
Multi-Compartmental Dissolution Method, an Efficient Tool for the Development of Enhanced Bioavailability Formulations Containing Poorly Soluble Acidic Drugs
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Equilibrium Solubility Measurements
2.3. Dissolution Testing
2.3.1. USP Dissolution Method
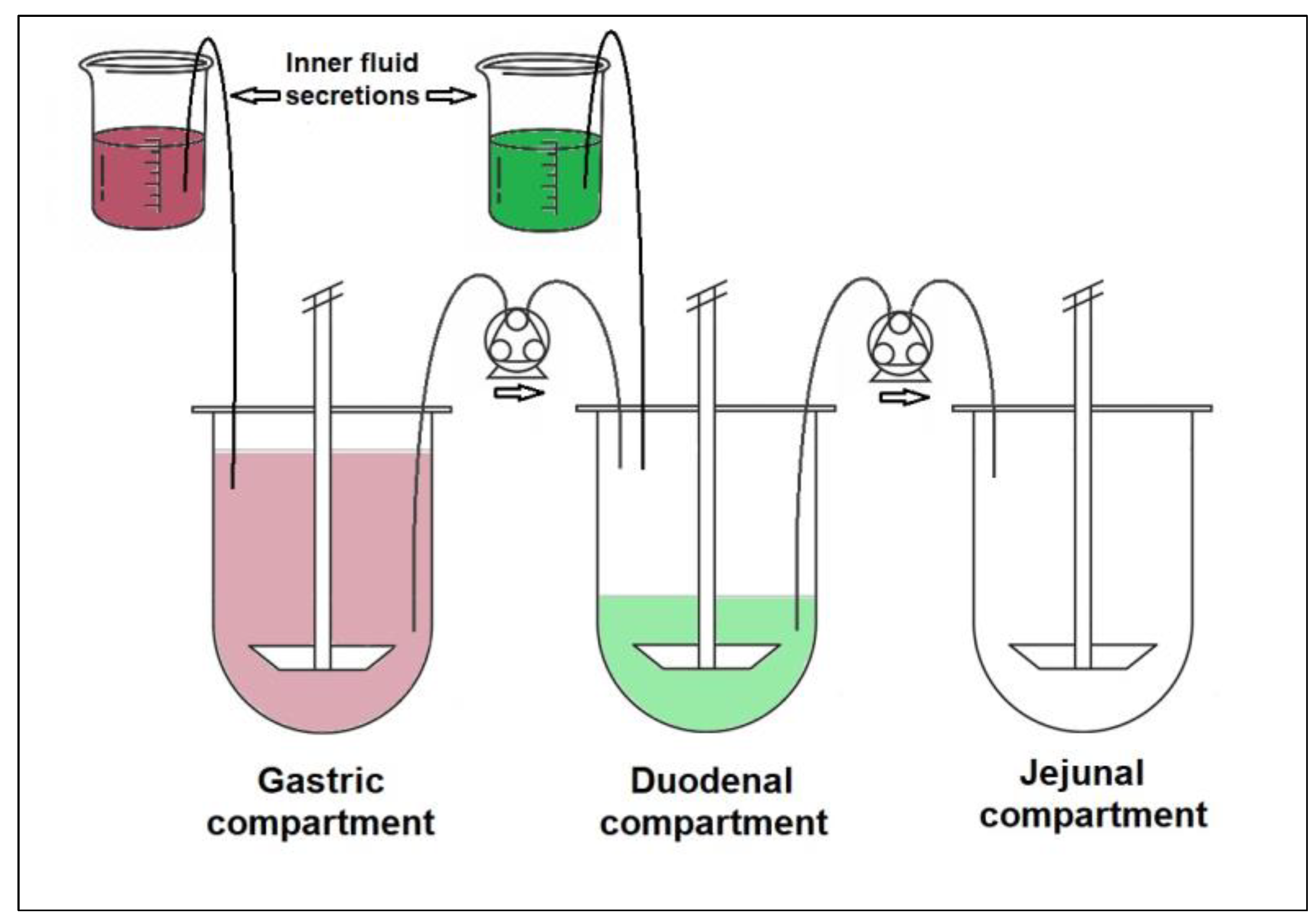
2.3.2. GIS Dissolution Method
2.4. Determination of Dissolved Drug Content by High-Performance Liquid Chromatography (HPLC)
2.5. In Vitro In Vivo Correlation (IVIVC)
3. Results and Discussion
3.1. Thermodynamic Equilibrium Solubility Measurements
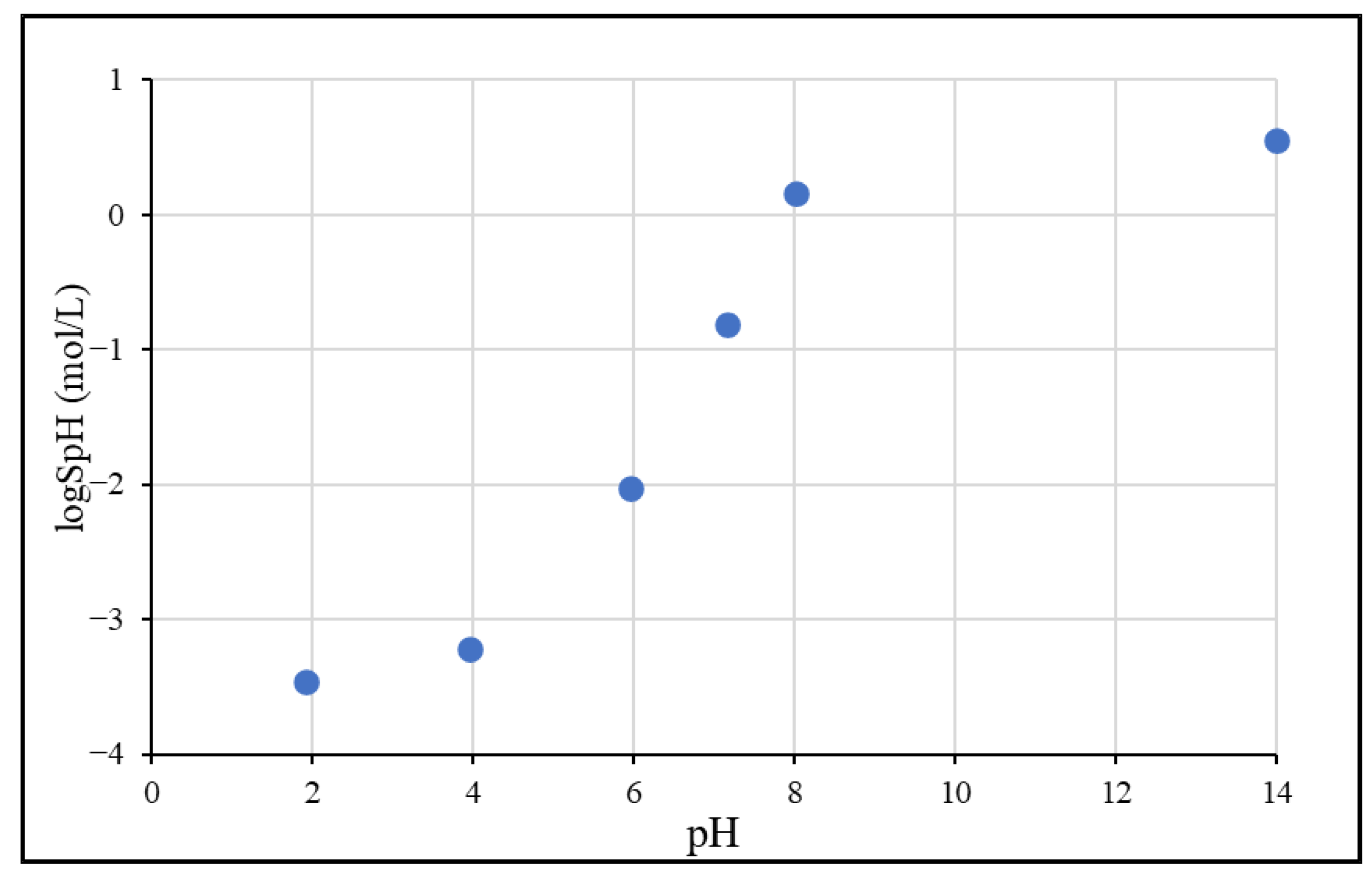
3.1.1. pH-Dependent Solubility
3.1.2. Solubility in Biorelevant Media
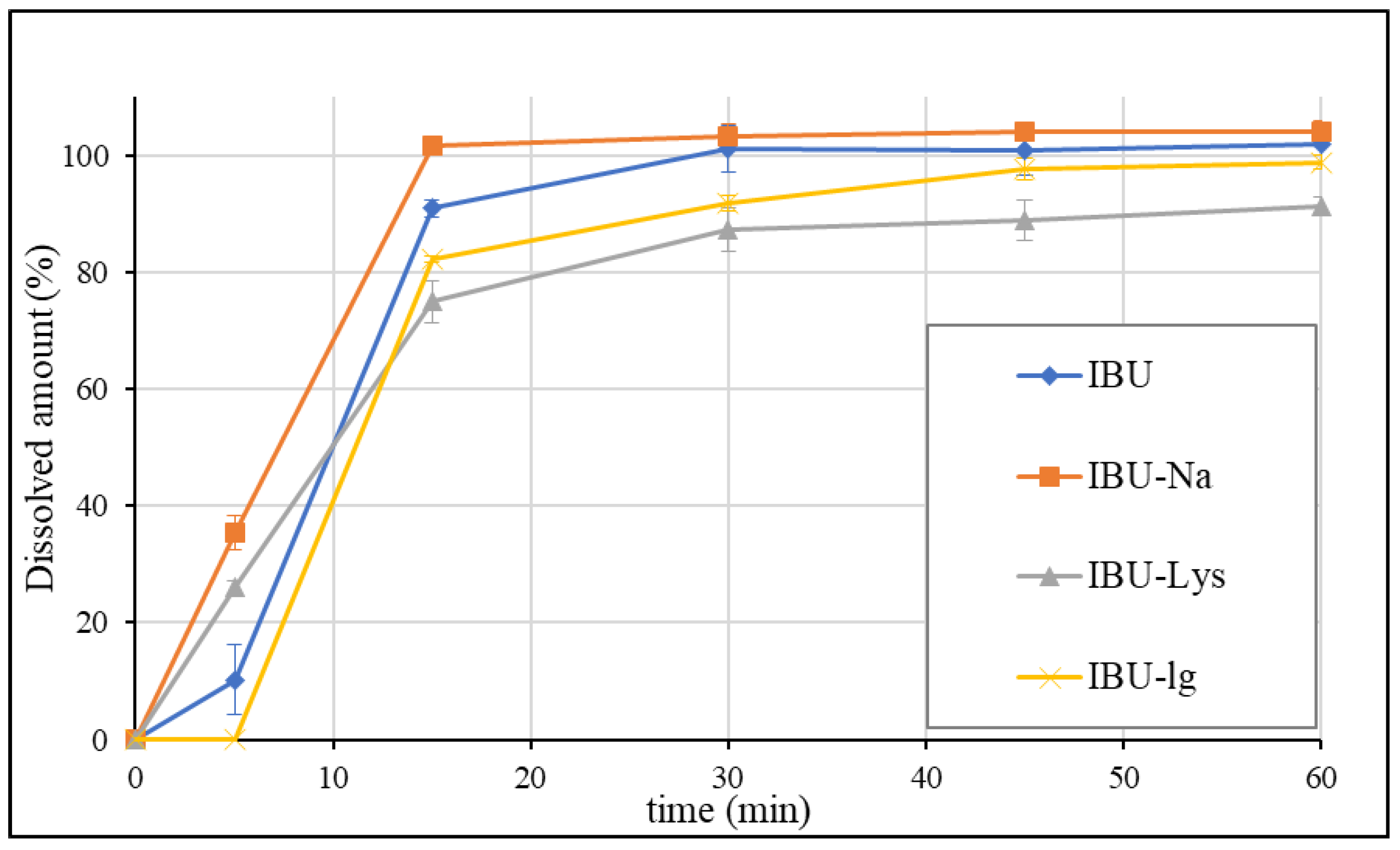
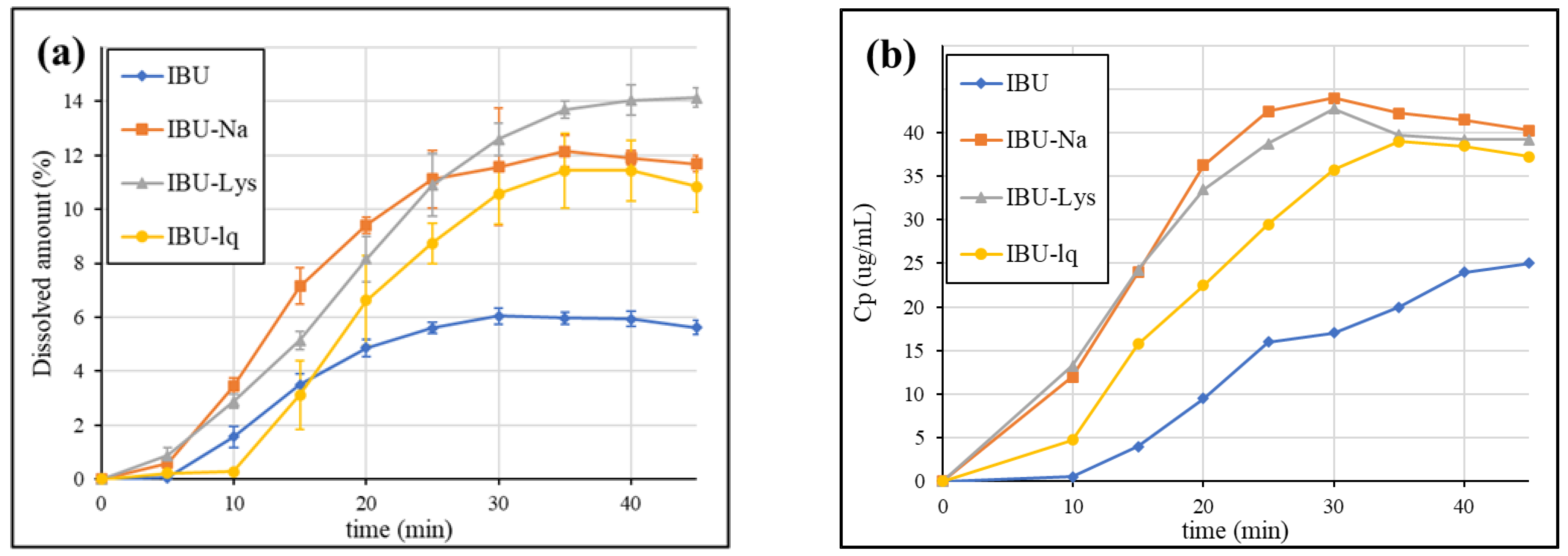
3.2. Dissolution Results Obtained by the USP Method
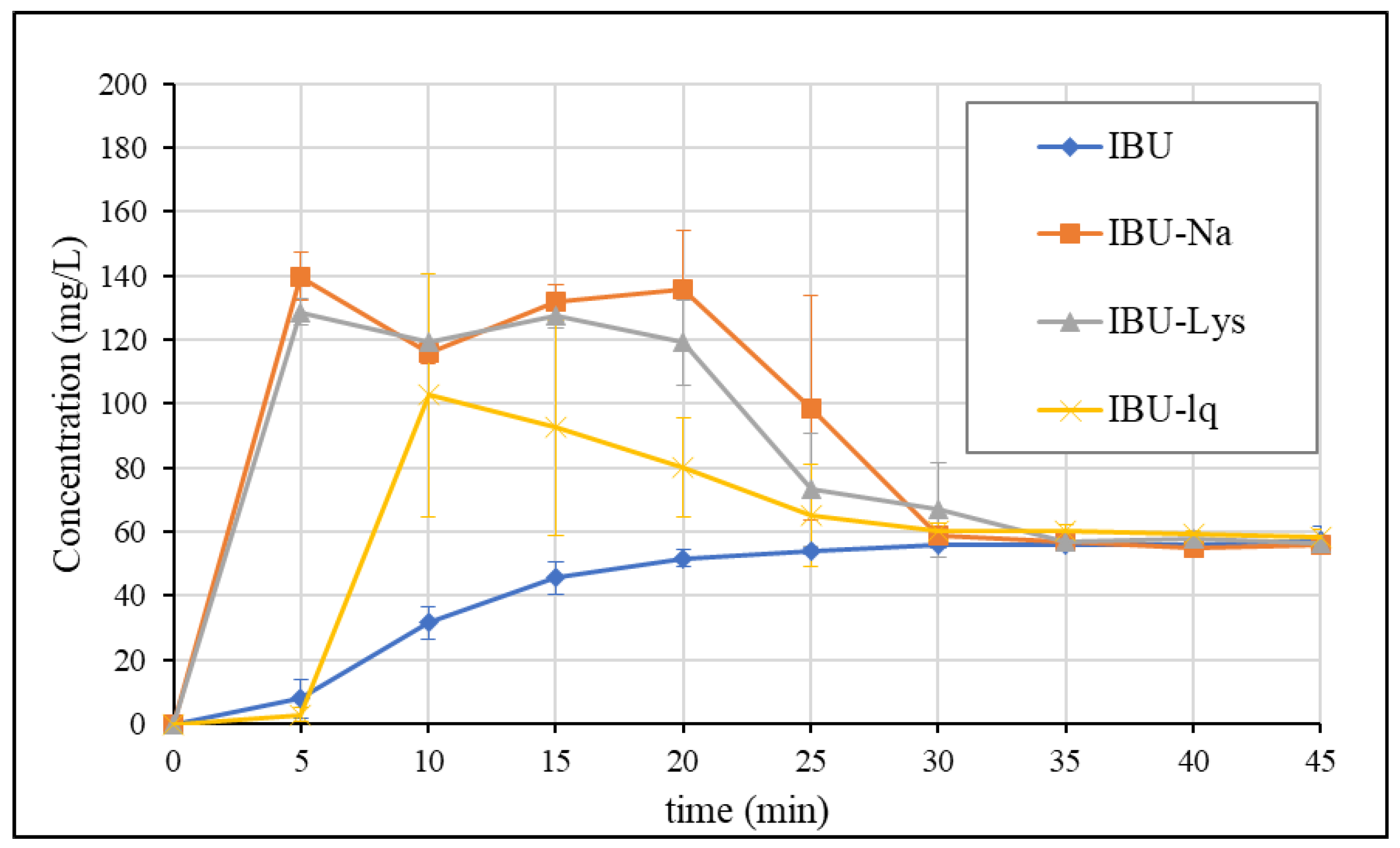
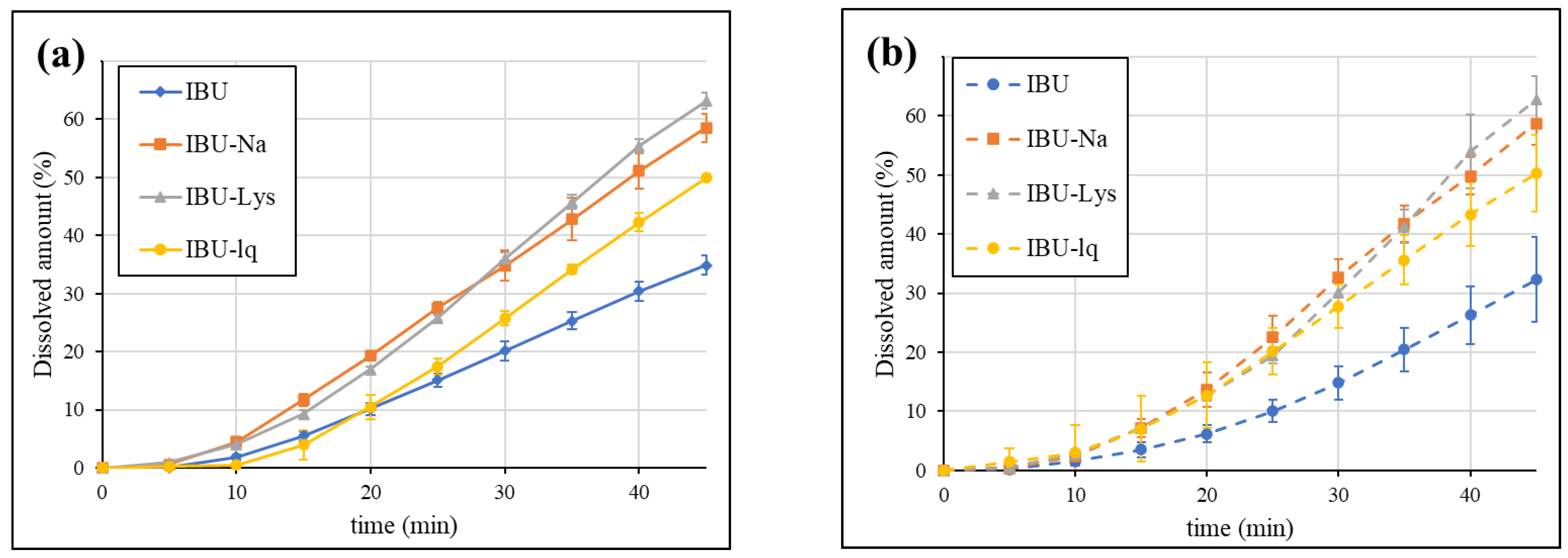
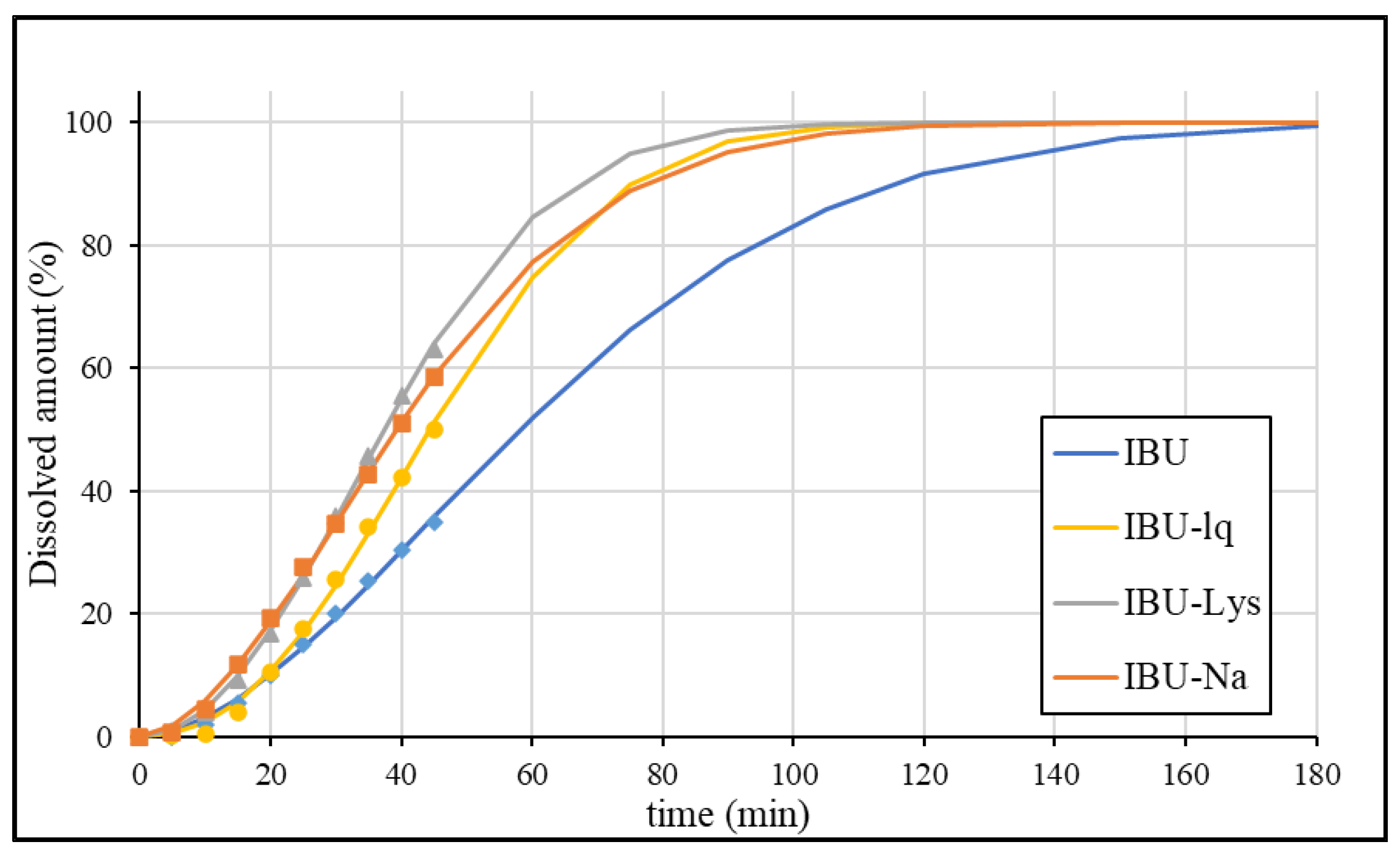
3.3. Dissolution Results Obtained by GIS Method
3.4. Establishment of the IVIVC Model
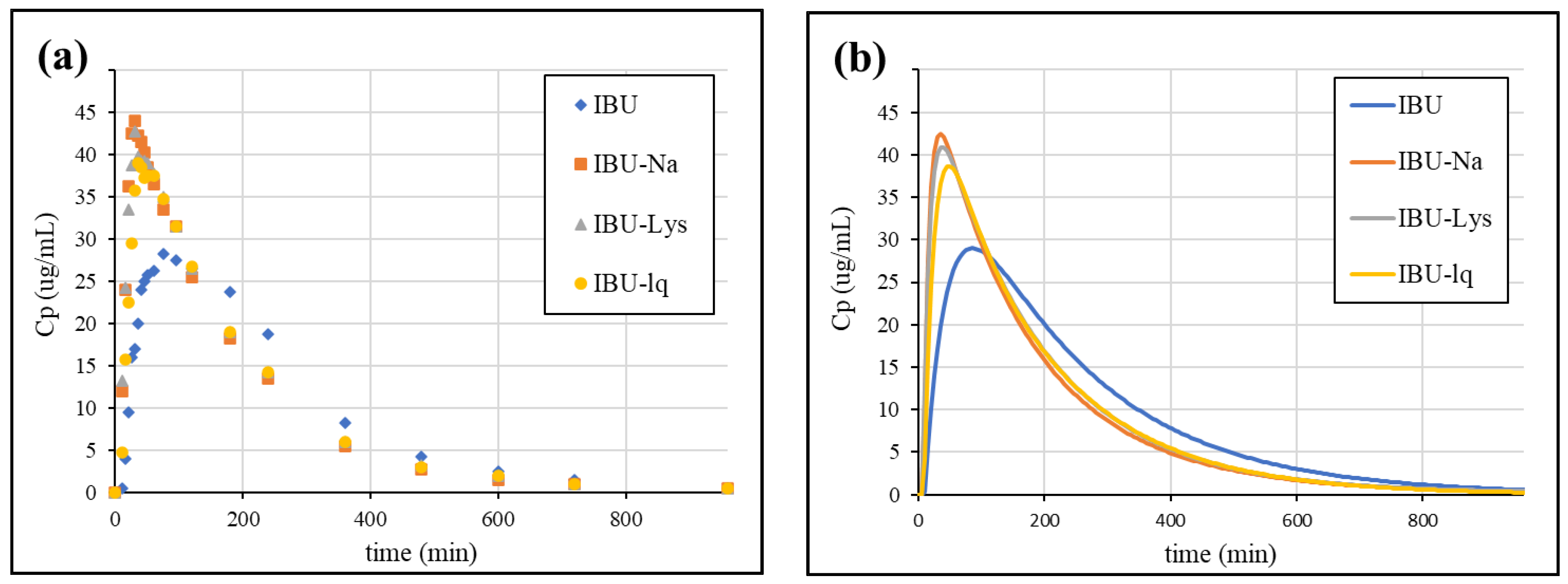
3.4.1. Analysis of In Vivo Data
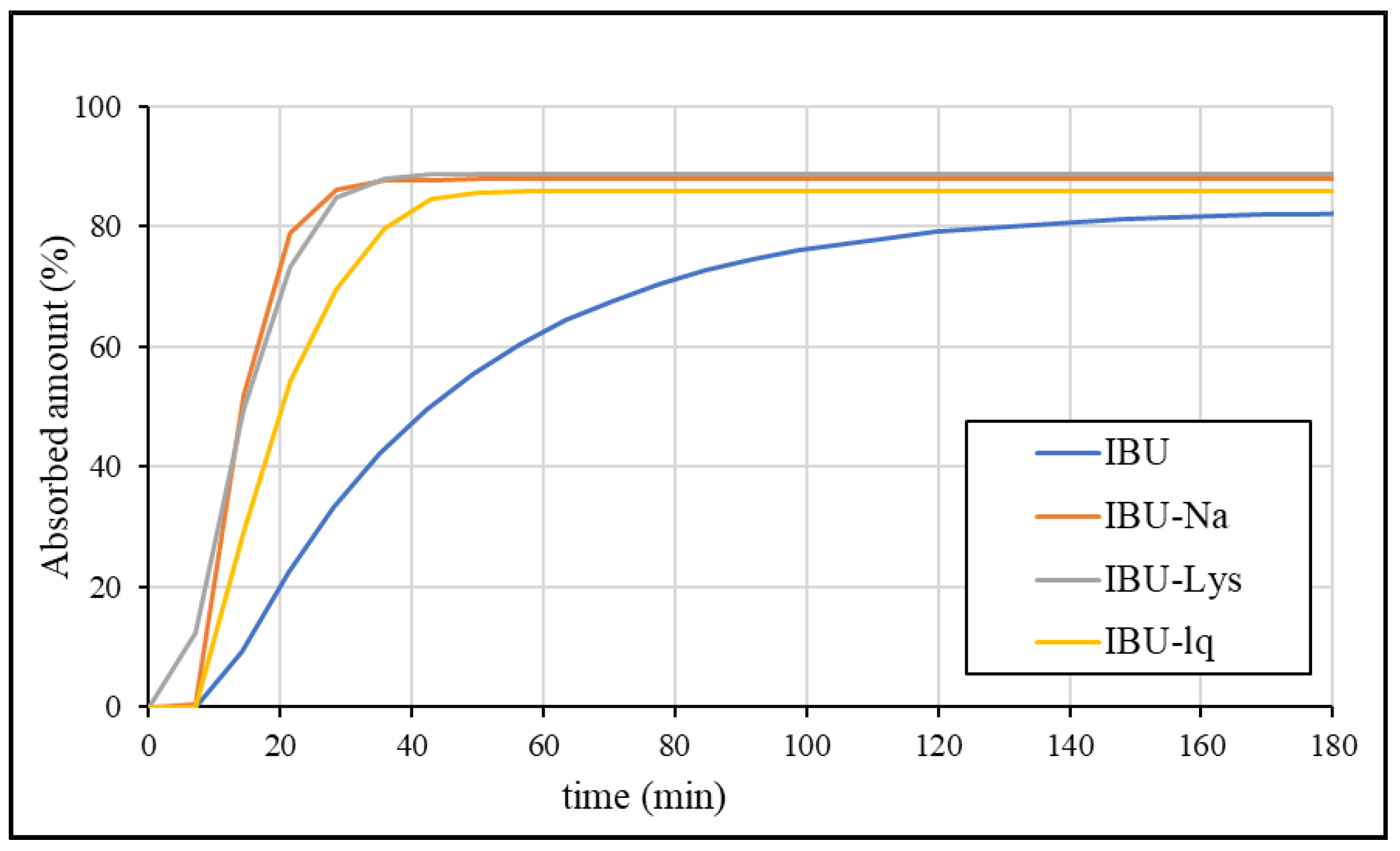
3.4.2. Fitting of In Vitro Dissolution Data
3.4.3. Correlation
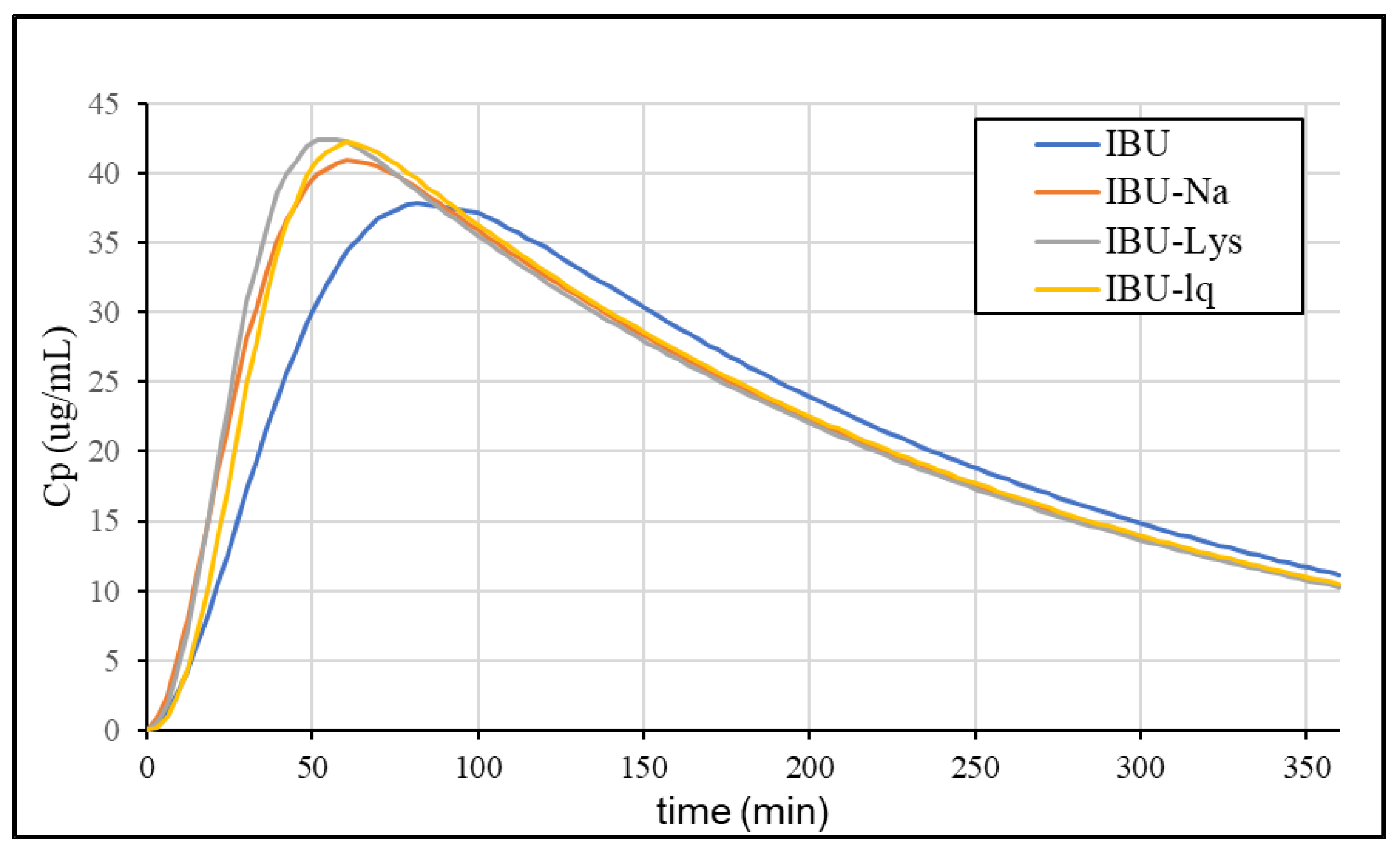
3.4.4. Simulation of Plasma Concentration Profiles
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| API | Active pharmaceutical ingredient |
| ASD | Artificial stomach duodenal model |
| BCS | Biopharmaceutics Classification System |
| BR | Britton–Robinson |
| DGM | Dynamic Gastric Model |
| GIS | Gastrointestinal Simulator |
| HPLC | High-Performance Liquid Chromatography |
| HGM | Human Gastric Simulator |
| IVIVC | In vitro–In vivo correlation |
| NSAID | Non-Steroidal Anti-Inflammatory Drug |
| SpH | PH-dependent solubility |
| TIM | TNO gastro-Intestinal Model |
| UIR | Unit impulse response |
| USP | United States Pharmacopoeia |
References
- Stegemann, S.; Leveiller, F.; Franchi, D.; de Jong, H.; Linden, H. When poor solubility becomes an issue: From early stage to proof of concept. Eur. J. Pharm. Sci. 2007, 31, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Amidon, G.L.; Lennernäs, H.; Shah, V.P.; Crison, J.R. A Theorical Basis for a Biopharmaceutic Drug Classification: The Correlation of In Vitro Drug Product Dissolution and In Vivo Bioavailability. Pharm. Res. 1995, 12, 413–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahlgren, D.; Sjögren, E.; Lennernäs, H. Intestinal absorption of BCS class II drugs administered as nanoparticles: A review based on in vivo data from intestinal perfusion models. ADMET DMPK 2020, 8, 375–390. [Google Scholar] [CrossRef] [PubMed]
- Serajuddin, A.T.M. Salt formation to improve drug solubility. Adv. Drug Deliv. Rev. 2007, 59, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Kanaujia, P.; Poovizhi, P.; Ng, W.K.; Tan, R.B.H. Amorphous formulations for dissolution and bioavailability enhancement of poorly soluble APIs. Powder Technol. 2015, 285, 2–15. [Google Scholar] [CrossRef]
- Kumar, R.; Thakur, A.K.; Chaudhari, P.; Banerjee, N. Particle Size Reduction Techniques of Pharmaceutical Compounds for the Enhancement of Their Dissolution Rate and Bioavailability. J. Pharm. Innov. 2022, 17, 333–352. [Google Scholar] [CrossRef]
- Agilent Dissolution Seminar Series. Available online: https://www.agilent.com/cs/library/flyers/Public/Dissolution_Seminar_Series.pdf (accessed on 10 January 2023).
- USP Chapter 711—Dissolution. Available online: https://www.usp.org/sites/default/files/usp/document/harmonization/gen-method/stage_6_monograph_25_feb_2011.pdf (accessed on 9 February 2023).
- Kostewicz, E.S.; Abrahamsson, B.; Brewster, M.; Brouwers, J.; Butler, J.; Carlert, S.; Dickinson, P.A.; Dressman, J.; Holm, R.; Klein, S.; et al. In vitro models for the prediction of in vivo performance of oral dosage forms. Eur. J. Pharm. Sci. 2014, 57, 342–366. [Google Scholar] [CrossRef]
- Vatier, J.; Malikova-Sekera, E.; Vitre, M.T.; Mignon, M. An artificial stomach-duodenum model for the in-vitro evaluation of antacids. Aliment. Pharmacol. Ther. 1992, 6, 447–458. [Google Scholar] [CrossRef]
- Carino, S.R.; Sperry, D.C.; Hawley, M. Relative bioavailability estimation of carbamazepine crystal forms using an artificial stomach-duodenum model. J. Pharm. Sci. 2006, 95, 116–125. [Google Scholar] [CrossRef]
- Bhattachar, S.N.; Perkins, E.J.; Tan, J.S.; Burns, L.J. Effect of gastric pH on the pharmacokinetics of a BCS Class II compound in dogs: Utilization of an artificial stomach and duodenum dissolution model and GastroPlus,TM simulations to predict absorption. J. Pharm. Sci. 2011, 100, 4756–4765. [Google Scholar] [CrossRef]
- Minekus, M.; Smeets-Peeters, M.; Havenaar, R.; Bernalier, A.; Fonty, G.; Marol-Bonnin, S.; Alric, M.; Marteau, P.; Huis In’t Veld, J.H.J. A computer-controlled system to simulate conditions of the large intestine with peristaltic mixing, water absorption and absorption of fermentation products. Appl. Microbiol. Biotechnol. 1999, 53, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Wickham, M.J.S.; Faulks, R.M.; Mann, J.; Mandalari, G. The design, operation, and application of a dynamic gastric model. Dissolution Technol. 2012, 19, 15–22. [Google Scholar] [CrossRef]
- Kong, F.; Singh, R.P. A Human Gastric Simulator (HGS) to Study Food Digestion in Human Stomach. J. Food Sci. 2010, 75, E627–E635. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, S.; Tsume, Y.; Amidon, G.E.; Amidon, G.L. Evaluation of a three compartment in Vitro gastrointestinal simulator dissolution apparatus to predict in Vivo dissolution. J. Pharm. Sci. 2014, 103, 3416–3422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsume, Y.; Takeuchi, S.; Matsui, K.; Amidon, G.E.; Amidon, G.L. In vitro dissolution methodology, mini-Gastrointestinal Simulator (mGIS), predicts better in vivo dissolution of a weak base drug, dasatinib. Eur. J. Pharm. Sci. 2015, 76, 203–212. [Google Scholar] [CrossRef]
- Tsume, Y.; Matsui, K.; Searls, A.L.; Takeuchi, S.; Amidon, G.E.; Sun, D.; Amidon, G.L. The impact of supersaturation level for oral absorption of BCS class IIb drugs, dipyridamole and ketoconazole, using in vivo predictive dissolution system: Gastrointestinal Simulator (GIS). Eur. J. Pharm. Sci. 2017, 102, 126–139. [Google Scholar] [CrossRef]
- Tsume, Y.; Igawa, N.; Drelich, A.J.; Amidon, G.E.; Amidon, G.L. The Combination of GIS and Biphasic to Better Predict In Vivo Dissolution of BCS Class IIb Drugs, Ketoconazole and Raloxifene. J. Pharm. Sci. 2018, 107, 307–316. [Google Scholar] [CrossRef] [Green Version]
- Tsume, Y.; Igawa, N.; Drelich, A.J.; Ruan, H.; Amidon, G.E.; Amidon, G.L. The in vivo predictive dissolution for immediate release dosage of donepezil and danazol, BCS class IIc drugs, with the GIS and the USP II with biphasic dissolution apparatus. J. Drug Deliv. Sci. Technol. 2020, 56, 1–9. [Google Scholar] [CrossRef]
- Hens, B.; Bermejo, M.; Tsume, Y.; Gonzalez-Alvarez, I.; Ruan, H.; Matsui, K.; Amidon, G.E.; Cavanagh, K.L.; Kuminek, G.; Benninghoff, G.; et al. Evaluation and optimized selection of supersaturating drug delivery systems of posaconazole (BCS class 2b) in the gastrointestinal simulator (GIS): An in vitro-in silico-in vivo approach. Eur. J. Pharm. Sci. 2018, 115, 258–269. [Google Scholar] [CrossRef]
- Day, R.O.; Graham, G.G. NSAIDs and their Indications. In Encyclopedia of Pain; Springer: Berlin/Heidelberg, Germany, 2007; pp. 1460–1463. [Google Scholar]
- Legg, T.J.; Laurent, A.L.; Leyva, R.; Kellstein, D. Ibuprofen Sodium Is Absorbed Faster than Standard Ibuprofen Tablets: Results of Two Open-Label, Randomized, Crossover Pharmacokinetic Studies. Drugs R. D 2014, 14, 283–290. [Google Scholar] [CrossRef] [Green Version]
- Laska, E.M.; Sunshine, A.; Marrero, I.; Olson, N.; Siegel, C.; McCormick, N. The correlation between blood levels of ibuprofen and clinical analgesic response. Clin. Pharmacol. Ther. 1986, 40, 1–7. [Google Scholar] [CrossRef] [PubMed]
- DeRuiter, J. Non-Steroidal Antiinflammatory Drugs (NSAIDS). 2002. Available online: http://webhome.auburn.edu/~deruija/nsaids_2002.pdf (accessed on 21 February 2023).
- Van Den Abeele, J.; Brouwers, J.; Mattheus, R.; Tack, J.; Augustijns, P. Gastrointestinal Behavior of Weakly Acidic BCS Class II Drugs in Man—Case Study of Diclofenac Potassium. J. Pharm. Sci. 2016, 105, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Khalid, F.; Farid Hassan, S.M.; Noor, R.; Zaheer, K.; Hassan, F.; Muhammad, I.N. Possibility of extending biopharmaceutics classification system based biowaiver to BCS class IIa drug. Pak. J. Pharm. Sci. 2019, 32, 2065–2073. [Google Scholar] [PubMed]
- Fu, Q.; Lu, H.D.; Xie, Y.F.; Liu, J.Y.; Han, Y.; Gong, N.B.; Guo, F. Salt formation of two BCS II drugs (indomethacin and naproxen) with (1R, 2R)-1,2-diphenylethylenediamine: Crystal structures, solubility and thermodynamics analysis. J. Mol. Struct. 2019, 1185, 281–289. [Google Scholar] [CrossRef]
- Advil Package Insert. Available online: https://www.medicines.org.uk/emc/files/pil.11165.pdf (accessed on 15 January 2023).
- Brain, P.; Leyva, R.; Doyle, G.; Kellstein, D. Onset of analgesia and efficacy of ibuprofen sodium in postsurgical dental pain: Randomized, placebo-controlled study versus standard ibuprofen. Clin. J. Pain 2015, 31, 444–450. [Google Scholar] [CrossRef] [Green Version]
- Schettler, T.; Paris, S.; Pellett, M.; Kidner, S.; Wilkinson, D. Comparative pharmacokinetics of two fast-dissolving oral ibuprofen formulations and a regular-release ibuprofen tablet in healthy volunteers. Clin. Drug Investig. 2001, 21, 73–78. [Google Scholar] [CrossRef]
- Cámara-Martinez, I.; Blechar, J.A.; Ruiz-Picazo, A.; Garcia-Arieta, A.; Calandria, C.; Merino-Sanjuan, V.; Langguth, P.; Gonzalez-Alvarez, M.; Bermejo, M.; Al-Gousous, J.; et al. Level A IVIVC for immediate release tablets confirms in vivo predictive dissolution testing for ibuprofen. Int. J. Pharm. 2022, 614, 121415. [Google Scholar] [CrossRef]
- USP Monograph-Ibuprofen Tablets. Available online: http://www.pharmacopeia.cn/v29240/usp29nf24s0_m39890.html (accessed on 15 January 2023).
- Baka, E.; Comer, J.E.A.; Takács-Novák, K. Study of equilibrium solubility measurement by saturation shake-flask method using hydrochlorothiazide as model compound. J. Pharm. Biomed. Anal. 2008, 46, 335–341. [Google Scholar] [CrossRef]
- Avdeef, A.; Fuguet, E.; Llinàs, A.; Ràfols, C.; Bosch, E.; Völgyi, G.; Verbic, T.; Boldyreva, E.; Takács-Novák, K. Equilibrium solubility measurement of ionizable drugs—Consensus recommendations for improving data quality. ADMET DMPK 2016, 4, 117–178. [Google Scholar] [CrossRef] [Green Version]
- Biorelevant Media Preparation Protocol. Available online: https://biorelevant.com/#media_prep_tool_tab (accessed on 15 January 2023).
- Appendix X—Dissolution Test. In Chinese Pharmacopoeia; China Medical Science and Technology Press: Beijing, China, 2010; pp. A105–A107.
- Völgyi, G.; Baka, E.; Box, K.J.; Comer, J.E.A.; Takács-Novák, K. Study of pH-dependent solubility of organic bases. Revisit of Henderson-Hasselbalch relationship. Anal. Chim. Acta 2010, 673, 40–46. [Google Scholar] [CrossRef]
- Avdeef, A. Solubility. In Absorption and Drug Development; John Wiley & Sons: Hoboken, NJ, USA, 2012; pp. 252–308. ISBN 3175723993. [Google Scholar]
- Takács-Novák, K.; Szoke, V.; Völgyi, G.; Horváth, P.; Ambrus, R.; Szabó-Révész, P. Biorelevant solubility of poorly soluble drugs: Rivaroxaban, furosemide, papaverine and niflumic acid. J. Pharm. Biomed. Anal. 2013, 83, 279–285. [Google Scholar] [CrossRef] [PubMed]
- FDA Extended Release Oral Dosage Forms: Development, Evaluation, and Application of In Vitro/In Vivo Correlations. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/extended-release-oral-dosage-forms-development-evaluation-and-application-vitroin-in-vivo-correlations (accessed on 15 January 2023).













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Product | Manufacturer | API Form | Abbreviation |
|---|---|---|---|
| Advil 200 mg coated tablets | Pfizer Consumer Healthcare, Madison, NJ, USA | ibuprofen free acid | IBU |
| Advil 256 mg film-coated tablets | Pfizer Consumer Healthcare, Madison, NJ, USA | ibuprofen sodium | IBU-Na |
| Dolowill RAPID 342 mg film-coated tablets | Goodwill Pharma, Szeged, Hungary | ibuprofen lysinate | IBU-Lys |
| Advil ULTRA 200 mg soft-gelatin capsules | Pfizer Consumer Healthcare, Madison, NJ, USA | Ibuprofen in solution | IBU-lq |
| Blank Biorelevant Media | Biorelevant Media | |||||
|---|---|---|---|---|---|---|
| Blank FaSSGF | Blank FaSSIF | Blank FaSSIF conc. | Full FaSSGF | Full FaSSIF | Full FaSSIF conc. | |
| NaCl | 2.00 g | 6.19 g | 40.24 g | 2.00 g | 6.19 g | 40.24 g |
| NaOH | - | 0.40 g | 2.60 g | - | 0.40 g | 2.60 g |
| NaH2PO4. H2O | - | 3.96 g | 25.74 g | - | 3.96 g | 25.74 g |
| SIF powder | - | - | - | 0.06 g | 2.25 g | 14.63 g |
| Pepsin | - | - | - | 0.10 g | - | - |
| pH adjustment | cc. HCl:purified water = 1:1 | 1M NaOH | - | cc. HCl:purified water = 1:1 | 1M NaOH | - |
| pH | SpH ± SD (µg/mL) 1 | logSpH (mol/L) |
|---|---|---|
| 1.92 | 70.8 ± 3.0 | −3.46 |
| 3.96 | 124 ± 13 | −3.22 |
| 5.95 | 1910 ± 70 | −2.03 |
| 7.17 | 32,033 ± 4135 | −0.81 |
| 8.02 | 300,000 ± 6500 | 0.16 |
| 14 | 734,000 ± 30,500 | 0.55 |
| Solvent | SpH ± SD (µg/mL) 1 |
|---|---|
| FaSSGF blank, pH 1.6 | 56.3 ± 0.6 |
| FaSSGF, pH 1.6 | 56.0 ± 0.5 |
| FeSSGF-acetate, pH 4.5 | 194 ± 2 |
| FeSSIF blank, pH 5.0 | 416 ± 12 |
| FeSSIF, pH 5.0 | 2103 ± 56 |
| FaSSIF blank, pH 6.5 | 2513 ± 15 |
| FaSSIF, pH 6.5 | 3160 ± 31 |
| Formulation | B | MDT (min) |
|---|---|---|
| IBU | 1.759 | 71.55 |
| IBU-Na | 1.786 | 48.23 |
| IBU-Lys | 2.087 | 44.55 |
| IBU-lq | 2.274 | 52.18 |
| Clinical Data Statistical Analysis of Individual Profiles | Clinical Data Mean Plasma conc. Profiles | IVIVC Prediction from GIS Dissolution | ||||||
|---|---|---|---|---|---|---|---|---|
| Formulation | Cmax | Ratio | tmax | Cmax | Ratio | Cmax | Ratio | tmax |
| IBU | 37.70 | N/A | 82.1 | 28.25 | N/A | 37.80 | N/A | 81.7 |
| IBU-Na | 47.00 | 1.25 | 35.2 | 44.00 | 1.56 | 41.00 | 1.09 | 60.5 |
| IBU-Lys | 49.90 | 1.32 | 35.1 | 42.75 | 1.51 | 42.40 | 1.12 | 61.4 |
| IBU-lq | 46.80 | 1.24 | 40.0 | 39.00 | 1.38 | 42.20 | 1.12 | 60.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Katona, M.T.; Nagy-Katona, L.; Szabó, R.; Borbás, E.; Tonka-Nagy, P.; Takács-Novák, K. Multi-Compartmental Dissolution Method, an Efficient Tool for the Development of Enhanced Bioavailability Formulations Containing Poorly Soluble Acidic Drugs. Pharmaceutics 2023, 15, 753. https://doi.org/10.3390/pharmaceutics15030753
Katona MT, Nagy-Katona L, Szabó R, Borbás E, Tonka-Nagy P, Takács-Novák K. Multi-Compartmental Dissolution Method, an Efficient Tool for the Development of Enhanced Bioavailability Formulations Containing Poorly Soluble Acidic Drugs. Pharmaceutics. 2023; 15(3):753. https://doi.org/10.3390/pharmaceutics15030753
Chicago/Turabian StyleKatona, Miklós Tamás, Lili Nagy-Katona, Réka Szabó, Enikő Borbás, Péter Tonka-Nagy, and Krisztina Takács-Novák. 2023. "Multi-Compartmental Dissolution Method, an Efficient Tool for the Development of Enhanced Bioavailability Formulations Containing Poorly Soluble Acidic Drugs" Pharmaceutics 15, no. 3: 753. https://doi.org/10.3390/pharmaceutics15030753