
Nasal Tumor Vaccination Protects against Lung Tumor Development by Induction of Resident Effector and Memory Anti-Tumor Immune Responses
 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Cell Line and Tumor Antigen Preparation (Tag) Preparation
2.3. Formulation of Nasal Nano Vaccine (CpG-NP-Tag)
2.4. Characterization of Nanoparticles
2.5. Animal Models
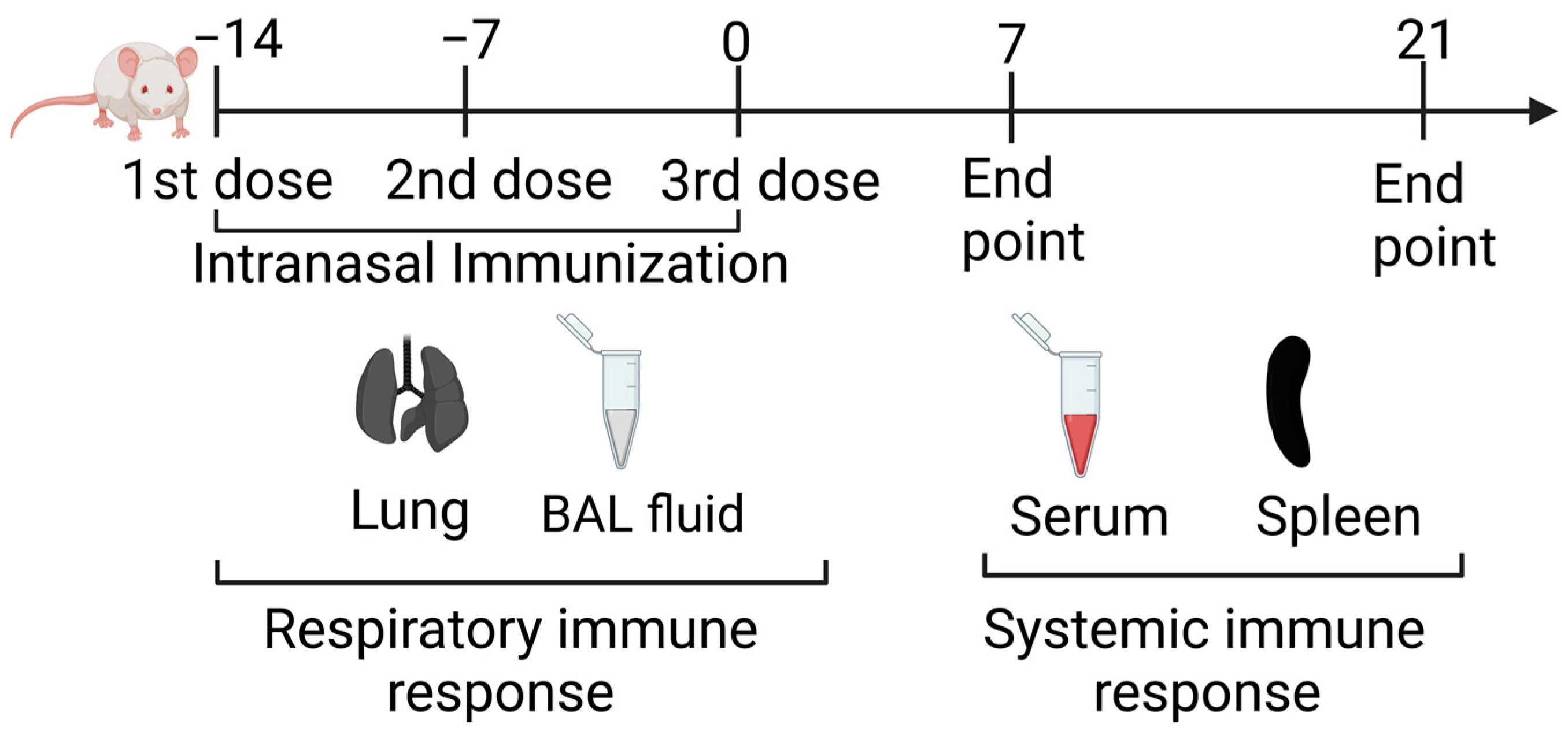
2.6. Intranasal Immunization
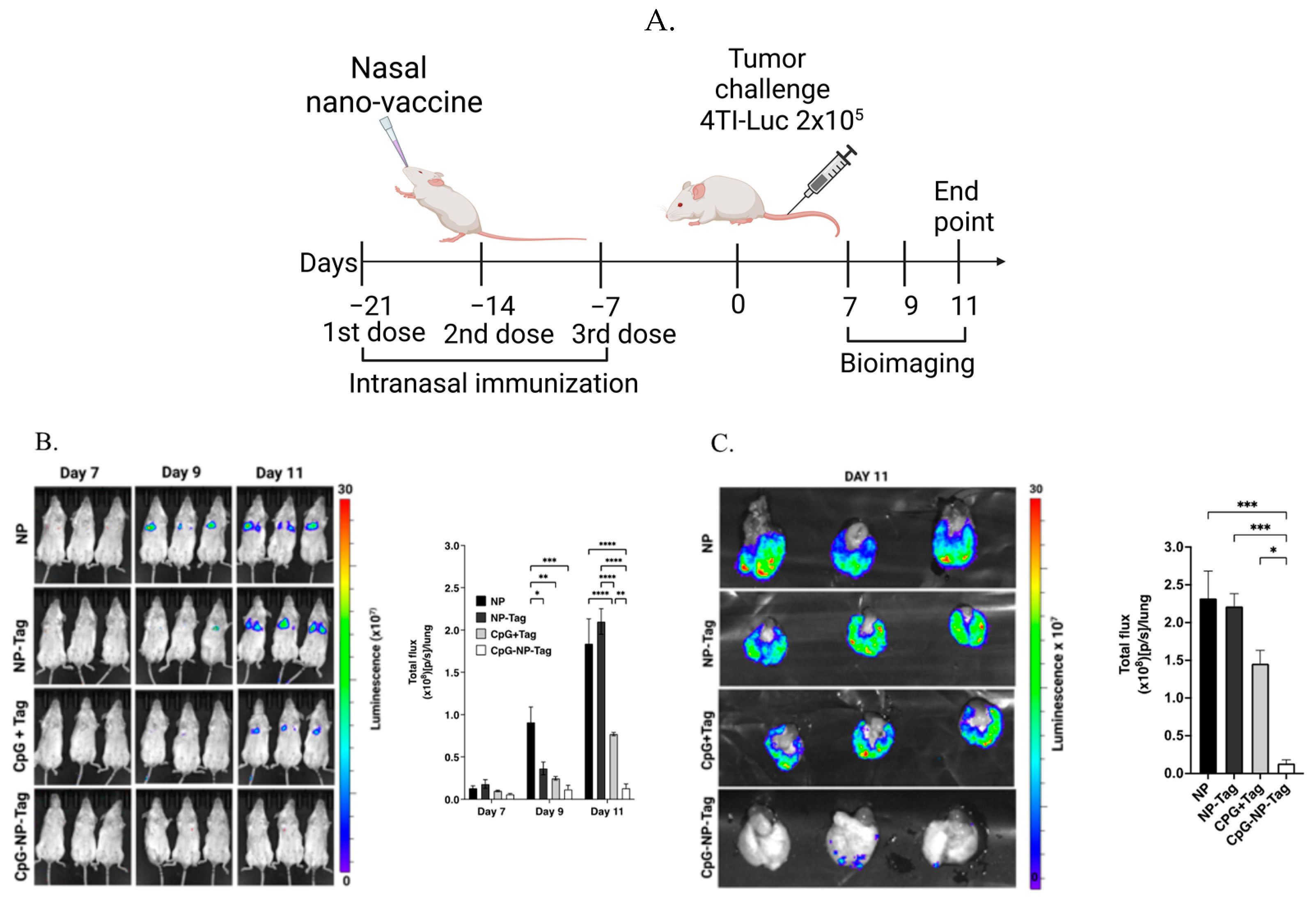
2.7. Tumor Challenge and Bioluminescence Imaging
2.8. Serum and BALF Collection
2.9. Isolation of Lung and Splenic Leukocytes
2.10. Flow Cytometry
2.11. Intracellular Staining
2.12. ELISA
2.13. Statistical Analysis
3. Results
3.1. Characterization of Intranasal CpG-NP-Tag NPs (Nasal Nano-Vaccine)
3.2. Nasal Nano-Vaccine, CpG-NP-Tag, Reduces Lung Colonization by 4T1 Breast Tumor Cells
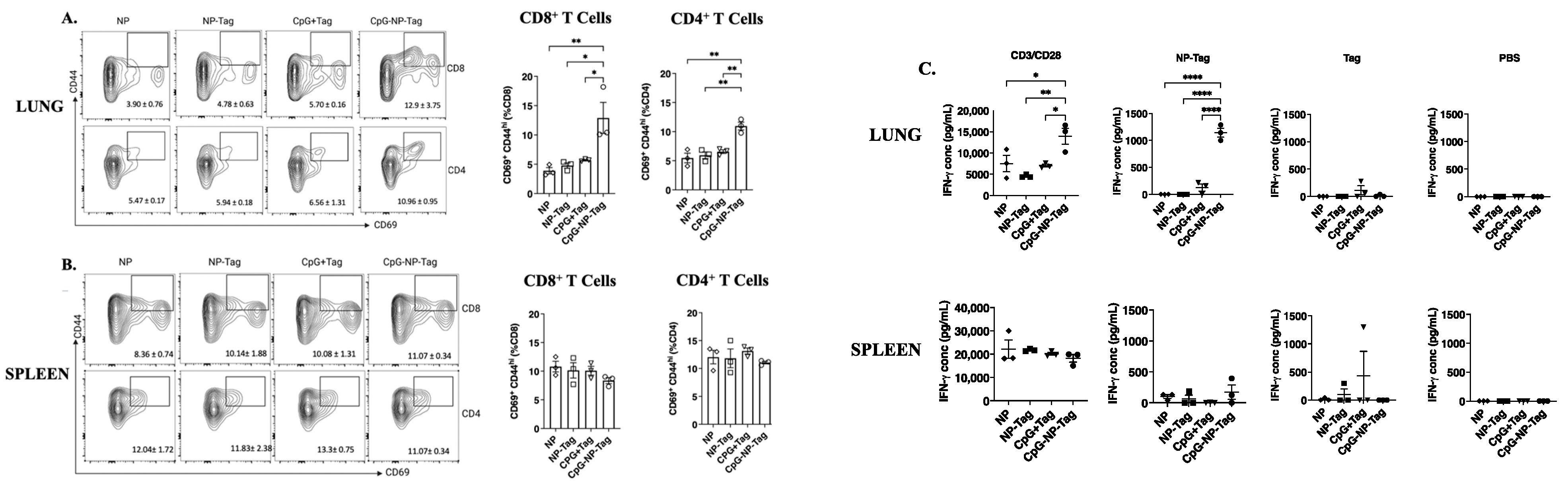
3.3. Nasal Nano-Vaccination Promotes Cellular and Humoral Immune Responses in the Lungs of Mice Challenged with 4T1 Tumor Cells
3.4. Intranasal Nano-Vaccine Induces Tumor-Specific Cell-Mediated and Humoral Antibody Immune Responses
3.5. Intranasal Nano-Vaccine Induces Preferential Expansion of Lung CD103+ CD69+ Resident Memory T Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jamil, A.; Kasi, A. Lung Metastasis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Donkor, M.; Jones, H.P. The Proposition of the Pulmonary Route as an Attractive Drug Delivery Approach of Nano-Based Immune Therapies and Cancer Vaccines to Treat Lung Tumors. Front. Nanotechnol. 2021, 3, 635194. [Google Scholar] [CrossRef]
- Nagrath, S.; Sequist, L.V.; Maheswaran, S.; Bell, D.W.; Irimia, D.; Ulkus, L.; Smith, M.R.; Kwak, E.L.; Digumarthy, S.; Muzikansky, A.; et al. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature 2007, 450, 1235–1239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambers, A.F.; Groom, A.C.; MacDonald, I.C. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2002, 2, 563–572. [Google Scholar] [CrossRef]
- Luzzi, K.J.; MacDonald, I.C.; Schmidt, E.E.; Kerkvliet, N.; Morris, V.L.; Chambers, A.F.; Groom, A.C. Multistep nature of metastatic inefficiency: Dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am. J. Pathol. 1998, 153, 865–873. [Google Scholar] [CrossRef]
- Cameron, M.D.; Schmidt, E.E.; Kerkvliet, N.; Nadkarni, K.V.; Morris, V.L.; Groom, A.C.; Chambers, A.F.; MacDonald, I.C. Temporal progression of metastasis in lung: Cell survival, dormancy, and location dependence of metastatic inefficiency. Cancer Res. 2000, 60, 2541–2546. [Google Scholar] [PubMed]
- Massagué, J.; Obenauf, A.C. Metastatic colonization by circulating tumour cells. Nature 2016, 529, 298–306. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Cao, X. Characteristics and Significance of the Pre-metastatic Niche. Cancer Cell 2016, 30, 668–681. [Google Scholar] [CrossRef] [Green Version]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317. [Google Scholar] [CrossRef]
- Chin, A.R.; Wang, S.E. Cancer Tills the Premetastatic Field: Mechanistic Basis and Clinical Implications. Clin. Cancer Res. 2016, 22, 3725–3733. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; MacDonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005, 438, 820–827. [Google Scholar] [CrossRef]
- Kitamura, T.; Qian, B.-Z.; Pollard, J.W. Immune cell promotion of metastasis. Nat. Rev. Immunol. 2015, 15, 73–86. [Google Scholar] [CrossRef] [Green Version]
- Hiam-Galvez, K.J.; Allen, B.M.; Spitzer, M.H. Systemic immunity in cancer. Nat. Rev. Cancer 2021, 21, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Tohme, S.; Simmons, R.L.; Tsung, A. Surgery for Cancer: A Trigger for Metastases. Cancer Res. 2017, 77, 1548–1552. [Google Scholar] [CrossRef] [Green Version]
- Staveley-O’Carroll, K.; Sotomayor, E.; Montgomery, J.; Borrello, I.; Hwang, L.; Fein, S.; Pardoll, D.; Levitsky, H. Induction of antigen-specific T cell anergy: An early event in the course of tumor progression. Proc. Natl. Acad. Sci. USA 1998, 95, 1178–1183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuenca, A.; Cheng, F.; Wang, H.; Brayer, J.; Horna, P.; Gu, L.; Bien, H.; Borrello, I.M.; Levitsky, H.I.; Sotomayor, E.M. Extra-lymphatic solid tumor growth is not immunologically ignored and results in early induction of antigen-specific T-cell anergy: Dominant role of cross-tolerance to tumor antigens. Cancer Res. 2003, 63, 9007–9015. [Google Scholar]
- Kokate, R.A.; Thamake, S.I.; Chaudhary, P.; Mott, B.; Raut, S.; Vishwanatha, J.K.; Jones, H.P. Enhancement of anti-tumor effect of particulate vaccine delivery system by ‘bacteriomimetic’ CpG functionalization of poly-lactic-co-glycolic acid nanoparticles. Nanomedicine 2015, 10, 915–929. [Google Scholar] [CrossRef] [Green Version]
- Kokate, R.A.; Chaudhary, P.; Sun, X.; Thamake, S.I.; Maji, S.; Chib, R.; Vishwanatha, J.K.; Jones, H.P. Rationalizing the use of functionalized poly-lactic-co-glycolic acid nanoparticles for dendritic cell-based targeted anticancer therapy. Nanomedicine 2016, 11, 479–494. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Cao, T.; Connolly, J.E.; Monnet, L.; Bennett, L.; Chapel, S.; Bagnis, C.; Mannoni, P.; Davoust, J.; Palucka, A.K.; et al. Hyperthermia enhances CTL cross-priming. J. Immunol. 2006, 176, 2134–2141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, T.; Mitsuhashi, N.; Sakurai, H.; Niibe, H. Modifications of tumor-associated antigen expression on human lung cancer cells by hyperthermia and cytokine. Anticancer Res. 1995, 15, 2601–2606. [Google Scholar]
- Couch, F.J.; Hart, S.N.; Sharma, P.; Toland, A.E.; Wang, X.; Miron, P.; Olson, J.E.; Godwin, A.K.; Pankratz, V.S.; Olswold, C.; et al. Inherited mutations in 17 breast cancer susceptibility genes among a large triple-negative breast cancer cohort unselected for family history of breast cancer. J. Clin. Oncol. 2015, 33, 304–311. [Google Scholar] [CrossRef] [Green Version]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lelekakis, M.; Moseley, J.M.; Martin, T.J.; Hards, D.; Williams, E.; Ho, P.; Lowen, D.; Javni, J.; Miller, F.R.; Slavin, J.; et al. A novel orthotopic model of breast cancer metastasis to bone. Clin. Exp. Metastasis 1999, 17, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Olkhanud, P.B.; Baatar, D.; Bodogai, M.; Hakim, F.; Gress, R.; Anderson, R.L.; Deng, J.; Xu, M.; Briest, S.; Biragyn, A. Breast cancer lung metastasis requires expression of chemokine receptor CCR4 and regulatory T cells. Cancer Res. 2009, 69, 5996–6004. [Google Scholar] [CrossRef] [Green Version]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [Green Version]
- Sasson, S.C.; Gordon, C.L.; Christo, S.N.; Klenerman, P.; Mackay, L.K. Local heroes or villains: Tissue-resident memory T cells in human health and disease. Cell Mol. Immunol. 2020, 17, 113–122. [Google Scholar] [CrossRef]
- Schenkel, J.M.; Masopust, D. Tissue-resident memory T cells. Immunity 2014, 41, 886–897. [Google Scholar] [CrossRef] [Green Version]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef] [PubMed]
- Kreutz, M.; Tacken, P.J.; Figdor, C.G. Targeting dendritic cells—Why bother? Blood 2013, 121, 2836–2844. [Google Scholar] [CrossRef] [PubMed]
- Palucka, K.; Banchereau, J. Dendritic-cell-based therapeutic cancer vaccines. Immunity 2013, 39, 38–48. [Google Scholar] [CrossRef] [Green Version]
- Cuzzubbo, S.; Mangsbo, S.; Nagarajan, D.; Habra, K.; Pockley, A.G.; McArdle, S.E.B. Cancer Vaccines: Adjuvant Potency, Importance of Age, Lifestyle, and Treatments. Front. Immunol. 2020, 11, 615240. [Google Scholar] [CrossRef] [PubMed]
- Dubensky, T.W., Jr.; Reed, S.G. Adjuvants for cancer vaccines. Semin. Immunol. 2010, 22, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Gutierro, I.; Hernández, R.M.; Igartua, M.; Gascón, A.R.; Pedraz, J.L. Size dependent immune response after subcutaneous, oral and intranasal administration of BSA loaded nanospheres. Vaccine 2002, 21, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Kurts, C.; Robinson, B.W.; Knolle, P.A. Cross-priming in health and disease. Nat. Rev. Immunol. 2010, 10, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Baumjohann, D.; Brossart, P. T follicular helper cells: Linking cancer immunotherapy and immune-related adverse events. J. Immunother Cancer 2021, 9, e002588. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Kim, J.Y.; Jeon, S.H.; Nam, H.; Jung, J.H.; Jeon, M.; Kim, E.S.; Bae, S.J.; Ahn, J.; Yoo, T.K.; et al. CD39(+) tissue-resident memory CD8(+) T cells with a clonal overlap across compartments mediate antitumor immunity in breast cancer. Sci. Immunol. 2022, 7, eabn8390. [Google Scholar] [CrossRef]
- Nizard, M.; Roussel, H.; Diniz, M.O.; Karaki, S.; Tran, T.; Voron, T.; Dransart, E.; Sandoval, F.; Riquet, M.; Rance, B.; et al. Induction of resident memory T cells enhances the efficacy of cancer vaccine. Nat. Commun. 2017, 8, 15221. [Google Scholar] [CrossRef]
- Egelston, C.A.; Avalos, C.; Tu, T.Y.; Rosario, A.; Wang, R.; Solomon, S.; Srinivasan, G.; Nelson, M.S.; Huang, Y.; Lim, M.H.; et al. Resident memory CD8+ T cells within cancer islands mediate survival in breast cancer patients. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [Green Version]
- Webb, J.R.; Milne, K.; Watson, P.; Deleeuw, R.J.; Nelson, B.H. Tumor-infiltrating lymphocytes expressing the tissue resident memory marker CD103 are associated with increased survival in high-grade serous ovarian cancer. Clin. Cancer Res. 2014, 20, 434–444. [Google Scholar] [CrossRef] [Green Version]
- Djenidi, F.; Adam, J.; Goubar, A.; Durgeau, A.; Meurice, G.; de Montpréville, V.; Validire, P.; Besse, B.; Mami-Chouaib, F. CD8+CD103+ tumor-infiltrating lymphocytes are tumor-specific tissue-resident memory T cells and a prognostic factor for survival in lung cancer patients. J. Immunol. 2015, 194, 3475–3486. [Google Scholar] [CrossRef] [Green Version]
- Malik, B.T.; Byrne, K.T.; Vella, J.L.; Zhang, P.; Shabaneh, T.B.; Steinberg, S.M.; Molodtsov, A.K.; Bowers, J.S.; Angeles, C.V.; Paulos, C.M.; et al. Resident memory T cells in the skin mediate durable immunity to melanoma. Sci. Immunol. 2017, 2, eaam6346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savas, P.; Virassamy, B.; Ye, C.; Salim, A.; Mintoff, C.P.; Caramia, F.; Salgado, R.; Byrne, D.J.; Teo, Z.L.; Dushyanthen, S.; et al. Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat. Med. 2018, 24, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.R.; Milne, K.; Nelson, B.H. PD-1 and CD103 Are Widely Coexpressed on Prognostically Favorable Intraepithelial CD8 T Cells in Human Ovarian Cancer. Cancer Immunol. Res. 2015, 3, 926–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanc, C.; Hans, S.; Tran, T.; Granier, C.; Saldman, A.; Anson, M.; Oudard, S.; Tartour, E. Targeting Resident Memory T Cells for Cancer Immunotherapy. Front. Immunol. 2018, 9, 1722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, T.; Hu, Y.; Lee, Y.T.; Bouchard, K.R.; Benechet, A.; Khanna, K.; Cauley, L.S. Lung-resident memory CD8 T cells (TRM) are indispensable for optimal cross-protection against pulmonary virus infection. J. Leukoc. Biol. 2014, 95, 215–224. [Google Scholar] [CrossRef] [Green Version]
- Zheng, M.Z.M.; Wakim, L.M. Tissue resident memory T cells in the respiratory tract. Mucosal. Immunol. 2022, 15, 379–388. [Google Scholar] [CrossRef]
- Vadalà, M.; Poddighe, D.; Laurino, C.; Palmieri, B. Vaccination and autoimmune diseases: Is prevention of adverse health effects on the horizon? EPMA J. 2017, 8, 295–311. [Google Scholar] [CrossRef] [Green Version]
- Hammerich, L.; Marron, T.U.; Upadhyay, R.; Svensson-Arvelund, J.; Dhainaut, M.; Hussein, S.; Zhan, Y.; Ostrowski, D.; Yellin, M.; Marsh, H.; et al. Systemic clinical tumor regressions and potentiation of PD1 blockade with in situ vaccination. Nat. Med. 2019, 25, 814–824. [Google Scholar] [CrossRef]
- Chen, A.; Wu, L.; Luo, Y.; Lu, S.; Wang, Y.; Zhou, Z.; Zhou, D.; Xie, Z.; Yue, J. Deep Tumor Penetrating Gold Nano-Adjuvant for NIR-II-Triggered In Situ Tumor Vaccination. Small 2022, 18, e2200993. [Google Scholar] [CrossRef]
- Ngamcherdtrakul, W.; Reda, M.; Nelson, M.A.; Wang, R.; Zaidan, H.Y.; Bejan, D.S.; Hoang, N.H.; Lane, R.S.; Luoh, S.W.; Leachman, S.A.; et al. In Situ Tumor Vaccination with Nanoparticle Co-Delivering CpG and STAT3 siRNA to Effectively Induce Whole-Body Antitumor Immune Response. Adv. Mater 2021, 33, e2100628. [Google Scholar] [CrossRef]
- Sandoval, F.; Terme, M.; Nizard, M.; Badoual, C.; Bureau, M.F.; Freyburger, L.; Clement, O.; Marcheteau, E.; Gey, A.; Fraisse, G.; et al. Mucosal imprinting of vaccine-induced CD8⁺ T cells is crucial to inhibit the growth of mucosal tumors. Sci. Transl. Med. 2013, 5, 172ra120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, T.; Shimosato, T.; Ueda, A.; Ishigatsubo, Y.; Klinman, D.M. Intrapulmonary Delivery of CpG Microparticles Eliminates Lung Tumors. Mol. Cancer Ther. 2015, 14, 2198–2205. [Google Scholar] [CrossRef] [PubMed]
- Perry, J.L.; Tian, S.; Sengottuvel, N.; Harrison, E.B.; Gorentla, B.K.; Kapadia, C.H.; Cheng, N.; Luft, J.C.; Ting, J.P.; DeSimone, J.M.; et al. Pulmonary Delivery of Nanoparticle-Bound Toll-like Receptor 9 Agonist for the Treatment of Metastatic Lung Cancer. ACS Nano 2020, 14, 7200–7215. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Particle Size (nm ± SD) | PDI | Zeta Potential (mV ± SD) | Encapsulation Efficiency (%) | µg Protein/mg NP |
|---|---|---|---|---|---|
| CpG-NP-Tag | 259.0 ± 2.10 | 0.138 | −14.56 ± 0.23 | 51 ± 7.70 | 23 |
| NP-Tag | 255.8 ± 1.84 | 0.248 | −6.98 ± 0.18 | 51 ± 7.70 | 23 |
| NP | 219.0 ± 0.82 | 0.040 | −5.52 ± 0.19 | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donkor, M.; Choe, J.; Reid, D.M.; Quinn, B.; Pulse, M.; Ranjan, A.; Chaudhary, P.; Jones, H.P. Nasal Tumor Vaccination Protects against Lung Tumor Development by Induction of Resident Effector and Memory Anti-Tumor Immune Responses. Pharmaceutics 2023, 15, 445. https://doi.org/10.3390/pharmaceutics15020445
Donkor M, Choe J, Reid DM, Quinn B, Pulse M, Ranjan A, Chaudhary P, Jones HP. Nasal Tumor Vaccination Protects against Lung Tumor Development by Induction of Resident Effector and Memory Anti-Tumor Immune Responses. Pharmaceutics. 2023; 15(2):445. https://doi.org/10.3390/pharmaceutics15020445
Chicago/Turabian StyleDonkor, Michael, Jamie Choe, Danielle Marie Reid, Byron Quinn, Mark Pulse, Amalendu Ranjan, Pankaj Chaudhary, and Harlan P. Jones. 2023. "Nasal Tumor Vaccination Protects against Lung Tumor Development by Induction of Resident Effector and Memory Anti-Tumor Immune Responses" Pharmaceutics 15, no. 2: 445. https://doi.org/10.3390/pharmaceutics15020445