
Nanosystems for Brain Targeting of Antipsychotic Drugs: An Update on the Most Promising Nanocarriers for Increased Bioavailability and Therapeutic Efficacy
 and
and
Abstract
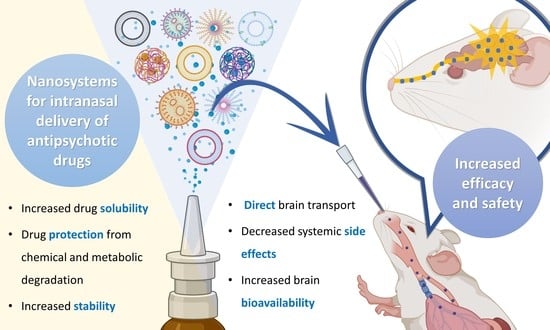
:
1. Introduction
1.1. Schizophrenia and Other Schizoaffective Diseases: Current Treatments and Challenges
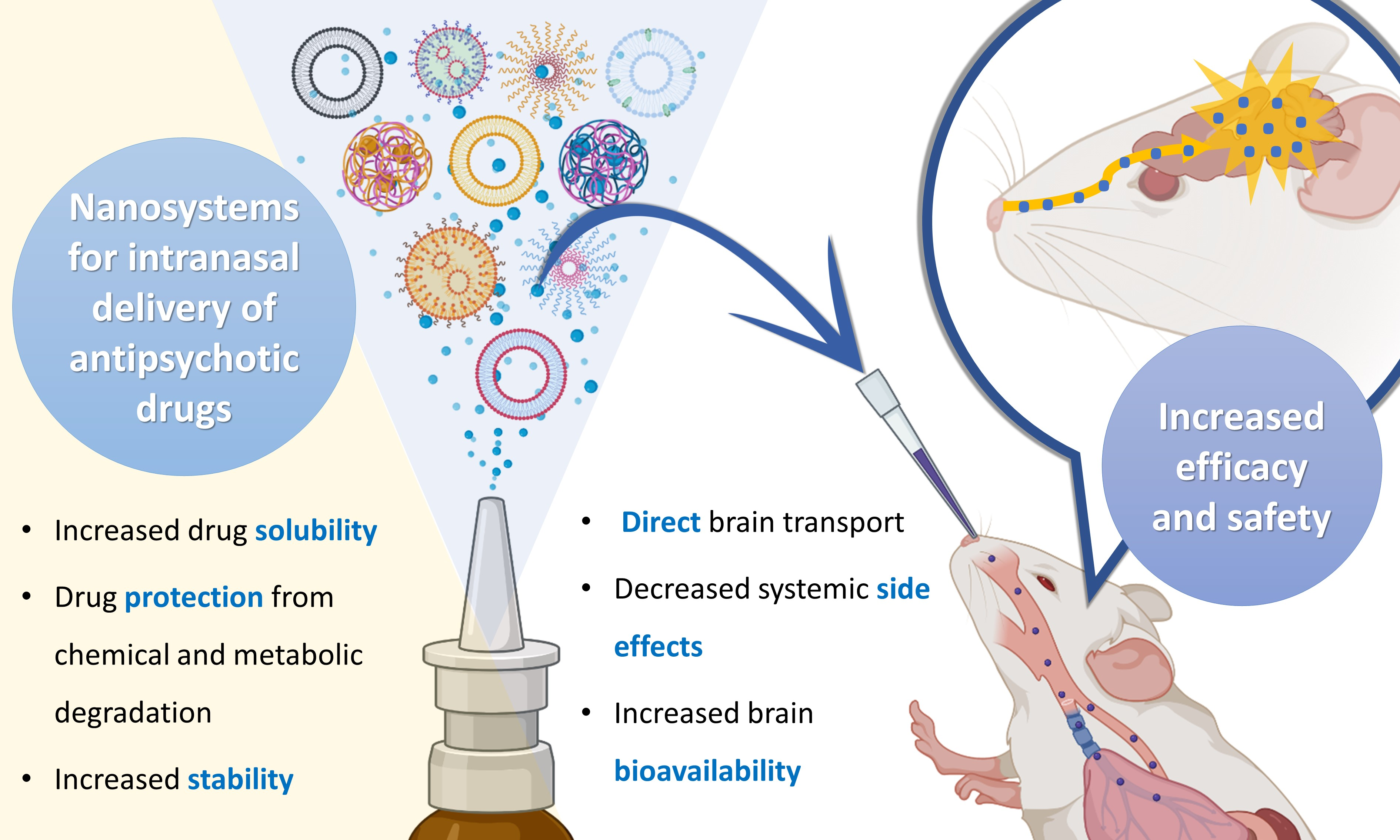
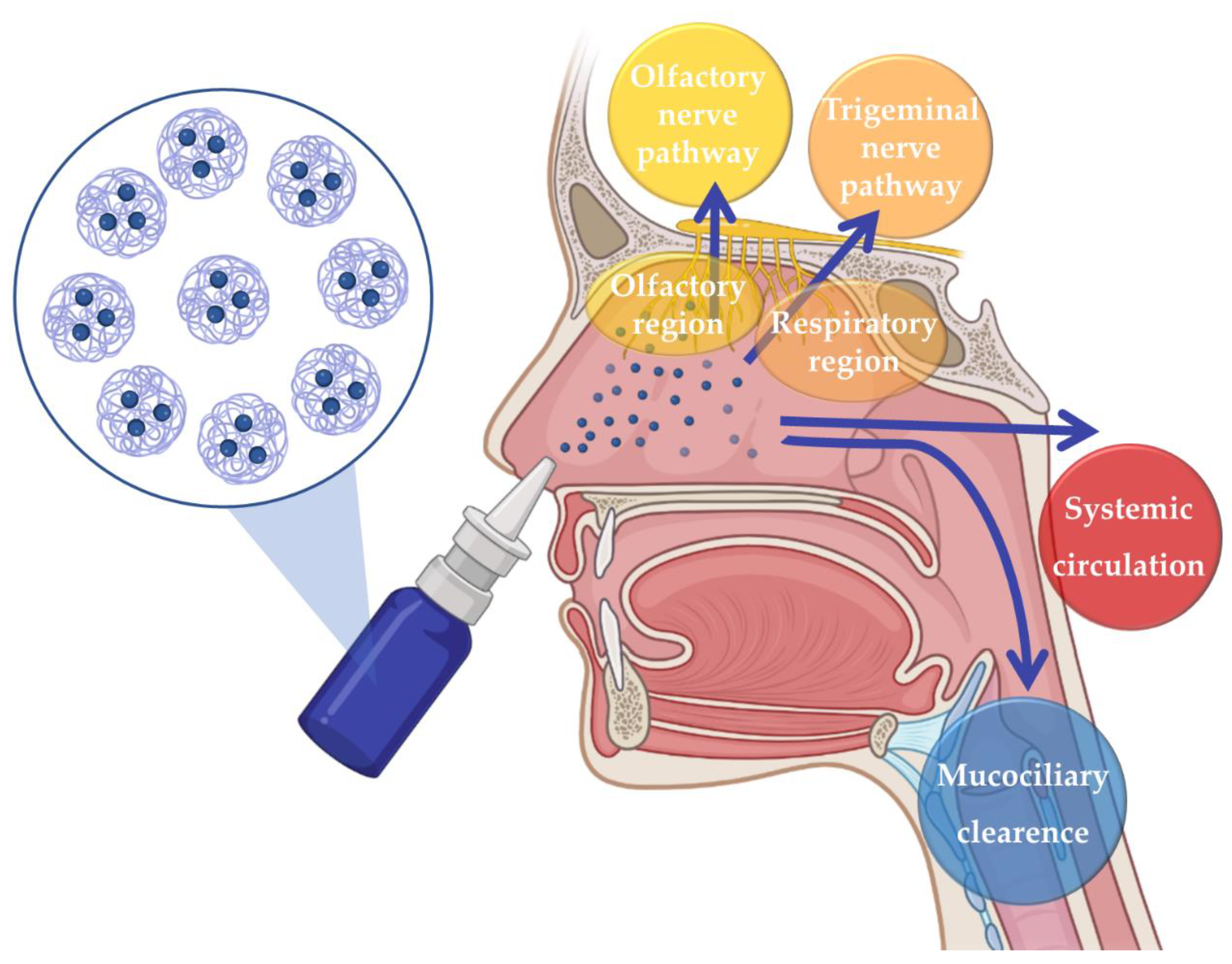
1.2. Nanosystems
2. 2nd Generation Antipsychotics
2.1. Quetiapine Nanoemulsion
2.2. Risperidone Spanlastics, Nanoemulsions and Solid Lipid Nanoparticles
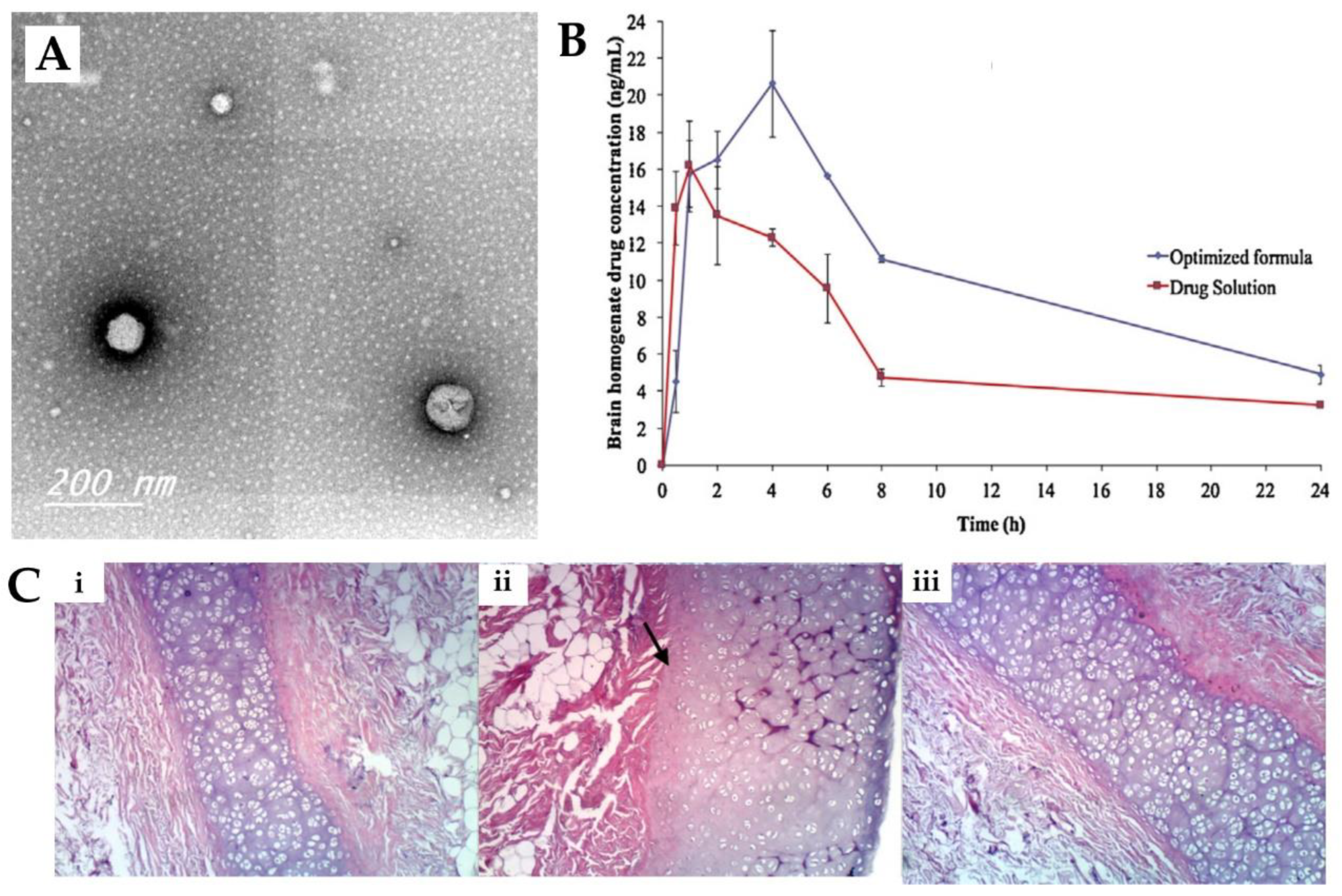
2.3. Olanzapine Solid Lipid Nanoparticles, Polymeric Nanoparticles, Nanostructured Lipid Carriers and Niosomes
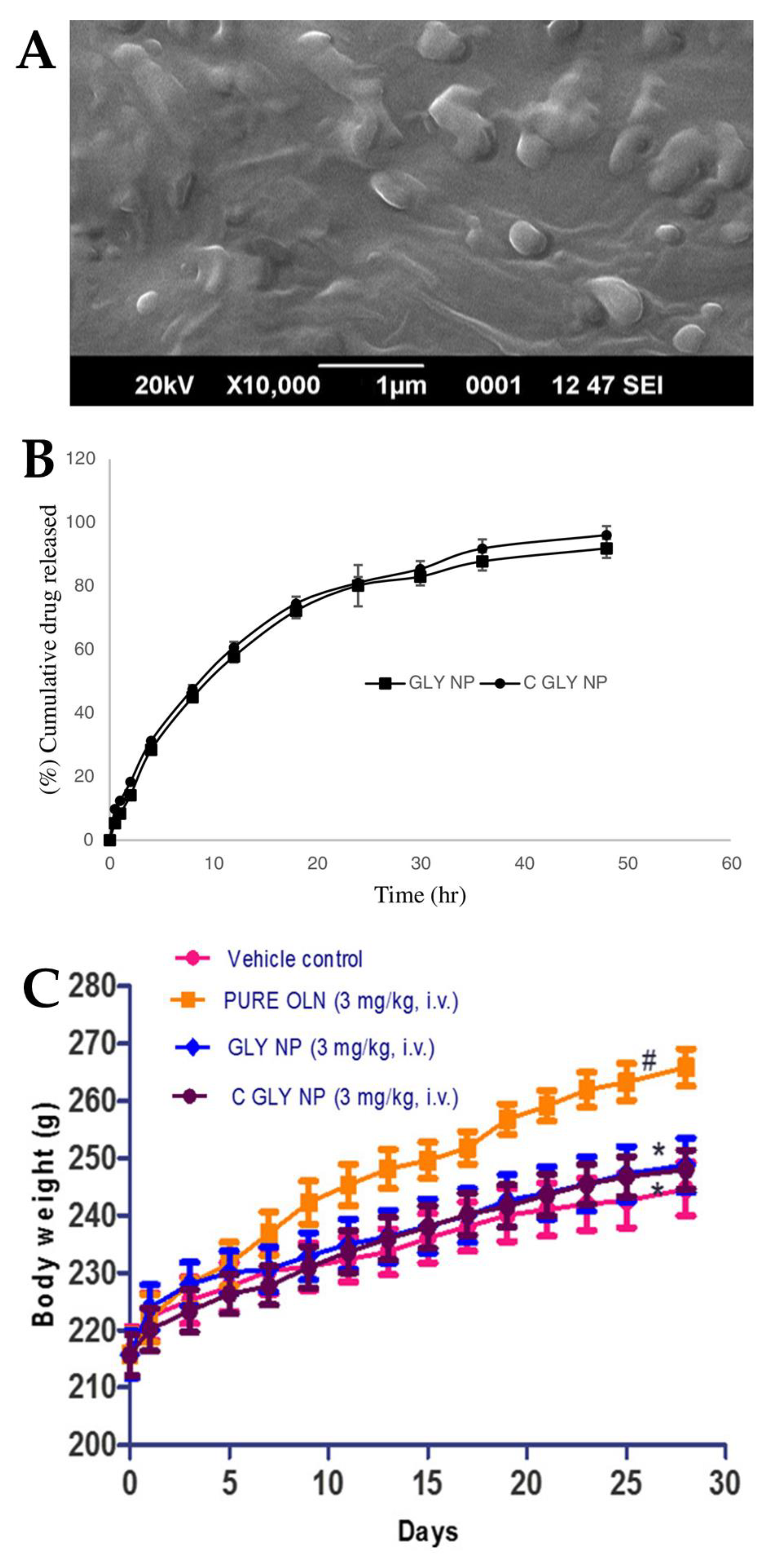
2.4. Asenapine Nanostructured Lipid Carriers and Nanoemulgel
2.5. Lurasidone Nanostructured Lipid Carriers and Mixed Polymeric Micelles
2.6. Zotepine Nanosuspension
2.7. Amisulpride Nanoemulsion and Nanoemulgel
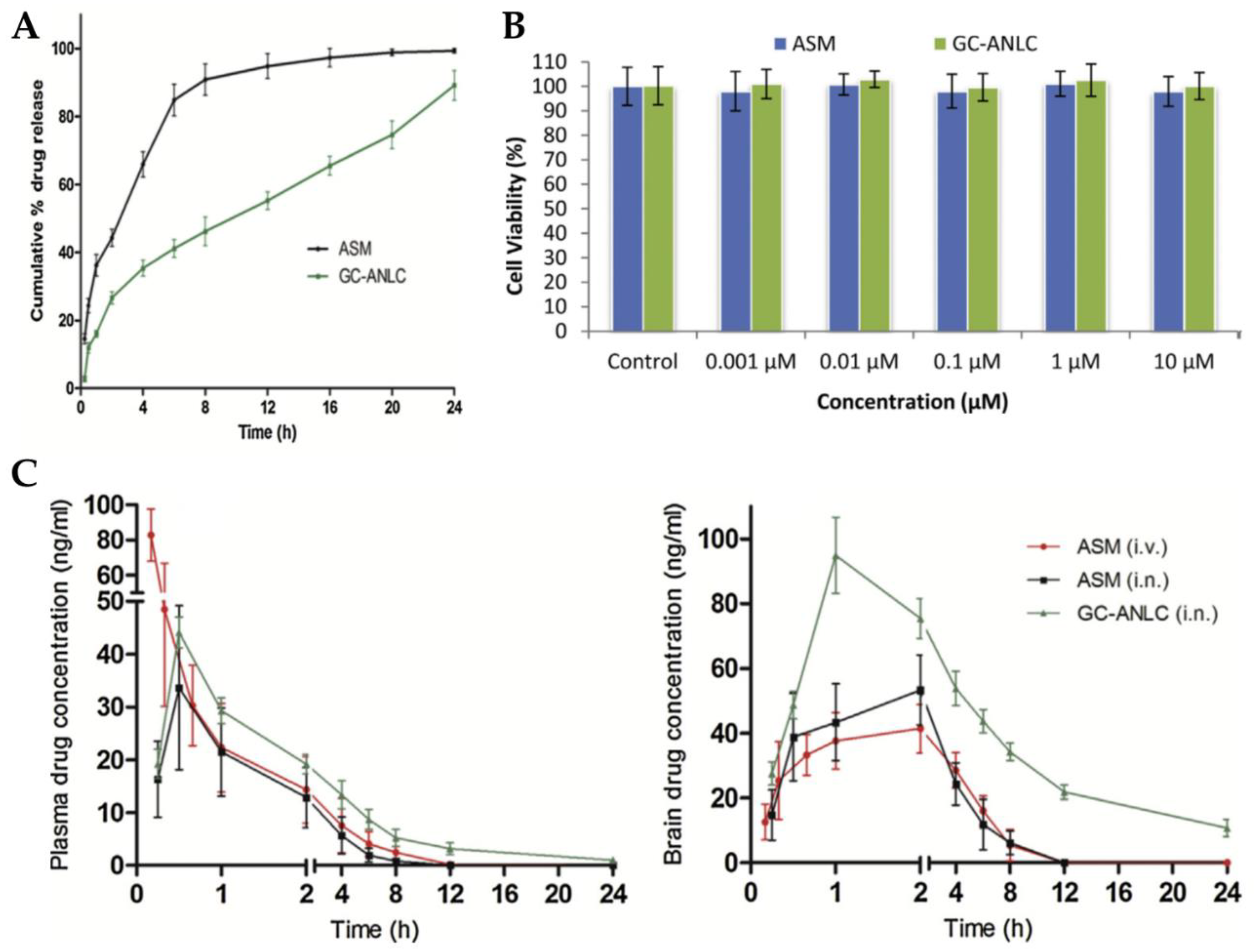
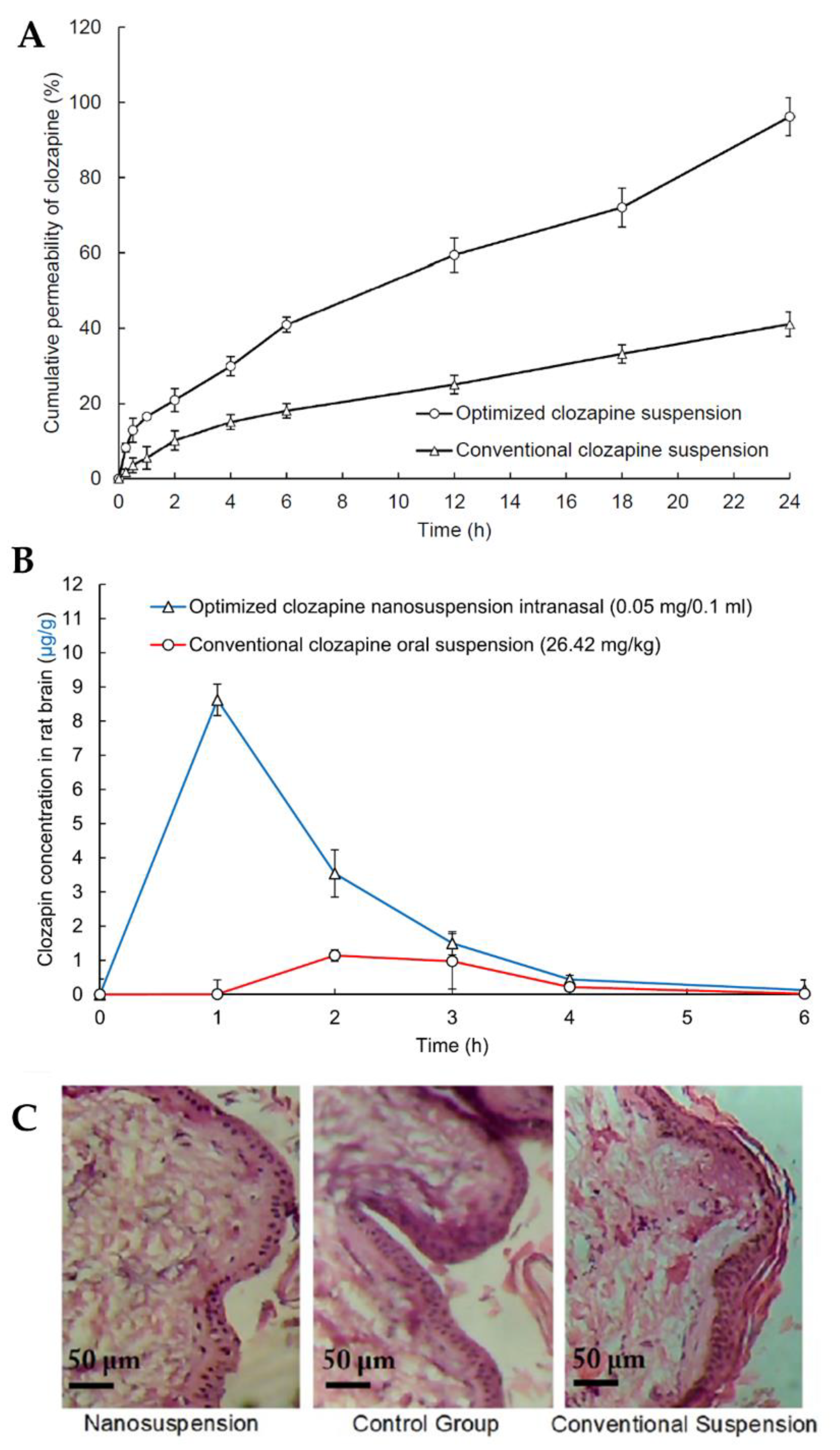
2.8. Clozapine Nanosuspension
3. 3rd Generation Antipsychotics
Aripiprazole Nanoemulgel
4. Final Discussion
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Stȩpień-Wyrobiec, O.; Nowak, M.; Wyrobiec, G.; Morawiec, E.; Wierzbik-Strońska, M.; Staszkiewicz, R.; Grabarek, B.O. Crossroad between current knowledge and new perspective of diagnostic and therapy of late-onset schizophrenia and very late-onset schizophrenia-like psychosis: An update. Front. Psychiatry 2022, 13, 1025414. [Google Scholar] [CrossRef]
- Ali, S.; Santomauro, D.; Ferrari, A.J.; Charlson, F. Excess mortality in severe mental disorders: A systematic review and meta-regression. J. Psychiatr. Res. 2022, 149, 97–105. [Google Scholar] [CrossRef]
- Bai, W.; Liu, Z.H.; Jiang, Y.Y.; Zhang, Q.E.; Rao, W.W.; Cheung, T.; Hall, B.J.; Xiang, Y.T. Worldwide prevalence of suicidal ideation and suicide plan among people with schizophrenia: A meta-analysis and systematic review of epidemiological surveys. Transl. Psychiatry 2021, 11, 552. [Google Scholar] [CrossRef]
- Vos, T.; Lim, S.S.; Abbafati, C.; Abbas, K.M.; Abbasi, M.; Abbasifard, M.; Abbasi-Kangevari, M.; Abbastabar, H.; Abd-Allah, F.; Abdelalim, A.; et al. Global Burden of 369 Diseases and Injuries in 204 Countries and Territories, 1990–2019: A Systematic Analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222. [Google Scholar] [CrossRef]
- Charlson, F.; van Ommeren, M.; Flaxman, A.; Cornett, J.; Whiteford, H.; Saxena, S. New WHO prevalence estimates of mental disorders in conflict settings: A systematic review and meta-analysis. Lancet 2019, 394, 240–248. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Li, Y.; Tian, B.; Liu, H.; Wu, S.; Wang, W. The effects and mechanism of environmental enrichment on MK-801 induced cognitive impairment in rodents with schizophrenia. Front. Cell. Neurosci. 2022, 16, 1024649. [Google Scholar] [CrossRef]
- Buizza, C.; Strozza, C.; Sbravati, G.; de Girolamo, G.; Ferrari, C.; Iozzino, L.; Macis, A.; Kennedy, H.G.; Candini, V. Positive and negative syndrome scale in forensic patients with schizophrenia spectrum disorders: A systematic review and meta-analysis. Ann. Gen. Psychiatry 2022, 21, 1–17. [Google Scholar] [CrossRef]
- Tsiglopoulos, J.; Pearson, N.; Mifsud, N.; Allott, K.; O’Donoghue, B. The association between vitamin D and symptom domains in psychotic disorders: A systematic review. Schizophr. Res. 2021, 237, 79–92. [Google Scholar] [CrossRef]
- Stone, W.S.; Phillips, M.R.; Yang, L.H.; Kegeles, L.S.; Susser, E.S.; Lieberman, J.A. Neurodegenerative model of schizophrenia: Growing evidence to support a revisit. Schizophr. Res. 2022, 243, 154–162. [Google Scholar] [CrossRef]
- Jauhar, S.; Johnstone, M.; McKenna, P.J. Schizophrenia. Lancet 2022, 399, 473–486. [Google Scholar] [CrossRef]
- Azorin, J.-M.; Simon, N. Dopamine Receptor Partial Agonists for the Treatment of Bipolar Disorder. Drugs 2019, 79, 1657–1677. [Google Scholar] [CrossRef]
- Elsayed, O.H.; Ercis, M.; Pahwa, M.; Singh, B. Treatment-Resistant Bipolar Depression: Therapeutic Trends, Challenges and Future Directions. Neuropsychiatr. Dis. Treat. 2022, 18, 2927–2943. [Google Scholar] [CrossRef]
- Bassett, D.; Boyce, P.; Lyndon, B.; Mulder, R.; Parker, G.; Porter, R.; Singh, A.; Bell, E.; Hamilton, A.; Morris, G.; et al. Guidelines for the management of psychosis in the context of mood disorders. Schizophr. Res. 2022, 241, 187–196. [Google Scholar] [CrossRef]
- McCutcheon, R.; Beck, K.; Jauhar, S.; Howes, O.D. Defining the Locus of Dopaminergic Dysfunction in Schizophrenia: A Meta-analysis and Test of the Mesolimbic Hypothesis. Schizophr. Bull. 2017, 44, 1301–1311. [Google Scholar] [CrossRef] [Green Version]
- Katthagen, T.; Kaminski, J.; Heinz, A.; Buchert, R.; Schlagenhauf, F. Striatal Dopamine and Reward Prediction Error Signaling in Unmedicated Schizophrenia Patients. Schizophr. Bull. 2020, 46, 1535–1546. [Google Scholar] [CrossRef] [Green Version]
- Maia, T.V.; Frank, M.J. An Integrative Perspective on the Role of Dopamine in Schizophrenia. Biol. Psychiatry 2016, 81, 52–66. [Google Scholar] [CrossRef] [Green Version]
- Howes, O.D.; Shatalina, E. Integrating the Neurodevelopmental and Dopamine Hypotheses of Schizophrenia and the Role of Cortical Excitation-Inhibition Balance. Biol. Psychiatry 2022, 92, 501–513. [Google Scholar] [CrossRef]
- Avram, M.; Brandl, F.; Cabello, J.; Leucht, C.; Scherr, M.; Mustafa, M.; Leucht, S.; Ziegler, S.; Sorg, C. Reduced striatal dopamine synthesis capacity in patients with schizophrenia during remission of positive symptoms. Brain 2019, 142, 1813–1826. [Google Scholar] [CrossRef]
- Sabe, M.; Pillinger, T.; Kaiser, S.; Chen, C.; Taipale, H.; Tanskanen, A.; Tiihonen, J.; Leucht, S.; Correll, C.U.; Solmi, M. Half a century of research on antipsychotics and schizophrenia: A scientometric study of hotspots, nodes, bursts, and trends. Neurosci. Biobehav. Rev. 2022, 136, 104608. [Google Scholar] [CrossRef]
- Kamei, H. Polypharmacy Management of Antipsychotics in Patients with Schizophrenia. Medicina 2022, 58, 1584. [Google Scholar] [CrossRef]
- Matsui, K.; Tokumasu, T.; Takekita, Y.; Inada, K.; Kanazawa, T.; Kishimoto, T.; Takasu, S.; Tani, H.; Tarutani, S.; Hashimoto, N.; et al. Switching to antipsychotic monotherapy vs. staying on antipsychotic polypharmacy in schizophrenia: A systematic review and meta-analysis. Schizophr. Res. 2019, 209, 50–57. [Google Scholar] [CrossRef]
- Seppälä, A.; Pylvänäinen, J.; Lehtiniemi, H.; Hirvonen, N.; Corripio, I.; Koponen, H.; Seppälä, J.; Ahmed, A.; Isohanni, M.; Miettunen, J.; et al. Predictors of response to pharmacological treatments in treatment-resistant schizophrenia—A systematic review and meta-analysis. Schizophr. Res. 2021, 236, 123–134. [Google Scholar] [CrossRef]
- Matthews, P.R.L.; Horder, J.; Pearce, M. Selective noradrenaline reuptake inhibitors for schizophrenia. Cochrane Database Syst. Rev. 2018, 2018, CD010219. [Google Scholar] [CrossRef] [Green Version]
- Lobo, M.C.; Whitehurst, T.S.; Kaar, S.J.; Howes, O.D. New and emerging treatments for schizophrenia: A narrative review of their pharmacology, efficacy and side effect profile relative to established antipsychotics. Neurosci. Biobehav. Rev. 2021, 132, 324–361. [Google Scholar] [CrossRef]
- Guerrin, C.G.; Doorduin, J.; Sommer, I.E.; de Vries, E.F. The dual hit hypothesis of schizophrenia: Evidence from animal models. Neurosci. Biobehav. Rev. 2021, 131, 1150–1168. [Google Scholar] [CrossRef]
- Mailman, R.; Murthy, R.B.M.A.V. Third Generation Antipsychotic Drugs: Partial Agonism or Receptor Functional Selectivity? Curr. Pharm. Des. 2010, 16, 488–501. [Google Scholar] [CrossRef] [Green Version]
- Lian, L.; Kim, D.D.; Procyshyn, R.M.; Cázares, D.; Honer, W.G.; Barr, A.M. Long-acting injectable antipsychotics for early psychosis: A comprehensive systematic review. PLoS ONE 2022, 17, e0267808. [Google Scholar] [CrossRef]
- Katona, L.; Bitter, I.; Czobor, P. A meta-analysis of effectiveness of real-world studies of antipsychotics in schizophrenia: Are the results consistent with the findings of randomized controlled trials? Transl. Psychiatry 2021, 11, 510. [Google Scholar] [CrossRef]
- Fabrazzo, M.; Cipolla, S.; Camerlengo, A.; Perris, F.; Catapano, F. Second-Generation Antipsychotics’ Effectiveness and Tolerability: A Review of Real-World Studies in Patients with Schizophrenia and Related Disorders. J. Clin. Med. 2022, 11, 4530. [Google Scholar] [CrossRef]
- Kishimoto, T.; Hagi, K.; Kurokawa, S.; Kane, J.M.; Correll, C.U. Long-acting injectable versus oral antipsychotics for the maintenance treatment of schizophrenia: A systematic review and comparative meta-analysis of randomised, cohort, and pre–post studies. Lancet Psychiatry 2021, 8, 387–404. [Google Scholar] [CrossRef]
- He, Y.; Tanaka, A.; Kishi, T.; Li, Y.; Matsunaga, M.; Tanihara, S.; Iwata, N.; Ota, A. Recent findings on subjective well-being and physical, psychiatric, and social comorbidities in individuals with schizophrenia: A literature review. Neuropsychopharmacol. Rep. 2022, 42, 430–436. [Google Scholar] [CrossRef]
- Suetani, S.; Honarparvar, F.; Siskind, D.; Hindley, G.; Veronese, N.; Vancampfort, D.; Allen, L.; Solmi, M.; Lally, J.; Gaughran, F.; et al. Increased rates of respiratory disease in schizophrenia: A systematic review and meta-analysis including 619,214 individuals with schizophrenia and 52,159,551 controls. Schizophr. Res. 2021, 237, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Rogowska, M.; Thornton, M.; Creese, B.; Velayudhan, L.; Aarsland, D.; Ballard, C.; Tsamakis, K.; Stewart, R.; Mueller, C. Implications of Adverse Outcomes Associated with Antipsychotics in Older Patients with Dementia: A 2011–2022 Update. Drugs Aging 2022, 40, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Grant, R.K.; Brindle, W.M.; Donnelly, M.C.; McConville, P.M.; Stroud, T.G.; Bandieri, L.; Plevris, J.N. Gastrointestinal and liver disease in patients with schizophrenia: A narrative review. World J. Gastroenterol. 2022, 28, 5515–5529. [Google Scholar] [CrossRef]
- Agrawal, M.; Saraf, S.; Saraf, S.; Antimisiaris, S.G.; Chougule, M.B.; Shoyele, S.A.; Alexander, A. Nose-to-brain drug delivery: An update on clinical challenges and progress towards approval of anti-Alzheimer drugs. J. Control. Release 2018, 281, 139–177. [Google Scholar] [CrossRef] [PubMed]
- Goel, H.; Kalra, V.; Verma, S.K.; Dubey, S.K.; Tiwary, A.K. Convolutions in the rendition of nose to brain therapeutics from bench to bedside: Feats & fallacies. J. Control. Release 2021, 341, 782–811. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.U.; Shehzad, A.; Ahmed, M.B.; Lee, Y.S. Intranasal Delivery of Nanoformulations: A potential way of treatment for neurological disorders. Molecules 2020, 25, 1929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kienitz, R.; Kay, L.; Beuchat, I.; Gelhard, S.; von Brauchitsch, S.; Mann, C.; Lucaciu, A.; Schäfer, J.-H.; Siebenbrodt, K.; Zöllner, J.-P.; et al. Benzodiazepines in the Management of Seizures and Status Epilepticus: A Review of Routes of Delivery, Pharmacokinetics, Efficacy, and Tolerability. CNS Drugs 2022, 36, 951–975. [Google Scholar] [CrossRef] [PubMed]
- Maeng, J.; Lee, K. Systemic and brain delivery of antidiabetic peptides through nasal administration using cell-penetrating peptides. Front. Pharmacol. 2022, 13, 1068495. [Google Scholar] [CrossRef]
- Rajput, A.; Pingale, P.; Dhapte-Pawar, V. Nasal delivery of neurotherapeutics via nanocarriers: Facets, aspects, and prospects. Front. Pharmacol. 2022, 13, 979682. [Google Scholar] [CrossRef]
- Alabsi, W.; Eedara, B.B.; Encinas-Basurto, D.; Polt, R.; Mansour, H.M. Nose-to-Brain Delivery of Therapeutic Peptides as Nasal Aerosols. Pharmaceutics 2022, 14, 1870. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.-T.; Maeng, H.-J. Pharmacokinetics and Pharmacodynamics of Intranasal Solid Lipid Nanoparticles and Nanostructured Lipid Carriers for Nose-to-Brain Delivery. Pharmaceutics 2022, 14, 572. [Google Scholar] [CrossRef] [PubMed]
- Pires, P.; Santos, A.O. Nanosystems in nose-to-brain drug delivery: A review of non-clinical brain targeting studies. J. Control. Release 2018, 270, 89–100. [Google Scholar] [CrossRef]
- Li, J.; Qiao, Y.; Wu, Z. Nanosystem trends in drug delivery using quality-by-design concept. J. Control. Release 2017, 256, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Iravani, S.; Varma, R.S. Advanced Drug Delivery Micro- and Nanosystems for Cardiovascular Diseases. Molecules 2022, 27, 5843. [Google Scholar] [CrossRef]
- Alhaj-Suliman, S.O.; Wafa, E.I.; Salem, A.K. Engineering nanosystems to overcome barriers to cancer diagnosis and treatment. Adv. Drug Deliv. Rev. 2022, 189, 114482. [Google Scholar] [CrossRef]
- Costa, C.P.; Moreira, J.; Amaral, M.H.; Lobo, J.M.S.; Silva, A. Nose-to-brain delivery of lipid-based nanosystems for epileptic seizures and anxiety crisis. J. Control. Release 2019, 295, 187–200. [Google Scholar] [CrossRef]
- Zorkina, Y.; Abramova, O.; Ushakova, V.; Morozova, A.; Zubkov, E.; Valikhov, M.; Melnikov, P.; Majouga, A.; Chekhonin, V. Nano Carrier Drug Delivery Systems for the Treatment of Neuropsychiatric Disorders: Advantages and Limitations. Molecules 2020, 25, 5294. [Google Scholar] [CrossRef]
- Rabaan, A.A.; Bukhamsin, R.; AlSaihati, H.; Alshamrani, S.A.; AlSihati, J.; Al-Afghani, H.M.; Alsubki, R.A.; Abuzaid, A.A.; Al-Abdulhadi, S.; Aldawood, Y.; et al. Recent Trends and Developments in Multifunctional Nanoparticles for Cancer Theranostics. Molecules 2022, 27, 8659. [Google Scholar] [CrossRef]
- Jampilek, J.; Kralova, K. Anticancer Applications of Essential Oils Formulated into Lipid-Based Delivery Nanosystems. Pharmaceutics 2022, 14, 2681. [Google Scholar] [CrossRef]
- Usharani, N.; Kanth, S.V.; Saravanan, N. Current nanotechnological strategies using lipids, carbohydrates, proteins and metal conjugates-based carrier systems for diagnosis and treatment of tuberculosis—A review. Int. J. Biol. Macromol. 2023, 227, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, M.I.; Lopes, C.M.; Amaral, M.H.; Costa, P.C. Surface-modified lipid nanocarriers for crossing the blood-brain barrier (BBB): A current overview of active targeting in brain diseases. Colloids Surf. B Biointerfaces 2023, 221, 112999. [Google Scholar] [CrossRef] [PubMed]
- Cagel, M.; Tesan, F.C.; Bernabeu, E.; Salgueiro, M.J.; Zubillaga, M.B.; Moretton, M.A.; Chiappetta, D.A. Polymeric mixed micelles as nanomedicines: Achievements and perspectives. Eur. J. Pharm. Biopharm. 2017, 113, 211–228. [Google Scholar] [CrossRef] [PubMed]
- Cabral, H.; Miyata, K.; Osada, K.; Kataoka, K. Block Copolymer Micelles in Nanomedicine Applications. Chem. Rev. 2018, 118, 6844–6892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Zhang, L.; Yang, T.; Wu, H. Stimuli-responsive polymeric micelles for drug delivery and cancer therapy. Int. J. Nanomed. 2018, 13, 2921–2942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thotakura, N.; Parashar, P.; Raza, K. Assessing the pharmacokinetics and toxicology of polymeric micelle conjugated therapeutics. Expert Opin. Drug Metab. Toxicol. 2020, 17, 323–332. [Google Scholar] [CrossRef]
- Hwang, D.; Ramsey, J.D.; Kabanov, A.V. Polymeric micelles for the delivery of poorly soluble drugs: From nanoformulation to clinical approval. Adv. Drug Deliv. Rev. 2020, 156, 80–118. [Google Scholar] [CrossRef]
- Alsaab, H.O.; Alharbi, F.D.; Alhibs, A.S.; Alanazi, N.B.; Alshehri, B.Y.; Saleh, M.A.; Alshehri, F.S.; Algarni, M.A.; Almugaiteeb, T.; Uddin, M.N.; et al. PLGA-Based Nanomedicine: History of Advancement and Development in Clinical Applications of Multiple Diseases. Pharmaceutics 2022, 14, 2728. [Google Scholar] [CrossRef]
- Pontes, A.P.; Welting, T.J.M.; Rip, J.; Creemers, L.B. Polymeric Nanoparticles for Drug Delivery in Osteoarthritis. Pharmaceutics 2022, 14, 2639. [Google Scholar] [CrossRef]
- Afzal, O.; Altamimi, A.S.A.; Nadeem, M.S.; Alzarea, S.I.; Almalki, W.H.; Tariq, A.; Mubeen, B.; Murtaza, B.N.; Iftikhar, S.; Riaz, N.; et al. Nanoparticles in Drug Delivery: From History to Therapeutic Applications. Nanomaterials 2022, 12, 4494. [Google Scholar] [CrossRef]
- Alabrahim, O.A.A.; Azzazy, H.M.E.-S. Polymeric nanoparticles for dopamine and levodopa replacement in Parkinson’s disease. Nanoscale Adv. 2022, 4, 5233–5244. [Google Scholar] [CrossRef]
- Alberto, M.; Paiva-Santos, A.C.; Veiga, F.; Pires, P.C. Lipid and Polymeric Nanoparticles: Successful Strategies for Nose-to-Brain Drug Delivery in the Treatment of Depression and Anxiety Disorders. Pharmaceutics 2022, 14, 2742. [Google Scholar] [CrossRef]
- Chaudhuri, A.; Kumar, D.N.; Shaik, R.A.; Eid, B.G.; Abdel-Naim, A.B.; Shadab; Ahmad, A.; Agrawal, A.K. Lipid-Based Nanoparticles as a Pivotal Delivery Approach in Triple Negative Breast Cancer (TNBC) Therapy. Int. J. Mol. Sci. 2022, 23, 10068. [Google Scholar] [CrossRef] [PubMed]
- Khairnar, S.V.; Pagare, P.; Thakre, A.; Nambiar, A.R.; Junnuthula, V.; Abraham, M.C.; Kolimi, P.; Nyavanandi, D.; Dyawanapelly, S. Review on the Scale-Up Methods for the Preparation of Solid Lipid Nanoparticles. Pharmaceutics 2022, 14, 1886. [Google Scholar] [CrossRef] [PubMed]
- Correia, A.; Monteiro, A.; Silva, R.; Moreira, J.; Lobo, J.S.; Silva, A. Lipid nanoparticles strategies to modify pharmacokinetics of central nervous system targeting drugs: Crossing or circumventing the blood–brain barrier (BBB) to manage neurological disorders. Adv. Drug Deliv. Rev. 2022, 189, 114485. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Pillai, J. Solid lipid matrix mediated nanoarchitectonics for improved oral bioavailability of drugs. Expert Opin. Drug Metab. Toxicol. 2019, 15, 499–515. [Google Scholar] [CrossRef] [PubMed]
- Paliwal, R.; Paliwal, S.R.; Kenwat, R.; Das Kurmi, B.; Sahu, M.K. Solid lipid nanoparticles: A review on recent perspectives and patents. Expert Opin. Ther. Pat. 2020, 30, 179–194. [Google Scholar] [CrossRef]
- Kalita, T.; Dezfouli, S.A.; Pandey, L.M.; Uludag, H. siRNA Functionalized Lipid Nanoparticles (LNPs) in Management of Diseases. Pharmaceutics 2022, 14, 2520. [Google Scholar] [CrossRef]
- Juhaščik, M.; Kováčik, A.; Huerta-Ángeles, G. Recent Advances of Hyaluronan for Skin Delivery: From Structure to Fabrication Strategies and Applications. Polymers 2022, 14, 4833. [Google Scholar] [CrossRef]
- Jacob, S.; Nair, A.B.; Shah, J.; Gupta, S.; Boddu, S.H.S.; Sreeharsha, N.; Joseph, A.; Shinu, P.; Morsy, M.A. Lipid Nanoparticles as a Promising Drug Delivery Carrier for Topical Ocular Therapy—An Overview on Recent Advances. Pharmaceutics 2022, 14, 533. [Google Scholar] [CrossRef]
- Annu; Rehman, S.; Shadab; Baboota, S.; Ali, J. Analyzing Nanotheraputics-Based Approaches for the Management of Psychotic Disorders. J. Pharm. Sci. 2019, 108, 3757–3768. [Google Scholar] [CrossRef] [PubMed]
- Markowicz-Piasecka, M.; Darłak, P.; Markiewicz, A.; Sikora, J.; Adla, S.K.; Bagina, S.; Huttunen, K.M. Current approaches to facilitate improved drug delivery to the central nervous system. Eur. J. Pharm. Biopharm. 2022, 181, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Pires, P.C.; Paiva-Santos, A.C.; Veiga, F. Nano and Microemulsions for the Treatment of Depressive and Anxiety Disorders: An Efficient Approach to Improve Solubility, Brain Bioavailability and Therapeutic Efficacy. Pharmaceutics 2022, 14, 2825. [Google Scholar] [CrossRef] [PubMed]
- Pires, P.C.; Paiva-Santos, A.C.; Veiga, F. Antipsychotics-Loaded Nanometric Emulsions for Brain Delivery. Pharmaceutics 2022, 14, 2174. [Google Scholar] [CrossRef]
- Choudhury, H.; Gorain, B.; Pandey, M.; Chatterjee, L.A.; Sengupta, P.; Das, A.; Molugulu, N.; Kesharwani, P. Recent Update on Nanoemulgel as Topical Drug Delivery System. J. Pharm. Sci. 2017, 106, 1736–1751. [Google Scholar] [CrossRef] [PubMed]
- Taha, M.; Alhakamy, N.A.; Shadab; Ahmad, M.Z.; Rizwanullah; Fatima, S.; Ahmed, N.; Alyazedi, F.M.; Karim, S.; Ahmad, J. Nanogels as Potential Delivery Vehicles in Improving the Therapeutic Efficacy of Phytopharmaceuticals. Polymers 2022, 14, 4141. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, P.; Chatterjee, B. Potential and future scope of nanoemulgel formulation for topical delivery of lipophilic drugs. Int. J. Pharm. 2017, 526, 353–365. [Google Scholar] [CrossRef]
- Pires, P.C.; Rodrigues, M.; Alves, G.; Santos, A.O. Strategies to Improve Drug Strength in Nasal Preparations for Brain Delivery of Low Aqueous Solubility Drugs. Pharmaceutics 2022, 14, 588. [Google Scholar] [CrossRef]
- Raza, F.; Evans, L.; Motallebi, M.; Zafar, H.; Pereira-Silva, M.; Saleem, K.; Peixoto, D.; Rahdar, A.; Sharifi, E.; Veiga, F.; et al. Liposome-based diagnostic and therapeutic applications for pancreatic cancer. Acta Biomater. 2023, 157, 1–23. [Google Scholar] [CrossRef]
- Losada-Barreiro, S.; Sezgin-Bayindir, Z.; Paiva-Martins, F.; Bravo-Díaz, C. Biochemistry of Antioxidants: Mechanisms and Pharmaceutical Applications. Biomedicines 2022, 10, 3051. [Google Scholar] [CrossRef]
- Guimarães, D.; Cavaco-Paulo, A.; Nogueira, E. Design of liposomes as drug delivery system for therapeutic applications. Int. J. Pharm. 2021, 601, 120571. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zhang, W.; Xiao, Q.; Fan, L.; Huang, D.; Chen, W.; He, W. Liposomes and liposome-like nanoparticles: From anti-fungal infection to the COVID-19 pandemic treatment. Asian J. Pharm. Sci. 2022, 17, 817–837. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Hanning, S.; Falconer, J.; Locke, M.; Wen, J. Recent advances in non-ionic surfactant vesicles (niosomes): Fabrication, characterization, pharmaceutical and cosmetic applications. Eur. J. Pharm. Biopharm. 2019, 144, 18–39. [Google Scholar] [CrossRef] [Green Version]
- Gomes, G.S.; Frank, L.A.; Contri, R.V.; Longhi, M.S.; Pohlmann, A.R.; Guterres, S.S. Nanotechnology-based alternatives for the topical delivery of immunosuppressive agents in psoriasis. Int. J. Pharm. 2023, 631, 122535. [Google Scholar] [CrossRef] [PubMed]
- Nemati, S.H.; Bigdeli, M.R.; Moghadam, F.M.; Sharifi, K. Neuroprotective effects of niosomes loaded with thymoquinone in the cerebral ischemia model of male Wistar rats. Nanomedicine: Nanotechnology, Biol. Med. 2023, 48, 102637. [Google Scholar] [CrossRef] [PubMed]
- Oransa, H.A.; Boughdady, M.F.; El-Sabbagh, H.M. Novel Mucoadhesive Chitosomes as a Platform for Enhanced Oral Bioavailability of Cinnarizine. Int. J. Nanomed. 2022, 17, 5641–5660. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Utreja, P. Vesicular nanocarrier based treatment of skin fungal infections: Potential and emerging trends in nanoscale pharmacotherapy. Asian J. Pharm. Sci. 2018, 14, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Lalu, L.; Tambe, V.; Pradhan, D.; Nayak, K.; Bagchi, S.; Maheshwari, R.; Kalia, K.; Tekade, R.K. Novel nanosystems for the treatment of ocular inflammation: Current paradigms and future research directions. J. Control. Release 2017, 268, 19–39. [Google Scholar] [CrossRef]
- Alharbi, W.S.; Hareeri, R.H.; Bazuhair, M.; Alfaleh, M.A.; Alhakamy, N.A.; Fahmy, U.A.; Alamoudi, A.A.; Badr-Eldin, S.M.; Ahmed, O.A.; AlGhamdi, S.A.; et al. Spanlastics as a Potential Platform for Enhancing the Brain Delivery of Flibanserin: In Vitro Response-Surface Optimization and In Vivo Pharmacokinetics Assessment. Pharmaceutics 2022, 14, 2627. [Google Scholar] [CrossRef]
- Ansari, M.D.; Saifi, Z.; Pandit, J.; Khan, I.; Solanki, P.; Sultana, Y.; Aqil, M. Spanlastics a Novel Nanovesicular Carrier: Its Potential Application and Emerging Trends in Therapeutic Delivery. AAPS PharmSciTech 2022, 23, 112. [Google Scholar] [CrossRef]
- Khan, K.U.; Minhas, M.U.; Badshah, S.F.; Suhail, M.; Ahmad, A.; Ijaz, S. Overview of nanoparticulate strategies for solubility enhancement of poorly soluble drugs. Life Sci. 2022, 291, 120301. [Google Scholar] [CrossRef] [PubMed]
- Akhter, H.; Ahmad, I.; Alshahrani, M.Y.; Al-Harbi, A.I.; Khalilullah, H.; Afzal, O.; Altamimi, A.S.A.; Ullah, S.N.M.N.; Ojha, A.; Karim, S. Drug Delivery Challenges and Current Progress in Nanocarrier-Based Ocular Therapeutic System. Gels 2022, 8, 82. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Wang, S.; Yu, Y.; Sun, W.; Fan, R.; Shi, J.; Gu, W.; Wang, Z.; Zhang, H.; Zheng, A. Review of nanosuspension formulation and process analysis in wet media milling using microhydrodynamic model and emerging characterization methods. Int. J. Pharm. 2022, 623, 121862. [Google Scholar] [CrossRef] [PubMed]
- Nagai, N.; Otake, H. Novel drug delivery systems for the management of dry eye. Adv. Drug Deliv. Rev. 2022, 191, 114582. [Google Scholar] [CrossRef]
- Boche, M.; Pokharkar, V. Quetiapine Nanoemulsion for Intranasal Drug Delivery: Evaluation of Brain-Targeting Efficiency. AAPS PharmSciTech 2016, 18, 686–696. [Google Scholar] [CrossRef] [PubMed]
- Abdelrahman, F.E.; Elsayed, I.; Gad, M.K.; Elshafeey, A.H.; Mohamed, M.I. Response surface optimization, Ex vivo and In vivo investigation of nasal spanlastics for bioavailability enhancement and brain targeting of risperidone. Int. J. Pharm. 2017, 530, 1–11. [Google Scholar] [CrossRef]
- Qureshi, M.; Aqil, M.; Imam, S.S.; Ahad, A.; Sultana, Y. Formulation and Evaluation of Neuroactive Drug Loaded Chitosan Nanoparticle for Nose to Brain Delivery: In-vitro Characterization and In-vivo Behavior Study. Curr. Drug Deliv. 2018, 16, 123–135. [Google Scholar] [CrossRef]
- Đorđević, S.M.; Santrač, A.; Cekić, N.D.; Marković, B.D.; Divović, B.; Ilić, T.M.; Savić, M.M.; Savić, S.D. Parenteral nanoemulsions of risperidone for enhanced brain delivery in acute psychosis: Physicochemical and in vivo performances. Int. J. Pharm. 2017, 533, 421–430. [Google Scholar] [CrossRef]
- Joseph, E.; Reddi, S.; Rinwa, V.; Balwani, G.; Saha, R. Design and in vivo evaluation of solid lipid nanoparticulate systems of Olanzapine for acute phase schizophrenia treatment: Investigations on antipsychotic potential and adverse effects. Eur. J. Pharm. Sci. 2017, 104, 315–325. [Google Scholar] [CrossRef]
- Natarajan, J.; Baskaran, M.; Humtsoe, L.C.; Vadivelan, R.; Justin, A. Enhanced brain targeting efficacy of Olanzapine through solid lipid nanoparticles. Artif. Cells, Nanomedicine, Biotechnol. 2016, 45, 364–371. [Google Scholar] [CrossRef] [Green Version]
- Joseph, E.; Reddi, S.; Rinwa, V.; Balwani, G.; Saha, R. DoE based Olanzapine loaded poly-caprolactone nanoparticles decreases extrapyramidal effects in rodent model. Int. J. Pharm. 2018, 541, 198–205. [Google Scholar] [CrossRef]
- Gadhave, D.G.; Tagalpallewar, A.A.; Kokare, C.R. Agranulocytosis-Protective Olanzapine-Loaded Nanostructured Lipid Carriers Engineered for CNS Delivery: Optimization and Hematological Toxicity Studies. AAPS PharmSciTech 2019, 20, 22. [Google Scholar] [CrossRef]
- Khallaf, R.A.; Aboud, H.M.; Sayed, O.M. Surface modified niosomes of olanzapine for brain targeting via nasal route; preparation, optimization, and in vivo evaluation. J. Liposome Res. 2019, 30, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Hidau, M.K.; Gautam, S.; Gupta, K.; Singh, K.P.; Singh, S.K. Glycol chitosan functionalized asenapine nanostructured lipid carriers for targeted brain delivery: Pharmacokinetic and teratogenic assessment. Int. J. Biol. Macromol. 2018, 108, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Kumbhar, S.A.; Kokare, C.R.; Shrivastava, B.; Gorain, B.; Choudhury, H. Preparation, characterization, and optimization of asenapine maleate mucoadhesive nanoemulsion using Box-Behnken design: In vitro and in vivo studies for brain targeting. Int. J. Pharm. 2020, 586, 119499. [Google Scholar] [CrossRef] [PubMed]
- Jazuli, I.; Annu; Nabi, B.; Moolakkadath, T.; Alam, T.; Baboota, S.; Ali, J. Optimization of Nanostructured Lipid Carriers of Lurasidone Hydrochloride Using Box-Behnken Design for Brain Targeting: In Vitro and In Vivo Studies. J. Pharm. Sci. 2019, 108, 3082–3090. [Google Scholar] [CrossRef]
- Pokharkar, V.; Suryawanshi, S.; Dhapte-Pawar, V. Exploring micellar-based polymeric systems for effective nose-to-brain drug delivery as potential neurotherapeutics. Drug Deliv. Transl. Res. 2019, 10, 1019–1031. [Google Scholar] [CrossRef]
- Pailla, S.R.; Talluri, S.; Rangaraj, N.; Ramavath, R.; Challa, V.S.; Doijad, N.; Sampathi, S. Intranasal Zotepine Nanosuspension: Intended for improved brain distribution in rats. DARU J. Pharm. Sci. 2019, 27, 541–556. [Google Scholar] [CrossRef]
- Gadhave, D.; Tupe, S.; Tagalpallewar, A.; Gorain, B.; Choudhury, H.; Kokare, C. Nose-to-brain delivery of amisulpride-loaded lipid-based poloxamer-gellan gum nanoemulgel: In vitro and in vivo pharmacological studies. Int. J. Pharm. 2021, 607, 121050. [Google Scholar] [CrossRef]
- Patel, H.P.; Chaudhari, P.S.; Gandhi, P.A.; Desai, B.V.; Desai, D.T.; Dedhiya, P.P.; Vyas, B.A.; Maulvi, F.A. Nose to brain delivery of tailored clozapine nanosuspension stabilized using (+)-alpha-tocopherol polyethylene glycol 1000 succinate: Optimization and in vivo pharmacokinetic studies. Int. J. Pharm. 2021, 600, 120474. [Google Scholar] [CrossRef]
- Kumbhar, S.A.; Kokare, C.R.; Shrivastava, B.; Gorain, B.; Choudhury, H. Antipsychotic Potential and Safety Profile of TPGS-Based Mucoadhesive Aripiprazole Nanoemulsion: Development and Optimization for Nose-To-Brain Delivery. J. Pharm. Sci. 2021, 110, 1761–1778. [Google Scholar] [CrossRef] [PubMed]
- Bhattamisra, S.K.; Shak, A.T.; Xi, L.W.; Safian, N.H.; Choudhury, H.; Lim, W.M.; Shahzad, N.; Alhakamy, N.A.; Anwer, K.; Radhakrishnan, A.K.; et al. Nose to brain delivery of rotigotine loaded chitosan nanoparticles in human SH-SY5Y neuroblastoma cells and animal model of Parkinson’s disease. Int. J. Pharm. 2020, 579, 119148. [Google Scholar] [CrossRef] [PubMed]
- Elsawy, H.; Sedky, A.; Taleb, M.F.A.; El-Newehy, M.H. Antidiabetic Wound Dressing Materials Based on Cellulosic Fabrics Loaded with Zinc Oxide Nanoparticles Synthesized by Solid-State Method. Polymers 2022, 14, 2168. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
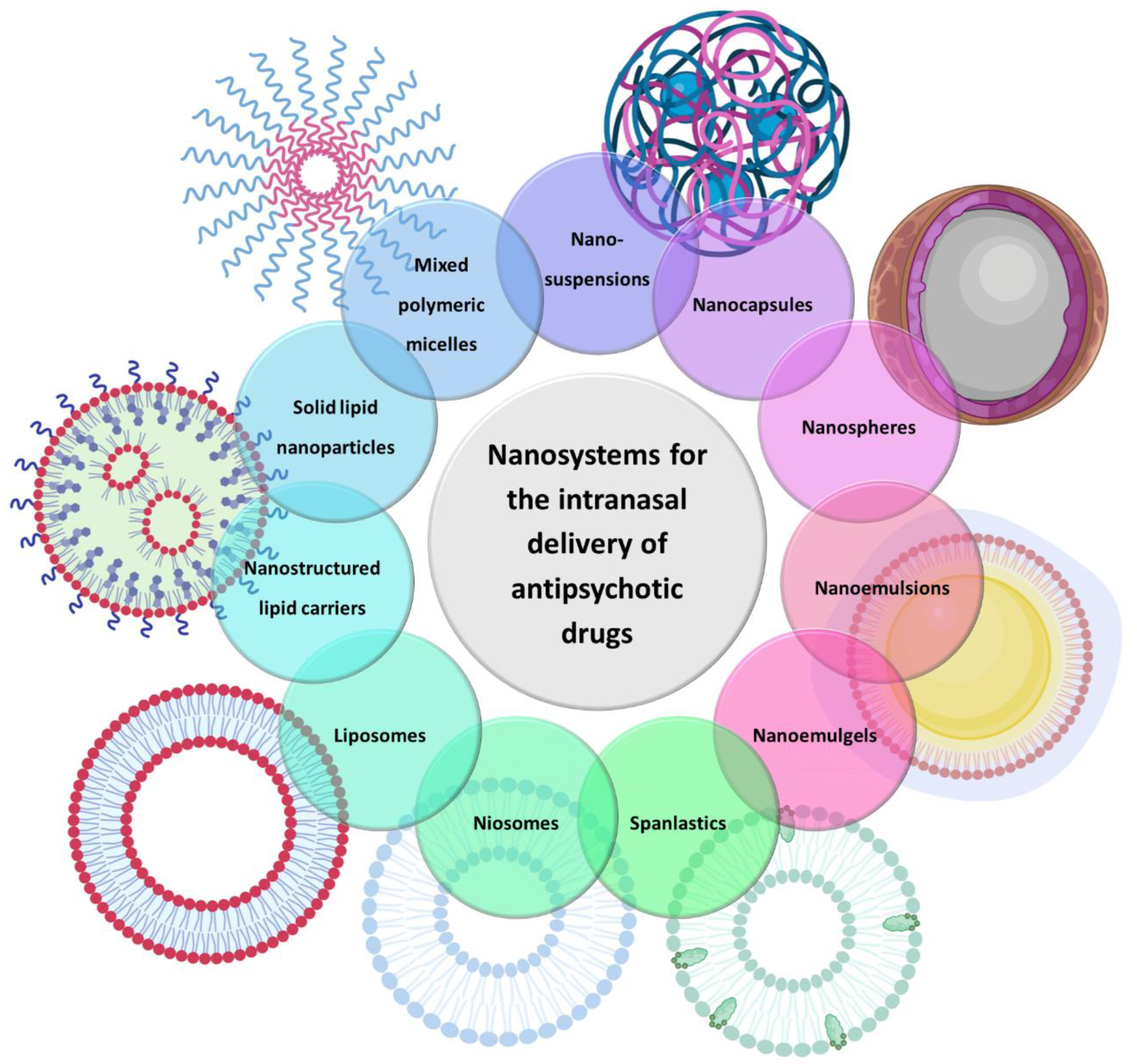
| Nanosystem Type | Size Range | Main Composition | Main Characteristics | Main Advantages | References |
|---|---|---|---|---|---|
| Mixed polymeric micelles | 20 to 200 nm | Amphiphilic copolymers | Hydrophobic inner core and a hydrophilic outer layer | High stability and encapsulation efficiency, good for the encapsulation of hydrophobic drugs | [53,54,55,56,57] |
| Nanocapsules | 10 to 1000 nm | Liquid lipids + polymers | Reservoir systems with liquid lipid core surrounded by a thin polymeric membrane | Controlled drug release, targeted drug delivery and enhanced drug permeation | [58,59,60,61,62] |
| Nanospheres | Polymers | No differentiated core, being formed by a dense polymeric matrix | |||
| Solid lipid nanoparticles | 50 to 1000 nm | Solid lipids + surfactants | Solid lipid core stabilized by a surfactant layer | Good for encapsulation of hydrophobic drugs, biocompatibility, controlled drug release | [63,64,65,66] |
| Nanostructured lipid carriers | Solid lipids + liquid lipds + surfactants | Liquid lipids included in an unstructured solid lipid matrix | Higher stability and encapsulation efficiency and lower tendency for crystallization than solid lipid nanoparticles, good for encapsulation of hydrophobic drugs, biocompatibility, controlled drug release | [67,68,69,70] | |
| Nanoemulsions | 10 to 200 nm | Oils, surfactants, cosurfactants, water | Liquid-in-liquid colloidal dispersions | Ease of preparation, enhanced drug permeation and increased drug solubility | [71,72,73,74] |
| Nanoemulgels | Oils, surfactants, cosurfactants, water, gelling polymer | Nanoemulsions with gel characteristics | High viscosity, ideal for retention at the administration site | [75,76,77,78] | |
| Liposomes | 20 to 1000 nm | Amphiphilic phospholipids | Spherical colloidal vesicles composed of one or more amphiphilic phospholipid bilayers delimiting an aqueous inner core | Versatility, since hydrophobic drugs can be incorporated into the membrane and hydrophilic drugs can be solubilized within the core, biodegradable | [79,80,81,82] |
| Niosomes | 10 to 1000 nm | Amphiphilic phospholipids + non-ionic amphiphilic surfactants | Modification of liposomes | Higher stability than liposomes, biodegradable | [83,84,85,86] |
| Spanlastics | 180 to 450 nm | Amphiphilic phospholipids + non-ionic amphiphilic surfactant Span® 60 (sorbitan monostearate 60) + edge activator | Modification of niosomes | Small size, deformable, increased encapsulation efficiency and permeation and biodegradable | [87,88,89,90] |
| Nanosuspensions | 1 to 1000 nm | Liquid vehicle + solid particles | Colloidal dispersions in which solid drug nanoparticles are suspended within a liquid | Ease of preparation, increased permeation/dissolution rate of hydrophobic drugs | [91,92,93,94] |
| Drug | Nanosystem Type | Administration Route | Excipients | Droplet Size (nm) | PDI | ZP (mV) | EE (%) | References |
|---|---|---|---|---|---|---|---|---|
| Quetiapine | Nanoemulsion | IN | Capmul® MCM, Tween® 80, Transcutol® P, water | 144 | 0.193 | −8.1 | 91 | [96] |
| Risperidone | Nanoemulsion | IV | Medium-chain triglycerides, soybean oil, soy lecithin, sodium oleate, benzyl alcohol, butylhydroxytoluene, glycerol, polysorbate 80, water | 184 | 0.110 | −56.0 | NR | [97] |
| Spanlastics | IN | Span® 60, ethanol, PVA | 103 | 0.341 | −45.9 | 64 | [98] | |
| SLN | IN | Oleic acid, stearic acid, Tween® 80, chitosan, water | 133 | 0.200 | +11.8 | 70 | [99] | |
| Olanzapine | SLN | IV | Water, glyceryl monostearate, poloxamer 188, | 157 | 0.411 | −37.3 | 73 | [100] |
| Water, glyceryl monostearate, poloxamer 188, Tween® 80 | 151 | 0.346 | −33.7 | 75 | ||||
| SLN | IV | Tripalmitin, stearyl amine, Tween® 80, water | 111 | 0.340 | +35.3 | 96 | [101] | |
| NP | IV | Polycaprolactone, poloxamer 188 | 73 | 0.231 | −32.5 | 79 | [102] | |
| Polycaprolactone, poloxamer 188, Tween® 80 | 81 | 0.312 | −27.8 | 77 | ||||
| NLC | IN | Labrafil® M 1944 CS, Compritol® 888 ATO, Gelucire® 44/14, Tween® 80, HPMC K4M, poloxamer 407 | 89 | 0.310 | −22.6 | 89 | [103] | |
| Niosomes | IN | Span® 80, cholesterol, chitosan | 250 | NR | NR | 72 | [104] | |
| Asenapine | NLC | IN | Oleic acid, glyceryl monostearate, water, Tween® 80, glycol chitosan | 184 | NR | +18.8 | 84 | [105] |
| Nanoemulgel | IN | Capmul® PG-8, Kolliphor® RH40, Transcutol® HP, water, Carbopol® 971 | 21 | 0.355 | −14.1 | NR | [106] | |
| Lurasidone | NLC | IN | Capryol® 90, GelotTM 64, Tween® 80, Transcutol® P | 207 | 0.392 | NR | 92 | [107] |
| MPM | IN | Pluronic® F127, Gelucire® 44/14 | 175 | NR | NR | 98 | [108] | |
| Zotepine | Nanosuspension | IN | Soy lecithin, Pluronic® F-127, HPMC E15 | 330 | 0.208 | +18.3 | NR | [109] |
| Amisulpride | Nanoemulsion | IN | Maisine® CC, Labrasol®, Transcutol® HP, water | 92 | 0.460 | −18.2 | 99 | [110] |
| Nanoemulgel | Maisine® CC, Labrasol®, Transcutol® HP, water, xanthan gum, poloxamer 407 | 106 | 0.510 | −16.0 | 99 | |||
| Clozapine | Nanosuspension | IN | TGPS, polyvinylpyrrolidone K30, water | 281 | NR | −0.8 | NR | [111] |
| Aripiprazole | Nanoemulgel | IN | Capmul® PG-8, TPGS, Transcutol® HP, water, Carbopol® 971 | 122 | 0.248 | −18.9 | NR | [112] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreira, M.D.; Duarte, J.; Veiga, F.; Paiva-Santos, A.C.; Pires, P.C. Nanosystems for Brain Targeting of Antipsychotic Drugs: An Update on the Most Promising Nanocarriers for Increased Bioavailability and Therapeutic Efficacy. Pharmaceutics 2023, 15, 678. https://doi.org/10.3390/pharmaceutics15020678
Ferreira MD, Duarte J, Veiga F, Paiva-Santos AC, Pires PC. Nanosystems for Brain Targeting of Antipsychotic Drugs: An Update on the Most Promising Nanocarriers for Increased Bioavailability and Therapeutic Efficacy. Pharmaceutics. 2023; 15(2):678. https://doi.org/10.3390/pharmaceutics15020678
Chicago/Turabian StyleFerreira, Maria Daniela, Joana Duarte, Francisco Veiga, Ana Cláudia Paiva-Santos, and Patrícia C. Pires. 2023. "Nanosystems for Brain Targeting of Antipsychotic Drugs: An Update on the Most Promising Nanocarriers for Increased Bioavailability and Therapeutic Efficacy" Pharmaceutics 15, no. 2: 678. https://doi.org/10.3390/pharmaceutics15020678