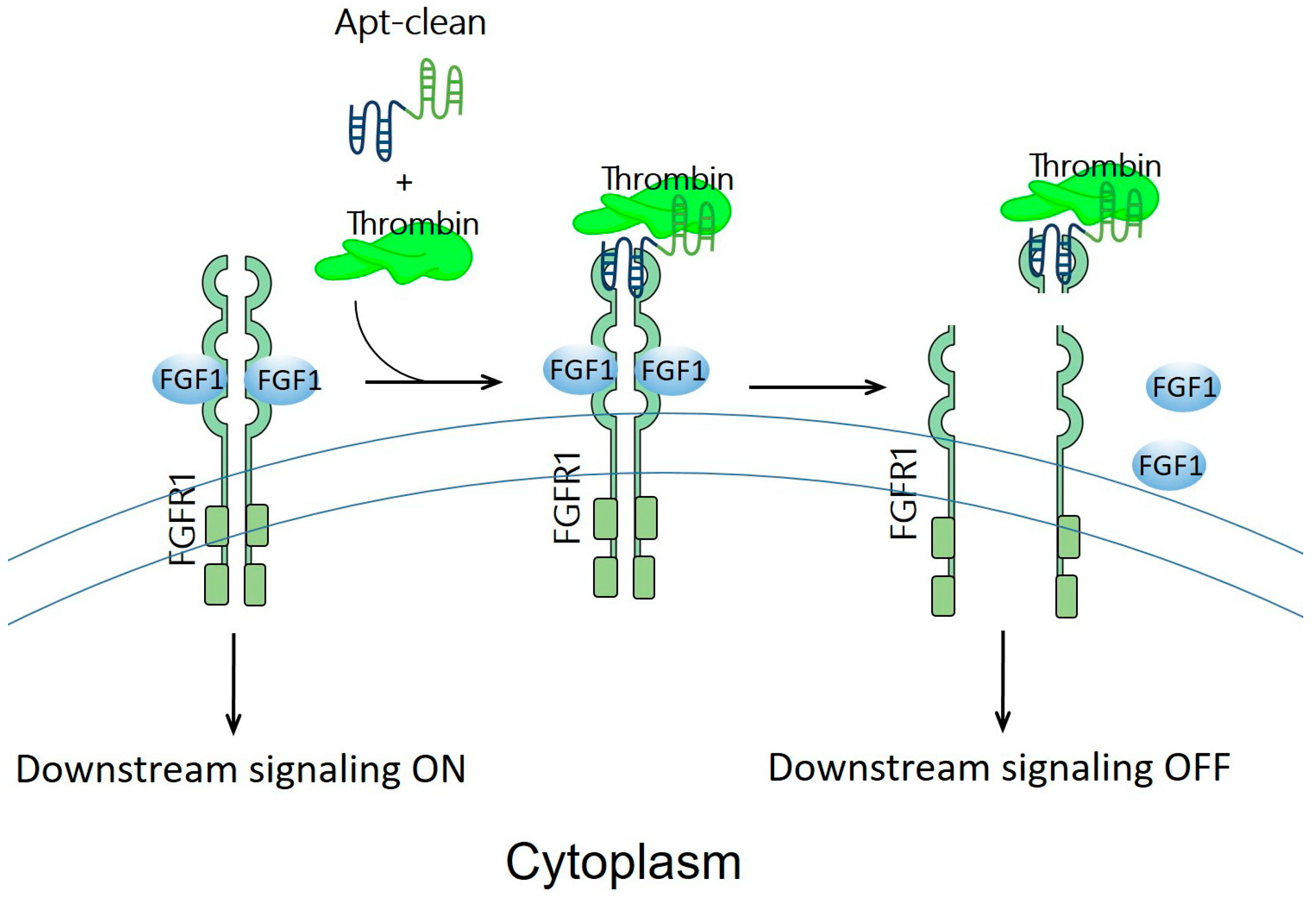
6. Apt-Clean: Bispecific Aptamers Act as Degraders for Targeting Extracellular Receptors
7. Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pettersson, M.; Crews, C.M. PROteolysis TArgeting Chimeras (PROTACs)—Past, Present and Future. Drug Discov. Today Technol. 2019, 31, 15–27. [Google Scholar] [CrossRef]
- Chen, A.; Koehler, A.N. Transcription Factor Inhibition: Lessons Learned and Emerging Targets. Trends Mol. Med. 2020, 26, 508–518. [Google Scholar] [CrossRef] [PubMed]
- Julio, A.R.; Backus, K.M. New Approaches to Target RNA Binding Proteins. Curr. Opin. Chem. Biol. 2021, 62, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Berg, T. Inhibition of Transcription Factors with Small Organic Molecules. Curr. Opin. Chem. Biol. 2008, 12, 464–471. [Google Scholar] [CrossRef]
- Slastnikova, T.A.; Ulasov, A.V.; Rosenkranz, A.A.; Sobolev, A.S. Targeted Intracellular Delivery of Antibodies: The State of the Art. Front. Pharmacol. 2018, 9, 1208. [Google Scholar] [CrossRef] [Green Version]
- Naito, M. Targeted protein degradation and drug discovery. J. Biochem. 2022, 172, 61–69. [Google Scholar] [CrossRef]
- Chen, Y.; Tandon, I.; Heelan, W.; Wang, Y.; Tang, W.; Hu, Q. Proteolysis-Targeting Chimera (PROTAC) Delivery System: Advancing Protein Degraders towards Clinical Translation. Chem. Soc. Rev. 2022, 51, 5330–5350. [Google Scholar] [CrossRef]
- Ohoka, N.; Okuhira, K.; Ito, M.; Nagai, K.; Shibata, N.; Hattori, T.; Ujikawa, O.; Shimokawa, K.; Sano, O.; Koyama, R.; et al. In Vivo Knockdown of Pathogenic Proteins via Specific and Nongenetic Inhibitor of Apoptosis Protein (IAP)-Dependent Protein Erasers (SNIPERs). J. Biol. Chem. 2017, 292, 4556–4570. [Google Scholar] [CrossRef] [Green Version]
- Burslem, G.M.; Song, J.; Chen, X.; Hines, J.; Crews, C.M. Enhancing Antiproliferative Activity and Selectivity of a FLT-3 Inhibitor by Proteolysis Targeting Chimera Conversion. J. Am. Chem. Soc. 2018, 140, 16428–16432. [Google Scholar] [CrossRef]
- Shibata, N.; Shimokawa, K.; Nagai, K.; Ohoka, N.; Hattori, T.; Miyamoto, N.; Ujikawa, O.; Sameshima, T.; Nara, H.; Cho, N.; et al. Pharmacological Difference between Degrader and Inhibitor against Oncogenic BCR-ABL Kinase. Sci. Rep. 2018, 8, 13549. [Google Scholar] [CrossRef]
- Burslem, G.M.; Schultz, A.R.; Bondeson, D.P.; Eide, C.A.; Stevens, S.L.S.; Druker, B.J.; Crews, C.M. Targeting BCR-ABL1 in Chronic Myeloid Leukemia by PROTAC-Mediated Targeted Protein Degradation. Cancer Res. 2019, 79, 4744–4753. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Crowley, V.M.; Wucherpfennig, T.G.; Dix, M.M.; Cravatt, B.F. Electrophilic PROTACs That Degrade Nuclear Proteins by Engaging DCAF16. Nat. Chem. Biol. 2019, 15, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhao, H.Y.; Xi, X.X.; Liu, Y.J.; Xin, M.; Mao, S.; Zhang, J.J.; Lu, A.X.; Zhang, S.Q. Discovery of Potent Epidermal Growth Factor Receptor (EGFR) Degraders by Proteolysis Targeting Chimera (PROTAC). Eur. J. Med. Chem. 2020, 189, 112061. [Google Scholar] [CrossRef] [PubMed]
- Ohoka, N.; Suzuki, M.; Uchida, T.; Tsuji, G.; Tsukumo, Y.; Yoshida, M.; Inoue, T.; Demizu, Y.; Ohki, H.; Naito, M. Development of Gilteritinib-Based Chimeric Small Molecules That Potently Induce Degradation of FLT3-ITD Protein. ACS Med. Chem. Lett. 2022, 13, 1885–1891. [Google Scholar] [CrossRef] [PubMed]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC Targeted Protein Degraders: The Past Is Prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef] [PubMed]
- Samarasinghe, K.T.G.; Jaime-Figueroa, S.; Burgess, M.; Nalawansha, D.A.; Dai, K.; Hu, Z.; Bebenek, A.; Holley, S.A.; Crews, C.M. Targeted Degradation of Transcription Factors by TRAFTACs: TRAnscription Factor TArgeting Chimeras. Cell Chem. Biol. 2021, 28, 648–661.e5. [Google Scholar] [CrossRef]
- Samarasinghe, K.T.G.; An, E.; Genuth, M.A.; Chu, L.; Holley, S.A.; Crews, C.M. OligoTRAFTACs: A Generalizable Method for Transcription Factor Degradation. RSC Chem. Biol. 2022, 3, 1144–1153. [Google Scholar] [CrossRef]
- Shao, J.; Yan, Y.; Ding, D.; Wang, D.; He, Y.; Pan, Y.; Yan, W.; Kharbanda, A.; Li, H.Y.; Huang, H. Destruction of DNA-Binding Proteins by Programmable Oligonucleotide PROTAC (O’PROTAC): Effective Targeting of LEF1 and ERG. Adv. Sci. 2021, 8, 2102555. [Google Scholar] [CrossRef]
- Liu, J.; Chen, H.; Kaniskan, H.Ü.; Xie, L.; Chen, X.; Jin, J.; Wei, W. TF-PROTACs Enable Targeted Degradation of Transcription Factors. J. Am. Chem. Soc. 2021, 143, 8902–8910. [Google Scholar] [CrossRef]
- Naganuma, M.; Ohoka, N.; Tsuji, G.; Tsujimura, H.; Matsuno, K.; Inoue, T.; Naito, M.; Demizu, Y. Development of Chimeric Molecules That Degrade the Estrogen Receptor Using Decoy Oligonucleotide Ligands. ACS Med. Chem. Lett. 2022, 13, 134–139. [Google Scholar] [CrossRef]
- He, S.; Gao, F.; Ma, J.; Ma, H.; Dong, G.; Sheng, C. Aptamer-PROTAC Conjugates (APCs) for Tumor-Specific Targeting in Breast Cancer. Angew. Chem. 2021, 60, 23299–23305. [Google Scholar] [CrossRef]
- Zhang, L.; Li, L.; Wang, X.; Zhang, Y.; Xie, T.; Zhang, H.; Li, X.; Peng, T.; Sun, X.; Dai, J.; et al. Development of a Novel PROTAC Using the Nucleic Acid Aptamer as a Targeting Ligand for Tumor-Selective Degradation of Nucleolin. Mol. Ther.-Nucleic Acids 2022, 13, 66–69. [Google Scholar] [CrossRef]
- Chen, M.; Zhou, P.; Kong, Y.; Li, J.; Li, Y.; Zhang, Y.; Ran, J.; Zhou, J.; Chen, Y.; Xie, S. Inducible Degradation of Oncogenic Nucleolin Using an Aptamer-Based PROTAC. J. Med. Chem. 2023, 66, 1339–1348. [Google Scholar] [CrossRef]
- Ghidini, A.; Cléry, A.; Halloy, F.; Allain, F.H.T.; Hall, J. RNA-PROTACs: Degraders of RNA-Binding Proteins. Angew. Chem. 2021, 60, 3163–3169. [Google Scholar] [CrossRef]
- Patil, K.M.; Chin, D.; Seah, H.L.; Shi, Q.; Lim, K.W.; Phan, A.T. G4-PROTAC: Targeted Degradation of a G-Quadruplex Binding Protein. Chem. Commun. 2021, 57, 12816–12819. [Google Scholar] [CrossRef]
- Hoshiyama, J.; Okada, Y.; Cho, S.; Ueki, S.; Sando, S. Apt-Clean: Aptamer-Mediated Cleavage of Extracellular Antigens for the Inhibition of Membrane Protein Functions. Biomater. Sci. 2023, 11, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Inukai, S.; Kock, K.H.; Bulyk, M.L. Transcription Factor–DNA Binding: Beyond Binding Site Motifs. Curr. Opin. Genet. Dev. 2017, 43, 110–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilanowska, A.; Studzińska, S. In Vivo and in Vitro studies of Antisense Oligonucleotides—A Review. RSC Adv. 2020, 10, 34501–34516. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Basiri, B.; Hassan, C.; Punt, C.; van der Hage, E.; den Besten, C.; Bartlett, M.G. Metabolite Profiling of the Antisense Oligonucleotide Eluforsen Using Liquid Chromatography-Mass Spectrometry. Mol. Ther.-Nucleic Acids 2019, 17, 714–725. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, Y.; Shi, L.; Yang, S.; Chang, J.; Zhong, Y.; Li, Q.; Xing, D. Recent Advances in IAP-Based PROTACs (SNIPERs) as Potential Therapeutic Agents. J. Enzyme Inhib. Med. Chem. 2022, 37, 1437–1453. [Google Scholar] [CrossRef]
- Naito, M.; Ohoka, N.; Shibata, N. SNIPERs—Hijacking IAP Activity to Induce Protein Degradation. Drug Discov. Today Technol. 2019, 31, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Ishikawa, M.; Naito, M.; Hashimoto, Y. Protein Knockdown Using Methyl Bestatin-Ligand Hybrid Molecules: Design and Synthesis of Inducers of Ubiquitination-Mediated Degradation of Cellular Retinoic Acid-Binding Proteins. J. Am. Chem. Soc. 2010, 132, 5820–5826. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Chambon, P. The Estrogen Receptor Binds Tightly to Its Responsive Element as a Ligand-Induced Homodimer. Cell 1988, 55, 145–156. [Google Scholar] [CrossRef]
- Yokoo, H.; Ohoka, N.; Naito, M.; Demizu, Y. Design and Synthesis of Peptide-Based Chimeric Molecules to Induce Degradation of the Estrogen and Androgen Receptors. Bioorg. Med. Chem. 2020, 28, 115595. [Google Scholar] [CrossRef] [PubMed]
- Ohoka, N.; Morita, Y.; Nagai, K.; Shimokawa, K.; Ujikawa, O.; Fujimori, I.; Ito, M.; Hayase, Y.; Okuhira, K.; Shibata, N.; et al. Derivatization of Inhibitor of Apoptosis Protein (IAP) Ligands Yields Improved Inducers of Estrogen Receptor Degradation. J. Biol. Chem. 2018, 293, 6776–6790. [Google Scholar] [CrossRef] [Green Version]
- Ye, M.; Hu, J.; Peng, M.; Liu, J.; Liu, J.; Liu, H.; Zhao, X.; Tan, W. Generating Aptamers by Cell-SELEX for Applications in Molecular Medicine. Int. J. Mol. Sci. 2012, 13, 3341–3353. [Google Scholar] [CrossRef] [Green Version]
- Nimjee, S.M.; White, R.R.; Becker, R.C.; Sullenger, B.A. Aptamers as Therapeutics. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 61–79. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Liu, B.; Lu, J.; Li, F.; Li, D.; Liang, C.; Dang, L.; Liu, J.; He, B.; Badshah, S.A.; et al. Progress and Challenges in Developing Aptamer-Functionalized Targeted Drug Delivery Systems. Int. J. Mol. Sci. 2015, 16, 23784–23822. [Google Scholar] [CrossRef]
- Yasmeen, F.; Seo, H.; Javaid, N.; Kim, M.S.; Choi, S. Therapeutic Interventions into Innate Immune Diseases by Means of Aptamers. Pharmaceutics 2020, 12, 955. [Google Scholar] [CrossRef]
- Yu, D.; Wang, D.; Zhu, F.G.; Bhagat, L.; Dai, M.; Kandimalla, E.R.; Agrawal, S. Modifications Incorporated in CpG Motifs of Oligodeoxynucleotides Lead to Antagonist Activity of Toll-like Receptors 7 and 9. J. Med. Chem. 2009, 52, 5108–5114. [Google Scholar] [CrossRef]
- Keefe, A.D.; Pai, S.; Ellington, A. Aptamers as Therapeutics. Nat. Rev. Drug Discov. 2010, 9, 537–550. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Reyes, E.M.; Teng, Y.; Bates, P.J. A New Paradigm for Aptamer Therapeutic AS1411 Action: Uptake by Macropinocytosis and Its Stimulation by a Nucleolin-Dependent Mechanism. Cancer Res. 2010, 70, 8617–8629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soundararajan, S.; Wang, L.; Sridharan, V.; Chen, W.; Courtenay-Luck, N.; Jones, D.; Spicer, E.K.; Fernandes, D.J. Plasma Membrane Nucleolin Is a Receptor for the Anticancer Aptamer AS1411 in MV4-11 Leukemia Cells. Mol. Pharmacol. 2009, 76, 984–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, J.E.; Bambury, R.M.; VanAllen, E.M.; Drabkin, H.A.; Lara, P.N.; Harzstark, A.L.; Wagle, N.; Figlin, R.A.; Smith, G.W.; Garraway, L.A.; et al. A Phase II Trial of AS1411 (a Novel Nucleolin-Targeted DNA Aptamer) in Metastatic Renal Cell Carcinoma. Investig. New Drugs 2014, 32, 178–187. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Zhao, G.; Zhang, S.; Nigim, F.; Zhou, G.; Yu, Z.; Song, Y.; Chen, Y.; Li, Y. AS1411-Induced Growth Inhibition of Glioma Cells by up-Regulation of P53 and down-Regulation of Bcl-2 and Akt1 via Nucleolin. PLoS ONE 2016, 11, e0167094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soundararajan, S.; Chen, W.; Spicer, E.K.; Courtenay-Luck, N.; Fernandes, D.J. The Nucleolin Targeting Aptamer AS1411 Destabilizes Bcl-2 Messenger RNA in Human Breast Cancer Cells. Cancer Res. 2008, 68, 2358–2365. [Google Scholar] [CrossRef] [Green Version]
- Cheung, K.L.; Kim, C.; Zhou, M.M. The Functions of BET Proteins in Gene Transcription of Biology and Diseases. Front. Mol. Biosci. 2021, 8, 728777. [Google Scholar] [CrossRef]
- Wang, L.; Lee, J.Y.; Gao, L.; Yin, J.; Duan, Y.; Jimenez, L.A.; Adkins, G.B.; Ren, W.; Li, L.; Fang, J.; et al. A DNA Aptamer for Binding and Inhibition of DNA Methyltransferase 1. Nucleic Acids Res. 2019, 47, 11527–11537. [Google Scholar] [CrossRef] [Green Version]
- Gerstberger, S.; Hafner, M.; Tuschl, T. A Census of Human RNA-Binding Proteins. Nat. Rev. Genet. 2014, 15, 829–845. [Google Scholar] [CrossRef]
- Quinones-Valdez, G.; Tran, S.S.; Jun, H.I.; Bahn, J.H.; Yang, E.W.; Zhan, L.; Brümmer, A.; Wei, X.; VanNostrand, E.L.; Pratt, G.A.; et al. Regulation of RNA Editing by RNA-Binding Proteins in Human Cells. Commun. Biol. 2019, 2, 19. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.L.; Li, B.; Luo, Y.X.; Lin, Q.; Liu, S.R.; Zhang, X.Q.; Zhou, H.; Yang, J.H.; Qu, L.H. Comprehensive Genomic Characterization of RNA-Binding Proteins across Human Cancers. Cell Rep. 2018, 22, 286–298. [Google Scholar] [CrossRef] [Green Version]
- Hentze, M.W.; Castello, A.; Schwarzl, T.; Preiss, T. A Brave New World of RNA-Binding Proteins. Nat. Rev. Mol. Cell Biol. 2018, 19, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Roos, M.; Pradère, U.; Ngondo, R.P.; Behera, A.; Allegrini, S.; Civenni, G.; Zagalak, J.A.; Marchand, J.R.; Menzi, M.; Towbin, H.; et al. A Small-Molecule Inhibitor of Lin28. ACS Chem. Biol. 2016, 11, 2773–2781. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; White, S.B.; Zhao, Q.; Lee, F.S. HIF-1α Binding to VHL Is Regulated by Stimulus-Sensitive Proline Hydroxylation. Proc. Natl. Acad. Sci. USA 2001, 98, 9630–9635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, M.; Oh, B.; Choi, J.; Sulistio, Y.A.; Woo, H.; Jo, A.; Kim, J.; Kim, E.; Kim, S.W.; Hwang, J.; et al. LIN 28A Loss of Function Is Associated with Parkinson’s Disease Pathogenesis. EMBO J. 2019, 38, e101196. [Google Scholar] [CrossRef]
- O’Leary, A.; Fernàndez-Castillo, N.; Gan, G.; Yang, Y.; Yotova, A.Y.; Kranz, T.M.; Grünewald, L.; Freudenberg, F.; Antón-Galindo, E.; Cabana-Domínguez, J.; et al. Behavioural and Functional Evidence Revealing the Role of RBFOX1 Variation in Multiple Psychiatric Disorders and Traits. Mol. Psychiatry 2022, 27, 4464–4473. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.T. G-Quartets 40 Years Later: From 5′-GMP to Molecular Biology and Supramolecular Chemistry. Angew. Chem. 2004, 43, 668–698. [Google Scholar] [CrossRef]
- Bryan, T.M. Mechanisms of DNA Replication and Repair: Insights from the Study of G-Quadruplexes. Molecules 2019, 24, 3439. [Google Scholar] [CrossRef] [Green Version]
- Spiegel, J.; Cuesta, S.M.; Adhikari, S.; Hänsel-Hertsch, R.; Tannahill, D.; Balasubramanian, S. G-Quadruplexes Are Transcription Factor Binding Hubs in Human Chromatin. Genome Biol. 2021, 22, 117. [Google Scholar] [CrossRef]
- Fleming, A.M.; Burrows, C.J. Interplay of Guanine Oxidation and G-Quadruplex Folding in Gene Promoters. J. Am. Chem. Soc. 2020, 142, 1115–1136. [Google Scholar] [CrossRef]
- Sauer, M.; Juranek, S.A.; Marks, J.; DeMagis, A.; Kazemier, H.G.; Hilbig, D.; Benhalevy, D.; Wang, X.; Hafner, M.; Paeschke, K. DHX36 Prevents the Accumulation of Translationally Inactive MRNAs with G4-Structures in Untranslated Regions. Nat. Commun. 2019, 10, 2421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.C.; Tippana, R.; Demeshkina, N.A.; Murat, P.; Balasubramanian, S.; Myong, S.; Ferré-D’amaré, A.R. Structural Basis of G-Quadruplex Unfolding by the DEAH/RHA Helicase DHX36. Nature 2018, 558, 465–483. [Google Scholar] [CrossRef] [PubMed]
- Heddi, B.; Cheong, V.V.; Martadinata, H.; Phan, A.T. Insights into G-Quadruplex Specific Recognition by the DEAH-Box Helicase RHAU: Solution Structure of a Peptide-Quadruplex Complex. Proc. Natl. Acad. Sci. USA 2015, 112, 9608–9613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giri, B.; Smaldino, P.J.; Thys, R.G.; Creacy, S.D.; Routh, E.D.; Hantgan, R.R.; Lattmann, S.; Nagamine, Y.; Akman, S.A.; Vaughn, J.P. G4 Resolvase 1 Tightly Binds and Unwinds Unimolecular G4-DNA. Nucleic Acids Res. 2011, 39, 7161–7178. [Google Scholar] [CrossRef] [Green Version]
- Do, N.Q.; Phan, A.T. Monomer-Dimer Equilibrium for the 5′-5′ Stacking of Propeller-Type Parallel-Stranded g-Quadruplexes: NMR Structural Study. Chem. Eur. J. 2012, 18, 14752–14759. [Google Scholar] [CrossRef]
- Coughlin, S.R. How the Protease Thrombin Talks to Cells. Proc. Natl. Acad. Sci. USA 1999, 96, 11023–11027. [Google Scholar] [CrossRef] [Green Version]
- Petrera, N.S.; Stafford, A.R.; Leslie, B.A.; Kretz, C.A.; Fredenburgh, J.C.; Weitz, J.I. Long Range Communication between Exosites 1 and 2 Modulates Thrombin Function. J. Biol. Chem. 2009, 284, 25620–25629. [Google Scholar] [CrossRef] [Green Version]
- Troisi, R.; Balasco, N.; Autiero, I.; Vitagliano, L.; Sica, F. Exosite Binding in Thrombin: A Global Structural/Dynamic Overview of Complexes with Aptamers and Other Ligands. Int. J. Mol. Sci. 2021, 22, 803. [Google Scholar] [CrossRef]
- Gallwitz, M.; Enoksson, M.; Thorpe, M.; Hellman, L. The Extended Cleavage Specificity of Human Thrombin. PLoS ONE 2012, 7, e31756. [Google Scholar] [CrossRef] [Green Version]
- Kretz, C.A.; Tomberg, K.; VanEsbroeck, A.; Yee, A.; Ginsburg, D. High Throughput Protease Profiling Comprehensively Defines Active Site Specificity for Thrombin and ADAMTS13. Sci. Rep. 2018, 8, 2788. [Google Scholar] [CrossRef] [Green Version]
- Vermaas, E.H.; Toole, J.J. Selection of Single-Stranded DNA Molecules That Bind and Inhibit Human Thrombin. Nature 1992, 355, 564–566. [Google Scholar]
- Tasset, D.M.; Kubik, M.F.; Steiner, W. Oligonucleotide Inhibitors of Human Thrombin That Bind Distinct Epitopes. J. Mol. Biol. 1997, 272, 688–698. [Google Scholar] [CrossRef] [PubMed]
- Hoshiyama, J.; Okada, Y.; Hayata, Y.; Eguchi, A.; Ueki, R.; Sando, S. Characterization of a DNA Aptamer with High Specificity toward Fibroblast Growth Factor Receptor 1. Chem. Lett. 2021, 50, 1949–1952. [Google Scholar] [CrossRef]
- Cowan, A.D.; Ciulli, A. Driving E3 Ligase Substrate Specificity for Targeted Protein Degradation: Lessons from Nature and the Laboratory. Annu. Rev. Biochem. 2022, 91, 295–319. [Google Scholar] [CrossRef]
- Weirauch, M.T.; Yang, A.; Albu, M.; Cote, A.G.; Montenegro-Montero, A.; Drewe, P.; Najafabadi, H.S.; Lambert, S.A.; Mann, I.; Cook, K.; et al. Determination and Inference of Eukaryotic Transcription Factor Sequence Specificity. Cell 2014, 158, 1431–1443. [Google Scholar] [CrossRef] [Green Version]
- Nitta, K.R.; Jolma, A.; Yin, Y.; Morgunova, E.; Kivioja, T.; Akhtar, J.; Hens, K.; Toivonen, J.; Deplancke, B.; Furlong, E.E.M.; et al. Conservation of Transcription Factor Binding Specificities across 600 Million Years of Bilateria Evolution. Elife 2015, 2015, e04837. [Google Scholar] [CrossRef] [Green Version]
- Jolma, A.; Yan, J.; Whitington, T.; Toivonen, J.; Nitta, K.R.; Rastas, P.; Morgunova, E.; Enge, M.; Taipale, M.; Wei, G.; et al. DNA-Binding Specificities of Human Transcription Factors. Cell 2013, 152, 327–339. [Google Scholar] [CrossRef] [Green Version]
- Hume, M.A.; Barrera, L.A.; Gisselbrecht, S.S.; Bulyk, M.L. UniPROBE, Update 2015: New Tools and Content for the Online Database of Protein-Binding Microarray Data on Protein-DNA Interactions. Nucleic Acids Res. 2015, 43, D117–D122. [Google Scholar] [CrossRef] [PubMed]
- Meylan, P.; Dreos, R.; Ambrosini, G.; Groux, R.; Bucher, P. EPD in 2020: Enhanced Data Visualization and Extension to NcRNA Promoters. Nucleic Acids Res. 2020, 48, D65–D69. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Rossi, J. Erratum: Aptamers as Targeted Therapeutics: Current Potential and Challenges. Nat. Rev. Drug Discov. 2017, 16, 440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glazier, D.A.; Glazier, D.A.; Liao, J.; Roberts, B.L.; Li, X.; Yang, K.; Stevens, C.M.; Tang, W.; Tang, W. Chemical Synthesis and Biological Application of Modified Oligonucleotides. Bioconjug. Chem. 2020, 31, 1213–1233. [Google Scholar] [CrossRef]
- Kumar, P.; Caruthers, M.H. DNA Analogues Modified at the Nonlinking Positions of Phosphorus. Acc. Chem. Res. 2020, 53, 2152–2166. [Google Scholar] [CrossRef]
- Clavé, G.; Reverte, M.; Vasseur, J.J.; Smietana, M. Modified Internucleoside Linkages for Nuclease-Resistant Oligonucleotides. RSC Chem. Biol. 2021, 2, 94–150. [Google Scholar] [CrossRef] [PubMed]
- Pellestor, F.; Paulasova, P. The Peptide Nucleic Acids (PNAs), Powerful Tools for Molecular Genetics and Cytogenetics. Eur. J. Hum. Genet. 2004, 12, 694–700. [Google Scholar] [CrossRef] [PubMed]
- Demidov, V.V.; Potaman, V.N.; Frank-Kamenetskil, M.D.; Egholm, M.; Buchard, O.; Sönnichsen, S.H.; Nlelsen, P.E. Stability of Peptide Nucleic Acids in Human Serum and Cellular Extracts. Biochem. Pharmacol. 1994, 48, 1310–1313. [Google Scholar] [CrossRef] [PubMed]
- Dieckmann, A.; Hagedorn, P.H.; Burki, Y.; Brügmann, C.; Berrera, M.; Ebeling, M.; Singer, T.; Schuler, F. A Sensitive In Vitro Approach to Assess the Hybridization-Dependent Toxic Potential of High Affinity Gapmer Oligonucleotides. Mol. Ther.-Nucleic Acids 2018, 10, 45–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burel, S.A.; Hart, C.E.; Cauntay, P.; Hsiao, J.; Machemer, T.; Katz, M.; Watt, A.; Bui, H.H.; Younis, H.; Sabripour, M.; et al. Hepatotoxicity of High Affinity Gapmer Antisense Oligonucleotides Is Mediated by RNase H1 Dependent Promiscuous Reduction of Very Long Pre-MRNA Transcripts. Nucleic Acids Res. 2015, 44, 2093–2109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, W.; DeHoyos, C.L.; Migawa, M.T.; Vickers, T.A.; Sun, H.; Low, A.; Bell, T.A.; Rahdar, M.; Mukhopadhyay, S.; Hart, C.E.; et al. Chemical Modification of PS-ASO Therapeutics Reduces Cellular Protein-Binding and Improves the Therapeutic Index. Nat. Biotechnol. 2019, 37, 640–650. [Google Scholar] [CrossRef]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576.e16. [Google Scholar] [CrossRef] [Green Version]
- Inuzuka, H.; Liu, J.; Wei, W.; Rezaeian, A.-H. PROTAC Technology for the Treatment of Alzheimer’s Disease: Advances and Perspectives. Acta Mater. Med. 2022, 1, 24–41. [Google Scholar] [CrossRef]
- Visvikis-Siest, S.; Theodoridou, D.; Kontoe, M.S.; Kumar, S.; Marschler, M. Milestones in Personalized Medicine: From the Ancient Time to Nowadays—The Provocation of COVID-19. Front. Genet. 2020, 11, 569175. [Google Scholar] [CrossRef] [PubMed]
- Basu, P.; Mukhopadhyay, A.; Konishi, I. Targeted Therapy for Gynecologic Cancers: Toward the Era of Precision Medicine. Int. J. Gynecol. Obstet. 2018, 143, 131–136. [Google Scholar] [CrossRef]
- Jevtić, P.; Haakonsen, D.L.; Rapé, M. An E3 Ligase Guide to the Galaxy of Small-Molecule-Induced Protein Degradation. Cell Chem. Biol. 2021, 28, 1000–1013. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Li, S.; Shu, H.B. The Membrane-Associated MARCH E3 Ligase Family: Emerging Roles in Immune Regulation. Front. Immunol. 2019, 10, 1751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okamoto, T.; Imaizumi, K.; Kaneko, M. The Role of Tissue-Specific Ubiquitin Ligases, RNF183, RNF186, RNF182 and RNF152, in Disease and Biological Function. Int. J. Mol. Sci. 2020, 21, 3921. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.B.; Zhao, X.; Chen, L.J.; Qian, H.L.; Yan, X.P. Fabrication of G-Quadruplex/Porphyrin Conjugated Gold/Persistent Luminescence Theranostic Nanoprobe for Imaging-Guided Photodynamic Therapy. Talanta 2021, 233, 122567. [Google Scholar] [CrossRef]
- Vindigni, G.; Raniolo, S.; Iacovelli, F.; Unida, V.; Stolfi, C.; Desideri, A.; Biocca, S. AS1411 Aptamer Linked to DNA Nanostructures Diverts Its Traffic inside Cancer Cells and Improves Its Therapeutic Efficacy. Pharmaceutics 2021, 13, 1671. [Google Scholar] [CrossRef]
- Zhang, W.; Zhou, R.; Yang, Y.; Peng, S.; Xiao, D.; Kong, T.; Cai, X.; Zhu, B. Aptamer-Mediated Synthesis of Multifunctional Nano-Hydroxyapatite for Active Tumour Bioimaging and Treatment. Cell Prolif. 2021, 54, e13105. [Google Scholar] [CrossRef]
- Lopes-Nunes, J.; Agonia, A.S.; Rosado, T.; Gallardo, E.; Palmeira-De-oliveira, R.; Palmeira-De-oliveira, A.; Martinez-De-oliveira, J.; Fonseca-Moutinho, J.; Campello, M.P.C.; Paiva, A.; et al. Aptamer-Functionalized Gold Nanoparticles for Drug Delivery to Gynecological Carcinoma Cells. Cancers 2021, 13, 4038. [Google Scholar] [CrossRef]
- Dehghani, S.; Alibolandi, M.; Tehranizadeh, Z.A.; Oskuee, R.K.; Nosrati, R.; Soltani, F.; Ramezani, M. Self-Assembly of an Aptamer-Decorated Chimeric Peptide Nanocarrier for Targeted Cancer Gene Delivery. Colloids Surf. B Biointerfaces 2021, 208, 112047. [Google Scholar] [CrossRef] [PubMed]
- Mehrnia, S.S.; Hashemi, B.; Mowla, S.J.; Nikkhah, M.; Arbabi, A. Radiosensitization of Breast Cancer Cells Using AS1411 Aptamer-Conjugated Gold Nanoparticles. Radiat. Oncol. 2021, 16, 33. [Google Scholar] [CrossRef] [PubMed]
- Miranda, A.; Santos, T.; Largy, E.; Cruz, C. Locking up the As1411 Aptamer with a Flanking Duplex: Towards an Improved Nucleolin-Targeting. Pharmaceuticals 2021, 14, 121. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Ga, L.; Ai, J.; Wang, Y. Progress in Cancer Drug Delivery Based on AS1411 Oriented Nanomaterials. J. Nanobiotechnol. 2022, 20, 57. [Google Scholar] [CrossRef]
- Qi, S.M.; Dong, J.; Xu, Z.Y.; Cheng, X.D.; Zhang, W.D.; Qin, J.J. PROTAC: An Effective Targeted Protein Degradation Strategy for Cancer Therapy. Front. Pharmacol. 2021, 12, 692574. [Google Scholar] [CrossRef]
- Li, K.; Crews, C.M. PROTACs: Past, Present and Future. Chem. Soc. Rev. 2022, 51, 5214–5236. [Google Scholar] [CrossRef] [PubMed]
- Ge, Z.; Liu, J.; Guo, L.; Yao, G.; Li, Q.; Wang, L.; Li, J.; Fan, C. Programming Cell-Cell Communications with Engineered Cell Origami Clusters. J. Am. Chem. Soc. 2020, 142, 8800–8808. [Google Scholar] [CrossRef]
- Rosier, B.J.H.M.; Markvoort, A.J.; Gumí Audenis, B.; Roodhuizen, J.A.L.; den Hamer, A.; Brunsveld, L.; de Greef, T.F.A. Proximity-Induced Caspase-9 Activation on a DNA Origami-Based Synthetic Apoptosome. Nat. Catal. 2020, 3, 295–306. [Google Scholar] [CrossRef]
- Seeman, N.C.; Sleiman, H.F. DNA Nanotechnology. Nat. Rev. Mater. 2017, 3, 17068. [Google Scholar] [CrossRef]
- Fu, T.; Lyu, Y.; Liu, H.; Peng, R.; Zhang, X.; Ye, M.; Tan, W. DNA-Based Dynamic Reaction Networks. Trends Biochem. Sci. 2018, 43, 547–560. [Google Scholar] [CrossRef]
- Veneziano, R.; Moyer, T.J.; Stone, M.B.; Wamhoff, E.C.; Read, B.J.; Mukherjee, S.; Shepherd, T.R.; Das, J.; Schief, W.R.; Irvine, D.J.; et al. Role of Nanoscale Antigen Organization on B-Cell Activation Probed Using DNA Origami. Nat. Nanotechnol. 2020, 15, 716–723. [Google Scholar] [CrossRef]
- Bian, X.; Zhang, Z.; Xiong, Q.; DeCamilli, P.; Lin, C. A Programmable DNA-Origami Platform for Studying Lipid Transfer between Bilayers. Nat. Chem. Biol. 2019, 15, 830–837. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; Wu, W.; Wang, Y.; Song, K.; Chen, G.; Chen, Y.; Wang, R.; Xu, J.; Cui, K.; Chen, H.; et al. DNA-Modularized Construction of Bivalent Ligands Precisely Regulates Receptor Binding and Activation DNA-Modularized Construction of Bivalent Ligands Precisely Regulates Receptor Binding and Activation. Chem 2023. [Google Scholar] [CrossRef]
- Melancon, B.J.; Tarr, J.C.; Panarese, J.D.; Wood, M.R.; Lindsley, C.W. Allosteric Modulation of the M1 Muscarinic Acetylcholine Receptor: Improving Cognition and a Potential Treatment for Schizophrenia and Alzheimer’s Disease. Drug Discov. Today 2013, 18, 1185–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bender, A.M.; Jones, C.K.; Lindsley, C.W. Classics in Chemical Neuroscience: Xanomeline. ACS Chem. Neurosci. 2017, 8, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Jumpertz, S.; Zhang, Y.; Appenroth, D.; Fleck, C.; Mohr, K.; Tränkle, C.; Decker, M. Hybrid Molecules from Xanomeline and Tacrine: Enhanced Tacrine Actions on Cholinesterases and Muscarinic M1 Receptors. J. Med. Chem. 2010, 53, 2094–2103. [Google Scholar] [CrossRef] [PubMed]
- Bonifazi, A.; Yano, H.; Del Bello, F.; Farande, A.; Quaglia, W.; Petrelli, R.; Matucci, R.; Nesi, M.; Vistoli, G.; Ferré, S.; et al. Synthesis and Biological Evaluation of a Novel Series of Heterobivalent Muscarinic Ligands Based on Xanomeline and 1-[3-(4-Butylpiperidin-1-Yl)Propyl]-1,2,3,4-Tetrahydroquinolin-2-One (77-LH-28-1). J. Med. Chem. 2014, 57, 9065–9077. [Google Scholar] [CrossRef]
- Wess, J.; Eglen, R.M.; Gautam, D. Muscarinic Acetylcholine Receptors: Mutant Mice Provide New Insights for Drug Development. Nat. Rev. Drug Discov. 2007, 6, 721–733. [Google Scholar] [CrossRef]
- Edmondson, S.D.; Yang, B.; Fallan, C. Proteolysis Targeting Chimeras (PROTACs)in ‘beyond Rule-of-Five’ Chemical Space: Recent Progress and Future Challenges. Bioorganic Med. Chem. Lett. 2019, 29, 1555–1564. [Google Scholar] [CrossRef]
- Cantrill, C.; Chaturvedi, P.; Rynn, C.; Petrig Schaffland, J.; Walter, I.; Wittwer, M.B. Fundamental Aspects of DMPK Optimization of Targeted Protein Degraders. Drug Discov. Today 2020, 25, 969–982. [Google Scholar] [CrossRef]
- Garber, K. The PROTAC Gold Rush. Nat. Biotechnol. 2022, 40, 12–16. [Google Scholar] [CrossRef]
- Beck, H.; Härter, M.; Haß, B.; Schmeck, C.; Baerfacker, L. Small Molecules and Their Impact in Drug Discovery: A Perspective on the Occasion of the 125th Anniversary of the Bayer Chemical Research Laboratory. Drug Discov. Today 2022, 27, 1560–1574. [Google Scholar] [CrossRef] [PubMed]
- Roberts, T.C.; Langer, R.; Wood, M.J.A. Advances in Oligonucleotide Drug Delivery. Nat. Rev. Drug Discov. 2020, 19, 673–694. [Google Scholar] [CrossRef] [PubMed]
- Hopfner, K.P.; Hornung, V. Molecular Mechanisms and Cellular Functions of CGAS–STING Signalling. Nat. Rev. Mol. Cell Biol. 2020, 21, 501–521. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Names | E3 Ligases | POIs | Cell Lines | Ref. |
|---|---|---|---|---|
| Core DNA/RNA Sequences Used | ||||
| TRAFTAC | VHL | NF-ĸB and brachyury | HeLa | [16] |
| To generate double-stranded DNA for recruiting POIs, a NF-ĸB binding sequence 5′-GGGAATTTCC-3′ and a brachyury binding sequence 5′-AATTTCACACCT-3′ were referenced. | ||||
| OligoTRAFTAC | VHL | cMYC and brachyury | HeLa and HEK293T | [17] |
| To generate double-stranded warheads, a cMYC binding consensus sequence 5′-CACGTGGTTGCCACGTG-3′ and a brachyury binding DNA sequence 5′-AATTTCACACCTAGGTGTGAAATT-3′ were referenced. | ||||
| O’PROTAC | VHL and CRBN | binding factor 1 (LEF1) and ETS-related gene (ERG) | PC-3 | [18] |
| A 18-mer oligonucleotide, 5′-TACAAAGATCAAAGGGTT-3′, was referenced for generating double-stranded DNA specific for targeting LEF1.A 19-mer oligonucleotide, 5′-ACGGACCGGAAATCCGGTT-3′, was referenced for generating double-stranded DNA specific for targeting ERG. | ||||
| TF-PROTAC | VHL | E2F and the subunit of NF-ĸB, p65 | HeLa | [19] |
| A single-stranded DNA sequence of 5′-TGGGGACTTTCCAGTTTCTGGAAAGTCCCCA-3′ was used as a warhead to target the subunit of NF-ĸB, p65, while double-stranded DNA 15-mers (sense chain was 5′-CTAGATTTCCCGCG-3′ and the antisense chain was 5′-CTAGCGCGGAAAT-3′) were selected to bait cancer-related E2F. | ||||
| SNIPER | IAP | ERα | MCF7 | [20] |
| The 21-mer, ERα-specific, double-stranded DNA was referenced from 5′-GTCAGGTCACAGTGACCTGAT-3′. | ||||
| Aptamer-PROTAC conjugates | VHL | BRD2, 3, and 4 | MCF7 | [21] |
| The 26-mer AS1411 was 5′-GGTGGTGGTGGTTGTGGTGGTGGTGG-3′. | ||||
| Aptamer-warheaded PROTAC | VHL | nucleolin | MCF7 and BT474 | [22] |
| The same AS1411 sequence as above was used. | ||||
| Light controllable, aptamer-warheaded PROTAC | CRBN | nucleolin | MCF7 and MDA-MB-231 | [23] |
| The same AS1411 sequence as above was used. | ||||
| RNA-warheaded PROTAC | VHL/HIF-1α | LIN28 and RBFOX1 | K562 | [24] |
| 5′-AGGAGAU-3′ was for Lin28, while 5′-UGCAUGU-3′ was for RBFOX1. | ||||
| G4-PROTAC | VHL and CRBN | RHAU | HeLa | [25] |
| RHAU-targeting G4 sequence was TT(GGGT)4. | ||||
| Apt-clean | No E3 used; instead, thrombin protease | FGFR1 | 3T3-L1 | [26] |
| The aptamer sequence of HD1 was 5′-GGTTGGTGTGGTTGG-3′, while the aptamer sequence of HD22 was 5′-AGTCCGTGGTAGGGCAGGTTGGGGTGACT-3′.A 38-mer DNA aptamer of FGFR1-binding SL38.2 was 5′-CGATCGATGGATGGTAGCTCGGTCGGGGTGGGTGGGTTGGCAATCGATCG-3′. | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shih, P.-C.; Naganuma, M.; Demizu, Y.; Naito, M. Current Status of Oligonucleotide-Based Protein Degraders. Pharmaceutics 2023, 15, 765. https://doi.org/10.3390/pharmaceutics15030765
Shih P-C, Naganuma M, Demizu Y, Naito M. Current Status of Oligonucleotide-Based Protein Degraders. Pharmaceutics. 2023; 15(3):765. https://doi.org/10.3390/pharmaceutics15030765
Chicago/Turabian StyleShih, Po-Chang, Miyako Naganuma, Yosuke Demizu, and Mikihiko Naito. 2023. "Current Status of Oligonucleotide-Based Protein Degraders" Pharmaceutics 15, no. 3: 765. https://doi.org/10.3390/pharmaceutics15030765