
DLin-MC3-Containing mRNA Lipid Nanoparticles Induce an Antibody Th2-Biased Immune Response Polarization in a Delivery Route-Dependent Manner in Mice
 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. mRNA-Based Lipid Nanoparticle (LNP) Preparation and Characterization
2.3. Protein-Based Subunit Vaccine Formulation
2.4. In Vitro Luciferase Transfection Assay
2.5. In Vitro p55Gag Polyprotein Detection Assay
2.6. Immunization and Sampling
2.7. Anti-p24 Antibody ELISA
2.8. Statistical Analysis
3. Results
3.1. Effect of the N/P Ratio and the Purification Temperature on Colloidal Physicochemical Characteristics of mRNA-LNP Formulations in Different Dispersants
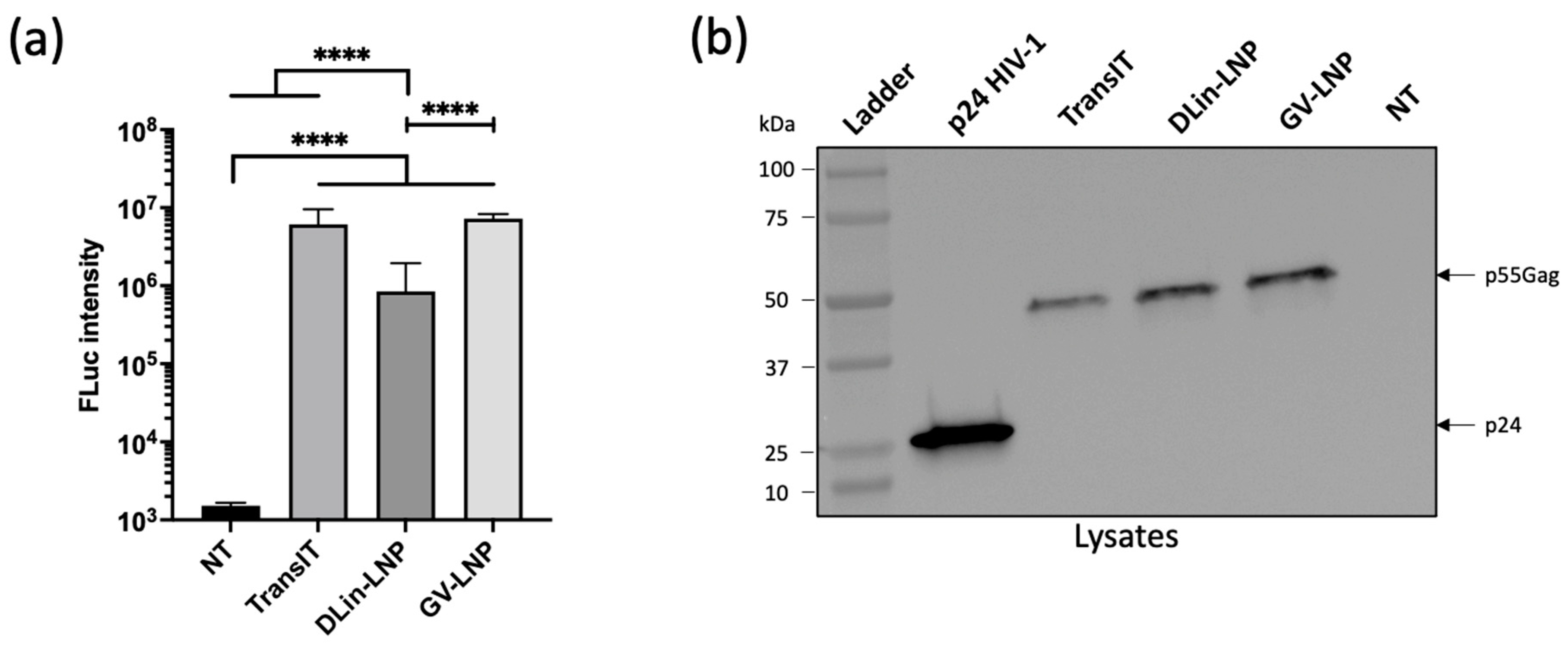
3.2. Both mRNA-LNPs Showed Efficient Cell Transfection and Translation In Vitro
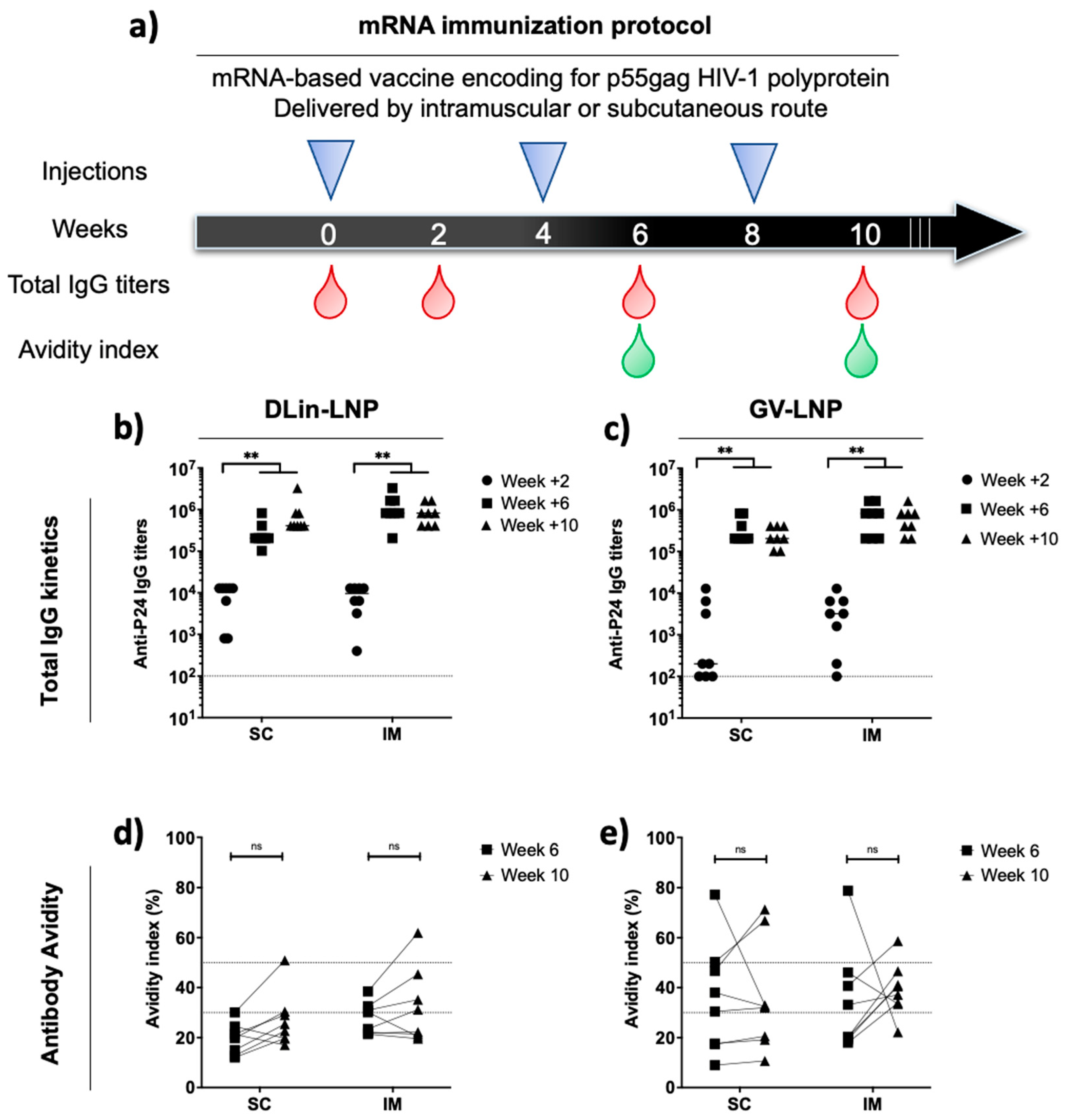
3.3. Administration Route of p55Gag mRNA-Based Vaccine Does Not Impact the Anti-p24 Humoral Immune Response Kinetics and Antibody Avidities
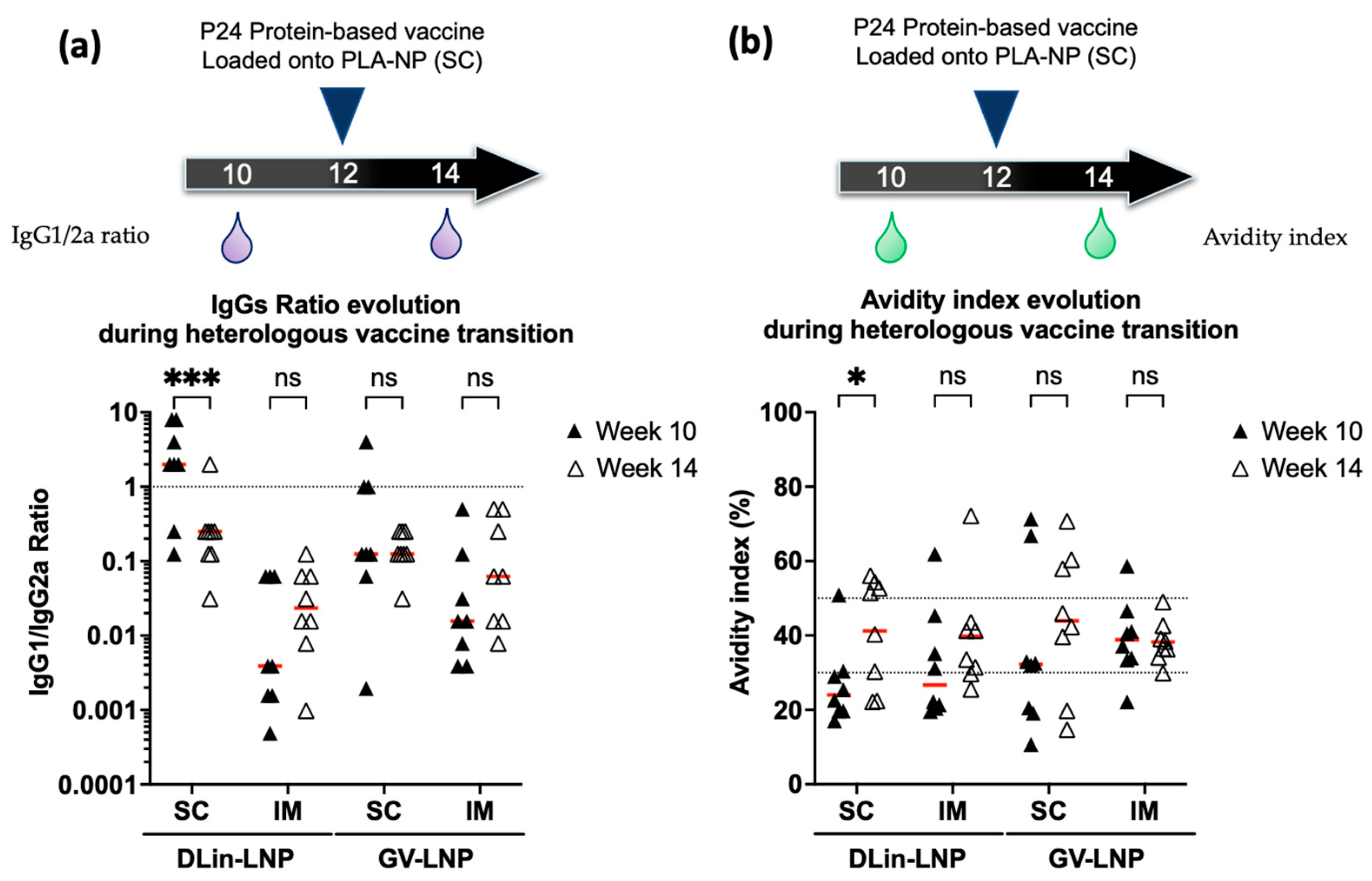
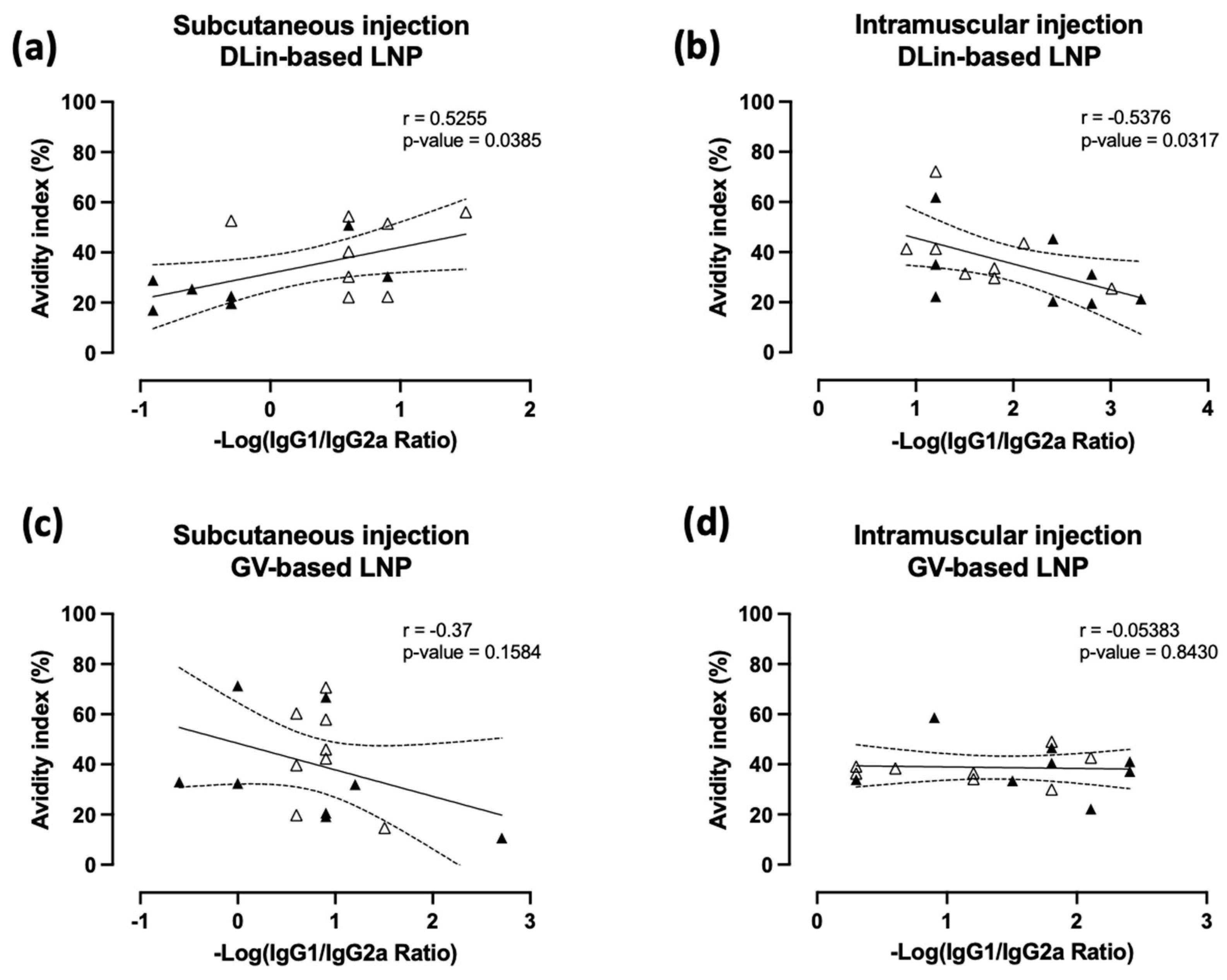
3.4. Immune Polarization Mediated by IgG1/2a Ratio Shows Contrasting Effect in an Ionizable Lipid- and Administration Route-Dependent Manner
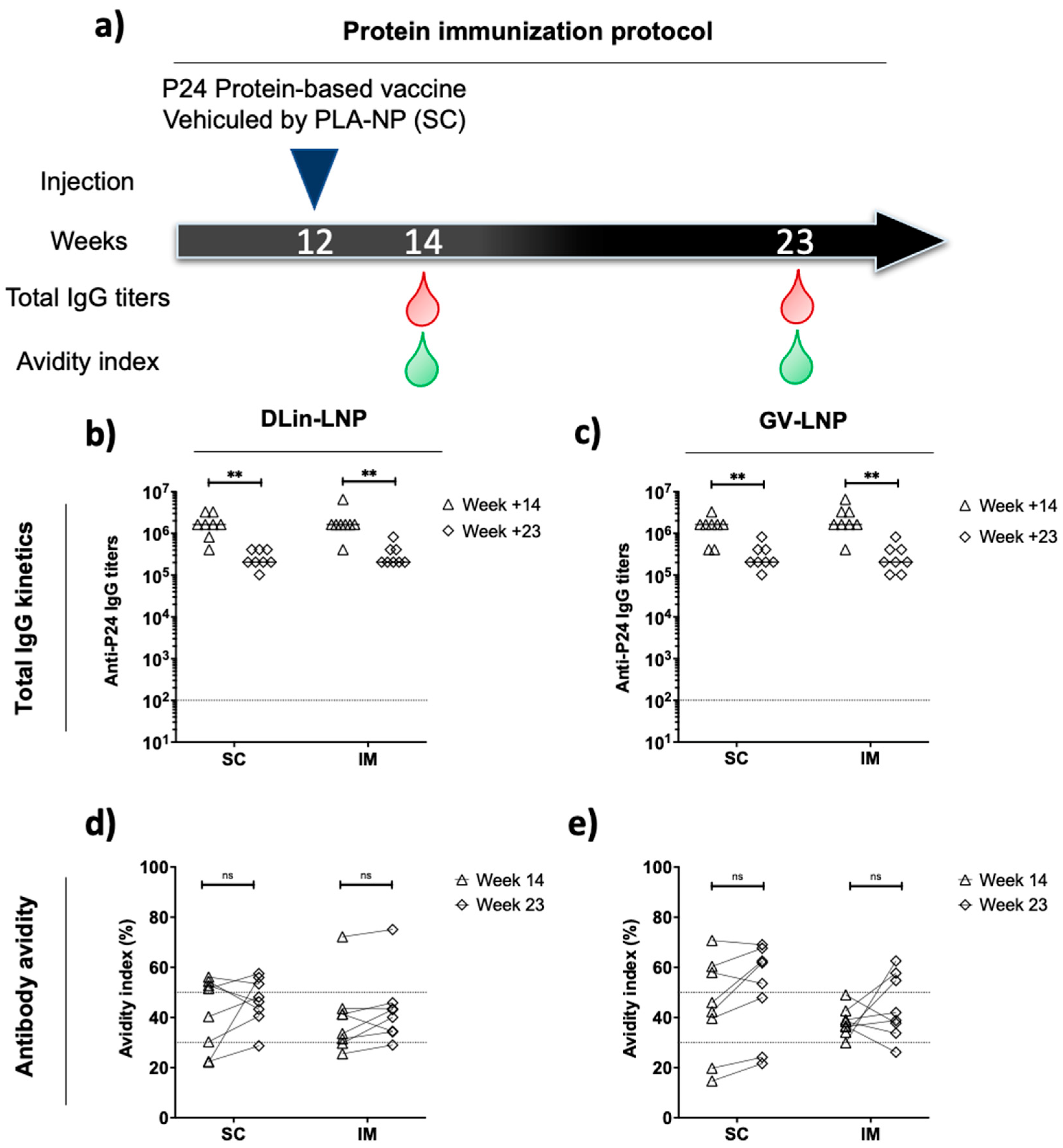
3.5. DLin-LNP SC and Heterologous Boost Combination
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wolff, J.A.; Malone, R.W.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P.L. Direct gene transfer into mouse muscle in vivo. Science 1990, 247, 1465–1468. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, Y.; Li, S.; Jia, L.; Wang, H.; Li, M.; Deng, J.; Zhu, A.; Ma, L.; Li, W.; et al. The nano delivery systems and applications of mRNA. Eur. J. Med. Chem. 2022, 227, 113910. [Google Scholar] [CrossRef]
- Semple, S.C.; Klimuk, S.K.; Harasym, T.O.; Dos Santos, N.; Ansell, S.M.; Wong, K.F.; Maurer, N.; Stark, H.; Cullis, P.R.; Hope, M.J.; et al. Efficient encapsulation of antisense oligonucleotides in lipid vesicles using ionizable aminolipids: Formation of novel small multilamellar vesicle structures. Biochim. Biophys. Acta (BBA)–Biomembr. 2001, 1510, 152–166. [Google Scholar] [CrossRef] [Green Version]
- Karikó, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA Recognition by Toll-like Receptors: The Impact of Nucleoside Modification and the Evolutionary Origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef] [Green Version]
- Fortner, A.; Freiburg, G.A.-L.; Schumacher, D. First COVID-19 Vaccines Receiving the US FDA and EMA Emergency Use Authorization. Discoveries 2021, 9, e122. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Tang, X.; Chen, Y.; Chen, K.; Na Fan, N.; Xiao, W.; Zheng, Q.; Li, G.; Teng, Y.; Wu, M.; et al. mRNA-based therapeutics: Powerful and versatile tools to combat diseases. Signal Transduct. Target. Ther. 2022, 7, 166. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, J.; Myhre, J.L.; Chen, S.; Tam, Y.Y.C.; Danescu, A.; Richman, J.; Cullis, P.R. Design of lipid nanoparticles for in vitro and in vivo delivery of plasmid DNA. Nanomed. Nanotechnol. Biol. Med. 2017, 13, 1377–1387. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, C.; Wang, C.; Jankovic, K.E.; Dong, Y. Lipids and Lipid Derivatives for RNA Delivery. Chem. Rev. 2021, 121, 12181–12277. [Google Scholar] [CrossRef]
- Roces, C.B.; Lou, G.; Jain, N.; Abraham, S.; Thomas, A.; Halbert, G.W.; Perrie, Y. Manufacturing Considerations for the Development of Lipid Nanoparticles Using Microfluidics. Pharmaceutics 2020, 12, 1095. [Google Scholar] [CrossRef]
- Albertsen, C.H.; Kulkarni, J.A.; Witzigmann, D.; Lind, M.; Petersson, K.; Simonsen, J.B. The role of lipid components in lipid nanoparticles for vaccines and gene therapy. Adv. Drug Deliv. Rev. 2022, 188, 114416. [Google Scholar] [CrossRef]
- Hassett, K.J.; Benenato, K.E.; Jacquinet, E.; Lee, A.; Woods, A.; Yuzhakov, O.; Himansu, S.; Deterling, J.; Geilich, B.M.; Ketova, T.; et al. Optimization of Lipid Nanoparticles for Intramuscular Administration of mRNA Vaccines. Mol. Ther. Nucleic Acids 2019, 15, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Q.; Wei, T.; Farbiak, L.; Johnson, L.T.; Dilliard, S.A.; Siegwart, D.J. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR–Cas gene editing. Nat. Nanotechnol. 2020, 15, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Tuyishime, S.; Muramatsu, H.; Kariko, K.; Mui, B.L.; Tam, Y.K.; Madden, T.D.; Hope, M.J.; Weissman, D. Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J. Control. Release 2015, 217, 345–351. [Google Scholar] [CrossRef] [Green Version]
- Sebastiani, F.; Yanez Arteta, M.; Lerche, M.; Porcar, L.; Lang, C.; Bragg, R.A.; Elmore, C.S.; Krishnamurthy, V.R.; Russell, R.A.; Darwish, T.; et al. Apolipoprotein E Binding Drives Structural and Compositional Rearrangement of mRNA-Containing Lipid Nanoparticles. ACS Nano 2021, 15, 6709–6722. [Google Scholar] [CrossRef]
- Khadke, S.; Roces, C.B.; Cameron, A.; Devitt, A.; Perrie, Y. Formulation and manufacturing of lymphatic targeting liposomes using microfluidics. J. Control. Release 2019, 307, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, M.J.; Alishetty, S.; Alameh, M.-G.; Said, H.; Wright, L.; Paige, M.; Soliman, O.; Weissman, D.; Cleveland, T.E.; Grishaev, A.; et al. Ionization and structural properties of mRNA lipid nanoparticles influence expression in intramuscular and intravascular administration. Commun. Biol. 2021, 4, 956. [Google Scholar] [CrossRef]
- Alameh, M.-G.; Tombácz, I.; Bettini, E.; Lederer, K.; Ndeupen, S.; Sittplangkoon, C.; Wilmore, J.R.; Gaudette, B.T.; Soliman, O.Y.; Pine, M.; et al. Lipid nanoparticles enhance the efficacy of mRNA and protein subunit vaccines by inducing robust T follicular helper cell and humoral responses. Immunity 2021, 54, 2877–2892.e7. [Google Scholar] [CrossRef]
- Connors, J.; Joyner, D.; Mege, N.J.; Cusimano, G.M.; Bell, M.R.; Marcy, J.; Taramangalam, B.; Kim, K.M.; Lin, P.J.C.; Tam, Y.K.; et al. Lipid nanoparticles (LNP) induce activation and maturation of antigen presenting cells in young and aged individuals. Commun. Biol. 2023, 6, 188. [Google Scholar] [CrossRef]
- Sidik, S.M. Vaccines protect against infection from Omicron subvariant—But not for long. Nature 2022. [Google Scholar] [CrossRef]
- Dolgin, E. COVID vaccine immunity is waning—How much does that matter? Nature 2021, 597, 606–607. [Google Scholar] [CrossRef]
- Liu, A.; Wang, X. The Pivotal Role of Chemical Modifications in mRNA Therapeutics. Front. Cell Dev. Biol. 2022, 10, 901510. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, S.; Satapathy, S.R.; Dutta, T. Delivery Strategies for mRNA Vaccines. Pharm. Med. 2022, 36, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zhou, Z.; Du, P.; Zhan, M.; Li, N.; Xiong, X.; Tang, S.; Man, M.; Baptista-Hon, D.T.; Lu, L. Heterologous mRNA vaccine booster increases neutralization of SARS-CoV-2 Omicron BA.2 variant. Signal Transduct. Target. Ther. 2022, 7, 17023. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.A.; Neshat, S.Y.; Green, J.J.; Santos, J.L.; Tuesca, A.D. Targeting strategies for mRNA delivery. Mater. Today Adv. 2022, 14, 100240. [Google Scholar] [CrossRef]
- Donnelly, R.F. Vaccine delivery systems. Hum. Vaccines Immunother. 2017, 13, 17–18. [Google Scholar] [CrossRef] [Green Version]
- Ng, J.Y. Inadvertent subcutaneous injection of COVID-19 vaccine. Postgrad. Med. J. 2021, 97, 400. [Google Scholar] [CrossRef]
- Friedensohn, L.; Zur, M.; Timofeyev, M.; Burshtein, S.; Ben Michael, Y.; Fink, N.; Glassberg, E. Sub-cutaneous Pfizer/BioNTech COVID-19 vaccine administration results in seroconversion among young adults. Vaccine 2021, 39, 6210–6212. [Google Scholar] [CrossRef]
- Mu, Z.; Haynes, B.; Cain, D. HIV mRNA Vaccines—Progress and Future Paths. Vaccines 2021, 9, 134. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S.G.; Archin, N.; Cannon, P.; Collins, S.; Jones, R.B.; de Jong, M.A.W.P.; Lambotte, O.; Lamplough, R.; Ndung’U, T.; Sugarman, J.; et al. Research priorities for an HIV cure: International AIDS Society Global Scientific Strategy 2021. Nat. Med. 2021, 27, 2085–2098. [Google Scholar] [CrossRef] [PubMed]
- Morris, L. mRNA vaccines offer hope for HIV. Nat. Med. 2021, 27, 2082–2084. [Google Scholar] [CrossRef]
- Saunders, K.O.; Pardi, N.; Parks, R.; Santra, S.; Mu, Z.; Sutherland, L.; Scearce, R.; Barr, M.; Eaton, A.; Hernandez, G.; et al. Lipid nanoparticle encapsulated nucleoside-modified mRNA vaccines elicit polyfunctional HIV-1 antibodies comparable to proteins in nonhuman primates. npj Vaccines 2021, 6, 50. [Google Scholar] [CrossRef]
- Zhang, P.; Narayanan, E.; Liu, Q.; Tsybovsky, Y.; Boswell, K.; Ding, S.; Hu, Z.; Follmann, D.; Lin, Y.; Miao, H.; et al. A multiclade env–gag VLP mRNA vaccine elicits tier-2 HIV-1-neutralizing antibodies and reduces the risk of heterologous SHIV infection in macaques. Nat. Med. 2021, 27, 2234–2245. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; LaBranche, C.C.; Ferrari, G.; Cain, D.W.; Tombácz, I.; Parks, R.J.; Muramatsu, H.; Mui, B.L.; Tam, Y.K.; Karikó, K.; et al. Characterization of HIV-1 Nucleoside-Modified mRNA Vaccines in Rabbits and Rhesus Macaques. Mol. Ther.–Nucleic Acids 2019, 15, 36–47. [Google Scholar] [CrossRef] [Green Version]
- Mu, Z.; Wiehe, K.; Saunders, K.O.; Henderson, R.; Cain, D.W.; Parks, R.; Martik, D.; Mansouri, K.; Edwards, R.J.; Newman, A.; et al. mRNA-encoded HIV-1 Env trimer ferritin nanoparticles induce monoclonal antibodies that neutralize heterologous HIV-1 isolates in mice. Cell Rep. 2022, 38, 110514. [Google Scholar] [CrossRef] [PubMed]
- Pavot, V.; Rochereau, N.; Primard, C.; Genin, C.; Perouzel, E.; Lioux, T.; Paul, S.; Verrier, B. Encapsulation of Nod1 and Nod2 receptor ligands into poly(lactic acid) nanoparticles potentiates their immune properties. J. Control. Release 2013, 167, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.F.S.; McKay, P.F.; Fiserova, A.; Klein, K.; Cope, A.; Rogers, P.; Swales, J.; Seaman, M.S.; Combadiere, B.; Shattock, R.J. Enhanced Immunogenicity of an HIV-1 DNA Vaccine Delivered with Electroporation via Combined Intramuscular and Intradermal Routes. J. Virol. 2014, 88, 6959–6969. [Google Scholar] [CrossRef] [Green Version]
- Ayad, C.; Yavuz, A.; Salvi, J.-P.; Libeau, P.; Exposito, J.-Y.; Ginet, V.; Monge, C.; Verrier, B.; Arruda, D.C. Comparison of Physicochemical Properties of LipoParticles as mRNA Carrier Prepared by Automated Microfluidic System and Bulk Method. Pharmaceutics 2022, 14, 1297. [Google Scholar] [CrossRef]
- Hassett, K.J.; Higgins, J.; Woods, A.; Levy, B.; Xia, Y.; Hsiao, C.J.; Acosta, E.; Almarsson, O.; Moore, M.J.; Brito, L.A. Impact of lipid nanoparticle size on mRNA vaccine immunogenicity. J. Control. Release 2021, 335, 237–246. [Google Scholar] [CrossRef]
- Trevaskis, N.; Kaminskas, L.; Porter, C. From sewer to saviour—Targeting the lymphatic system to promote drug exposure and activity. Nat. Rev. Drug Discov. 2015, 14, 781–803. [Google Scholar] [CrossRef]
- Rolland, M.; Heckerman, D.; Deng, W.; Rousseau, C.M.; Coovadia, H.; Bishop, K.; Goulder, P.J.R.; Walker, B.D.; Brander, C.; Mullins, J.I. Broad and Gag-Biased HIV-1 Epitope Repertoires Are Associated with Lower Viral Loads. PLoS ONE 2008, 3, e1424. [Google Scholar] [CrossRef] [Green Version]
- Zuñiga, R.; Lucchetti, A.; Galvan, P.; Sanchez, S.; Sanchez, C.; Hernandez, A.; Sanchez, H.; Frahm, N.; Linde, C.H.; Hewitt, H.S.; et al. Relative Dominance of Gag p24-Specific Cytotoxic T Lymphocytes Is Associated with Human Immunodeficiency Virus Control. J. Virol. 2006, 80, 3122–3125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rancan, F.; Amselgruber, S.; Hadam, S.; Munier, S.; Pavot, V.; Verrier, B.; Hackbarth, S.; Combadiere, B.; Blume-Peytavi, U.; Vogt, A. Particle-based transcutaneous administration of HIV-1 p24 protein to human skin explants and targeting of epidermal antigen presenting cells. J. Control. Release 2014, 176, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Hameed, S.A.; Paul, S.; Dellosa, G.K.Y.; Jaraquemada, D.; Bello, M.B. Towards the future exploration of mucosal mRNA vaccines against emerging viral diseases; lessons from existing next-generation mucosal vaccine strategies. npj Vaccines 2022, 7, 71. [Google Scholar] [CrossRef]
- Vuitika, L.; Prates-Syed, W.A.; Silva, J.D.Q.; Crema, K.P.; Côrtes, N.; Lira, A.; Lima, J.B.M.; Camara, N.O.S.; Schimke, L.F.; Cabral-Marques, O.; et al. Vaccines against Emerging and Neglected Infectious Diseases: An Overview. Vaccines 2022, 10, 1385. [Google Scholar] [CrossRef] [PubMed]
- Le, T.; Sun, C.; Chang, J.; Zhang, G.; Yin, X. mRNA Vaccine Development for Emerging Animal and Zoonotic Diseases. Viruses 2022, 14, 401. [Google Scholar] [CrossRef] [PubMed]
- Mora, C.; McKenzie, T.; Gaw, I.M.; Dean, J.M.; von Hammerstein, H.; Knudson, T.A.; Setter, R.O.; Smith, C.Z.; Webster, K.M.; Patz, J.A.; et al. Over half of known human pathogenic diseases can be aggravated by climate change. Nat. Clim. Chang. 2022, 12, 869–875. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, A.; Kamel, M. Climatic changes and their role in emergence and re-emergence of diseases. Environ. Sci. Pollut. Res. 2020, 27, 22336–22352. [Google Scholar] [CrossRef]
- Cheng, X.; Lee, R.J. The role of helper lipids in lipid nanoparticles (LNPs) designed for oligonucleotide delivery. Adv. Drug Deliv. Rev. 2016, 99, 129–137. [Google Scholar] [CrossRef]
- Cullis, P.R.; Hope, M.J. Lipid Nanoparticle Systems for Enabling Gene Therapies. Mol. Ther. 2017, 25, 1467–1475. [Google Scholar] [CrossRef] [Green Version]
- Barua, S.; Mitragotri, S. Challenges associated with penetration of nanoparticles across cell and tissue barriers: A review of current status and future prospects. Nano Today 2014, 9, 223–243. [Google Scholar] [CrossRef] [Green Version]
- Paunovska, K.; Sago, C.D.; Monaco, C.M.; Hudson, W.H.; Castro, M.G.; Rudoltz, T.G.; Kalathoor, S.; Vanover, D.A.; Santangelo, P.J.; Ahmed, R.; et al. A Direct Comparison of in Vitro and in Vivo Nucleic Acid Delivery Mediated by Hundreds of Nanoparticles Reveals a Weak Correlation. Nano Lett. 2018, 18, 2148–2157. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschi, C.; Terpos, E.; Rosati, M.; Angel, M.; Bear, J.; Stellas, D.; Karaliota, S.; Apostolakou, F.; Bagratuni, T.; Patseas, D.; et al. Systemic IL-15, IFN-γ and IP-10/CXCL10 Signature Associated with Effective Immune Response to SARS-CoV-2 in BNT162b2 mRNA Vaccine Recipients. Cell Rep. 2021, 36, 109504. [Google Scholar] [CrossRef] [PubMed]
- Parhiz, H.; Brenner, J.S.; Patel, P.N.; Papp, T.E.; Shahnawaz, H.; Li, Q.; Shi, R.; Zamora, M.E.; Yadegari, A.; Marcos-Contreras, O.A.; et al. Added to pre-existing inflammation, mRNA-lipid nanoparticles induce inflammation exacerbation (IE). J. Control. Release 2022, 344, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Valentin, A.; Bergamaschi, C.; Rosati, M.; Angel, M.; Burns, R.; Agarwal, M.; Gergen, J.; Petsch, B.; Oostvogels, L.; Loeliger, E.; et al. Comparative immunogenicity of an mRNA/LNP and a DNA vaccine targeting HIV gag conserved elements in macaques. Front. Immunol. 2022, 13, 945706. [Google Scholar] [CrossRef]
- Laczkó, D.; Hogan, M.J.; Toulmin, S.A.; Hicks, P.; Lederer, K.; Gaudette, B.T.; Castaño, D.; Amanat, F.; Muramatsu, H.; Oguin, T.H.; et al. A Single Immunization with Nucleoside-Modified mRNA Vaccines Elicits Strong Cellular and Humoral Immune Responses against SARS-CoV-2 in Mice. Immunity 2020, 53, 724–732.e7. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LNP-Based Vaccine | N/P Ratio | DSP Temperature | Hydrodynamic Diameter (nm) (1) | Polydispersity Index (PI) (1) | Zeta Potential (Zp) | mRNA Encapsulation Efficiency (%) |
|---|---|---|---|---|---|---|
| DLin-LNP | 7 | 8 °C | 75 ± 0.7 | 0.089 ± 0.015 | −3.7 ± 0.5 | 95.4 ± 0.8 |
| 8 | 68 ± 2 | 0.068 ± 0.016 | −6.6 ± 1.6 | 90.2 ± 1.4 | ||
| 7 | 20 °C | 88 ± 3 **** | 0.139 ± 0.007 ** | −5.2 ± 1.4 | 92.8 ± 1.7 | |
| 8 | 73 ± 0.9 * | 0.087 ± 0.004 | −6.5 ± 1.1 | 91.9 ± 1.8 | ||
| GV-LNP | 7 | 8 °C | 90 ± 3 | 0.142 ± 0.002 | −3.3 ± 1.0 | 95.4 ± 0.8 |
| 8 | 75.6 ± 1.5 | 0.085 ± 0.021 | −5.3 ± 0.2 | 90.3 ± 0.3 | ||
| 7 | 20 °C | 78 ± 2.8 **** | 0.130 ± 0.012 | −3.7 ± 0.4 | 92.8 ± 1.7 | |
| 8 | 74 ± 0.4 | 0.086 ± 0.009 | −4.4 ± 0.1 | 94.3 ± 1.8 |
| LNP-Based Vaccine | N/P Ratio | DSP Temperature | Hydrodynamic Diameter (nm) (1) | Polydispersity Index (PI) (1) | Zeta Potential (Zp) (1) |
|---|---|---|---|---|---|
| DLin-LNP | 7 | 8 °C | 86.5 ± 3.4 ** | 0.12 ± 0.051 | 2.6 ± 0.1 **** |
| 8 | 74.2 ± 1.7 * | 0.06 ± 0.003 | −6.6 ± 0.3 | ||
| 7 | 20 °C | 93.7 ± 3.8 * | 0.140 ± 0.069 | 3.1 ± 0.3 **** | |
| 8 | 81.1 ± 1.7 ** | 0.074 ± 0.010 | −5.7 ± 0.7 | ||
| GV-LNP | 7 | 8 °C | 128.1 ± 4.4 **** | 0.15 ± 0.013 | 4.4 ± 2.3 **** |
| 8 | 108.9 ± 2.64 **** | 0.19 ± 0.020 **** | 3.1 ± 1.7 **** | ||
| 7 | 20 °C | 110.1 ± 8.1 **** | 0.164 ± 0.031 * | 5.4 ± 1 **** | |
| 8 | 115.2 ± 2.2 **** | 0.162 ± 0.006 *** | 0.3 ± 0.5 **** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yavuz, A.; Coiffier, C.; Garapon, C.; Gurcan, S.; Monge, C.; Exposito, J.-Y.; Arruda, D.C.; Verrier, B. DLin-MC3-Containing mRNA Lipid Nanoparticles Induce an Antibody Th2-Biased Immune Response Polarization in a Delivery Route-Dependent Manner in Mice. Pharmaceutics 2023, 15, 1009. https://doi.org/10.3390/pharmaceutics15031009
Yavuz A, Coiffier C, Garapon C, Gurcan S, Monge C, Exposito J-Y, Arruda DC, Verrier B. DLin-MC3-Containing mRNA Lipid Nanoparticles Induce an Antibody Th2-Biased Immune Response Polarization in a Delivery Route-Dependent Manner in Mice. Pharmaceutics. 2023; 15(3):1009. https://doi.org/10.3390/pharmaceutics15031009
Chicago/Turabian StyleYavuz, Altan, Céline Coiffier, Cynthia Garapon, Serra Gurcan, Claire Monge, Jean-Yves Exposito, Danielle Campiol Arruda, and Bernard Verrier. 2023. "DLin-MC3-Containing mRNA Lipid Nanoparticles Induce an Antibody Th2-Biased Immune Response Polarization in a Delivery Route-Dependent Manner in Mice" Pharmaceutics 15, no. 3: 1009. https://doi.org/10.3390/pharmaceutics15031009