
mRNA-Based Therapeutics in Cancer Treatment

Abstract
:1. Introduction
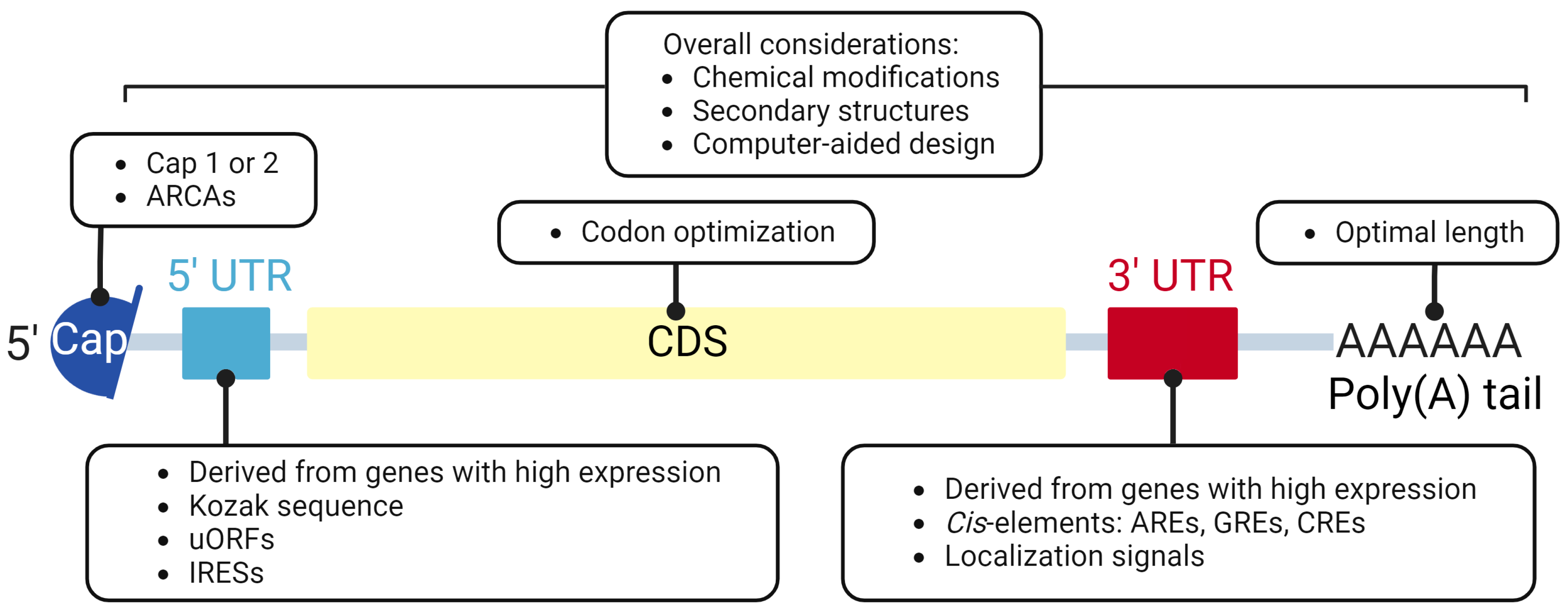
2. Components and Design of IVT mRNAs
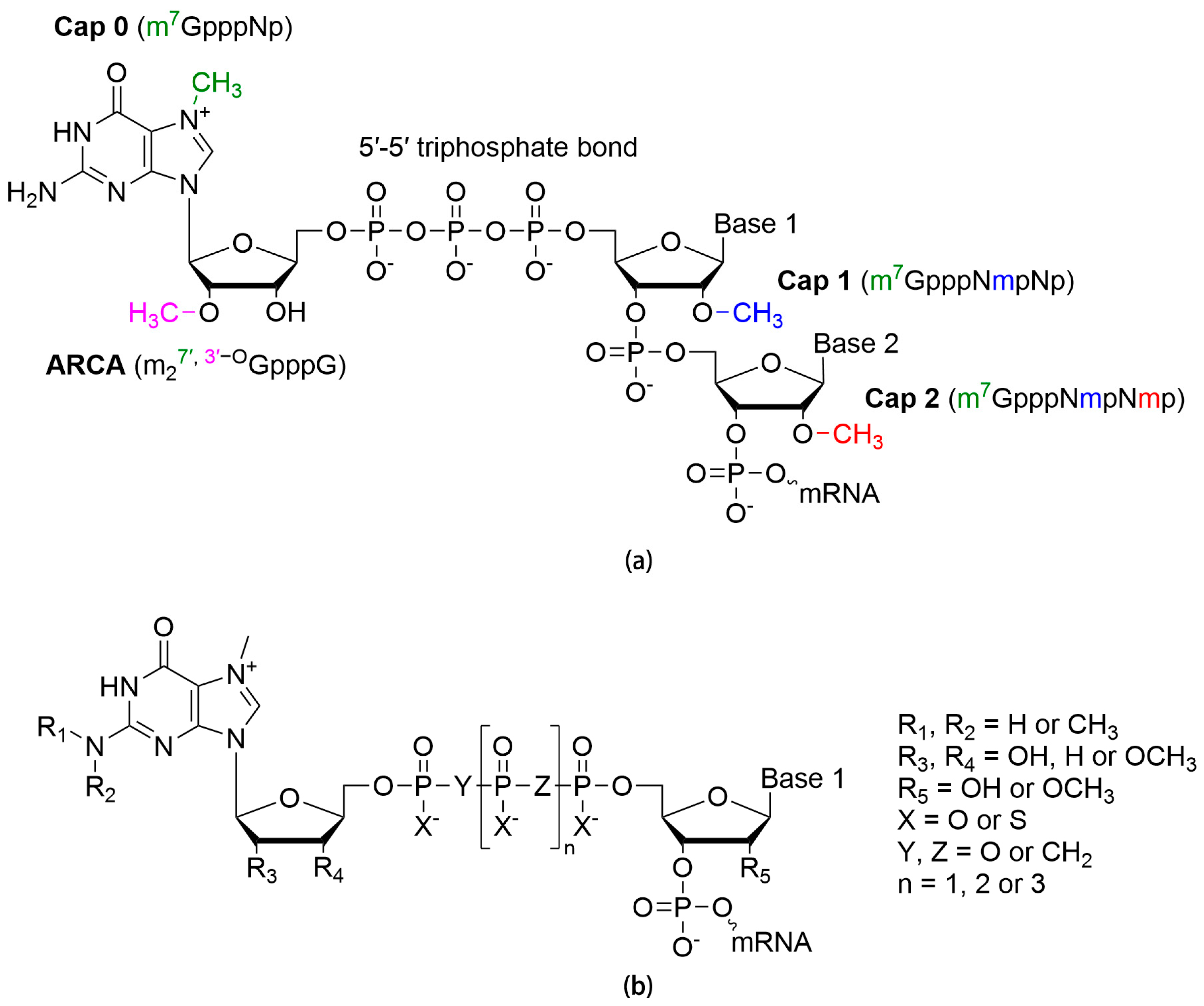
2.1. 5′ Cap
2.2. Untranslated Region (UTR)
2.3. Coding Sequence (CDS)
2.4. Poly(A) Tailing
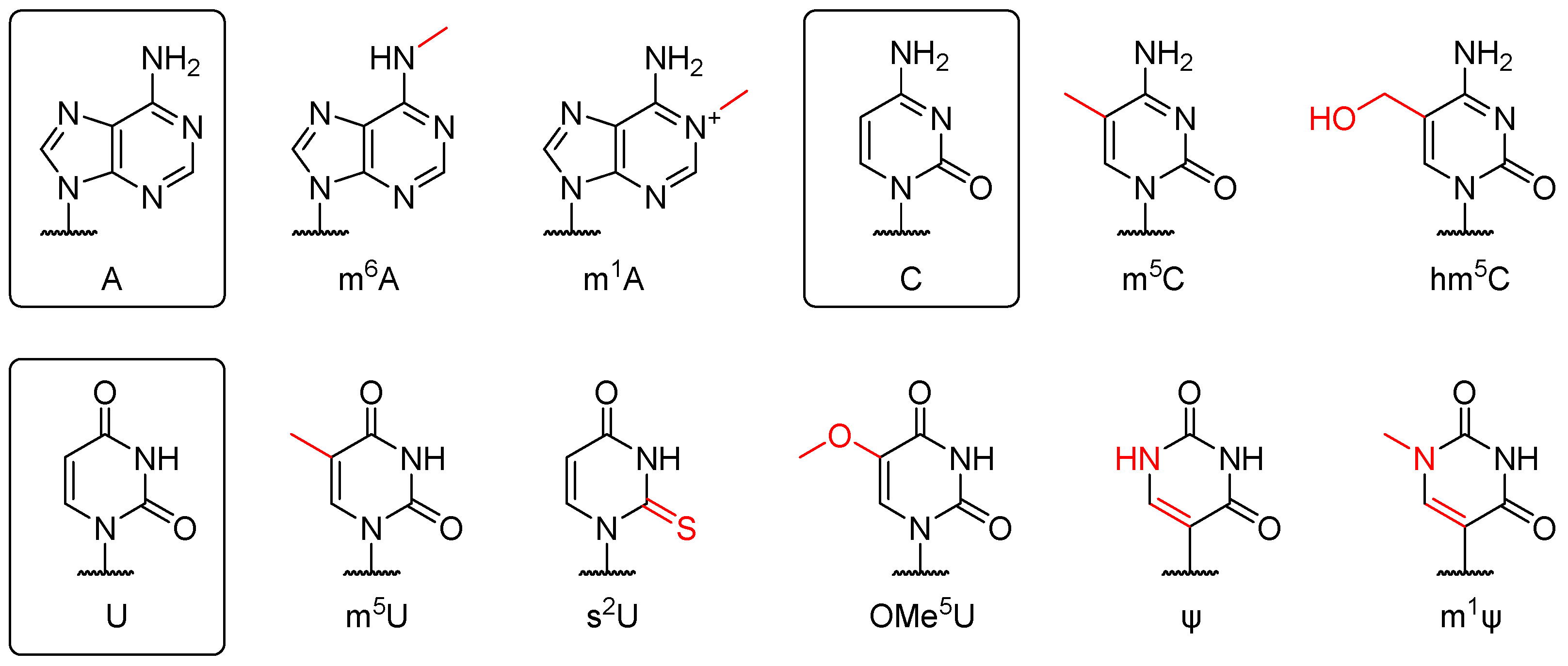
2.5. Chemical Modification
3. mRNA Delivery System
3.1. Naked mRNA Injection or Electroporation
3.2. Liposome and RNA Lipoplexes (LPX)
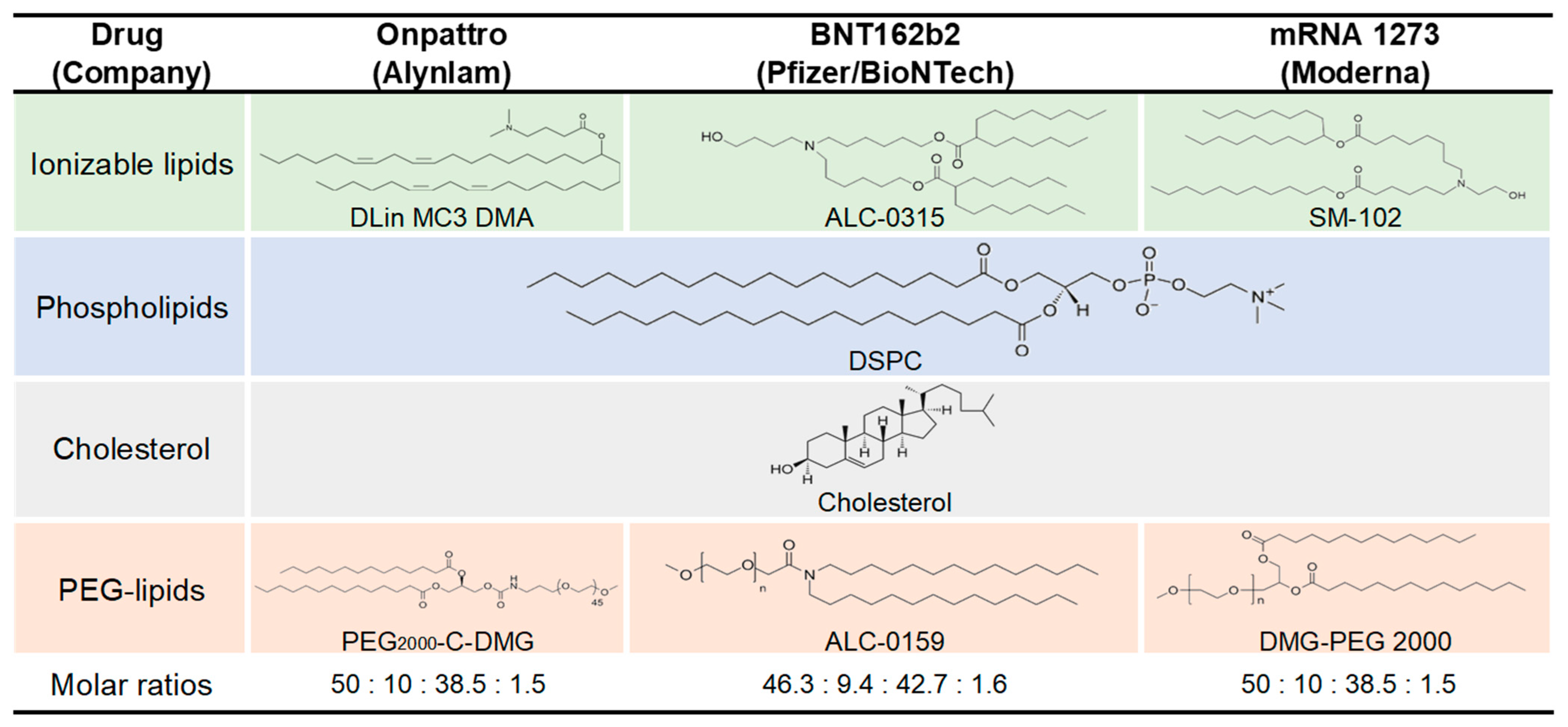
3.3. Lipid Nanoparticle (LNP)
3.4. Polymer-Based Nanoparticles
3.5. Cationic Nanoemulsions (CNEs)
3.6. Protamine-Based Delivery
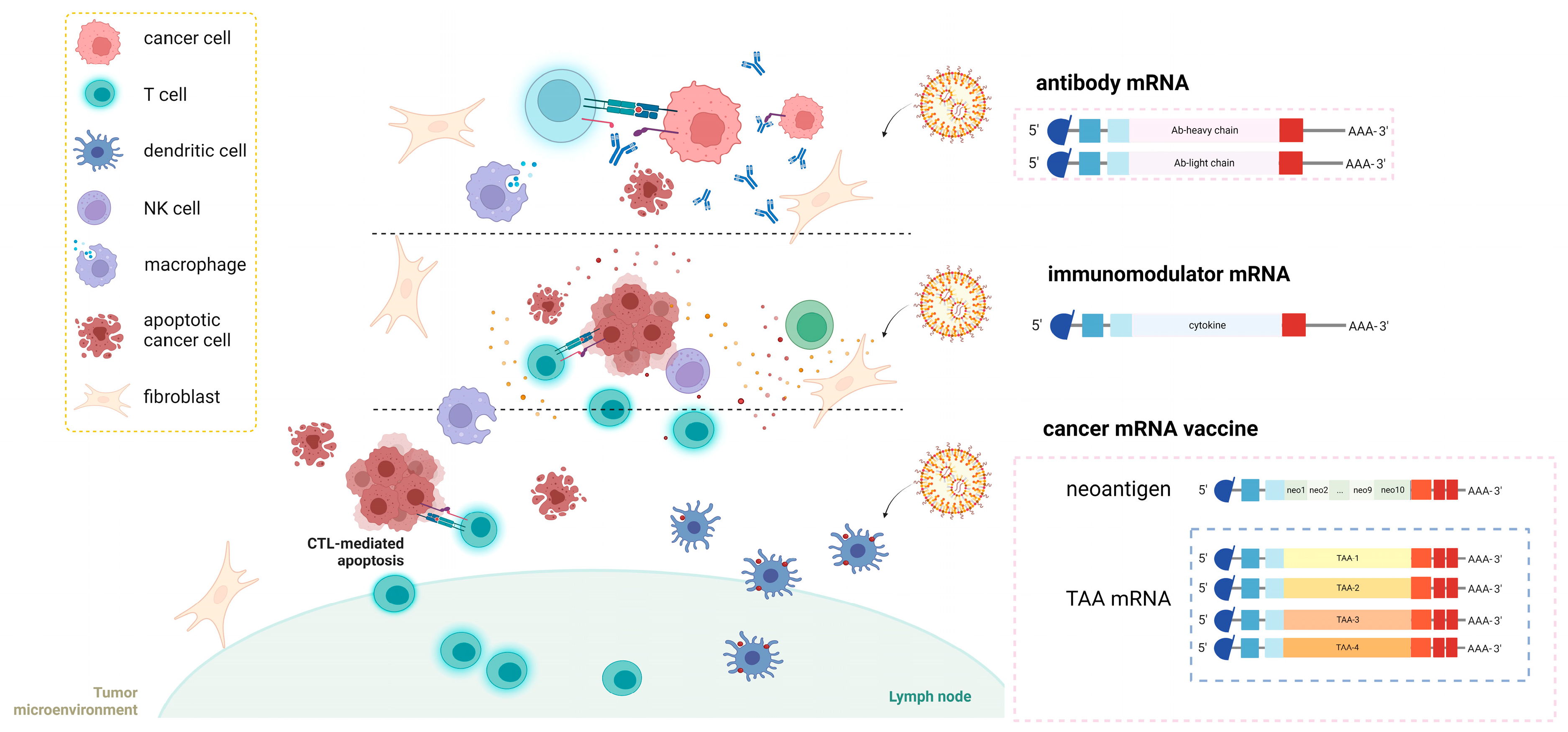
4. mRNA-Based Cancer Immunotherapies
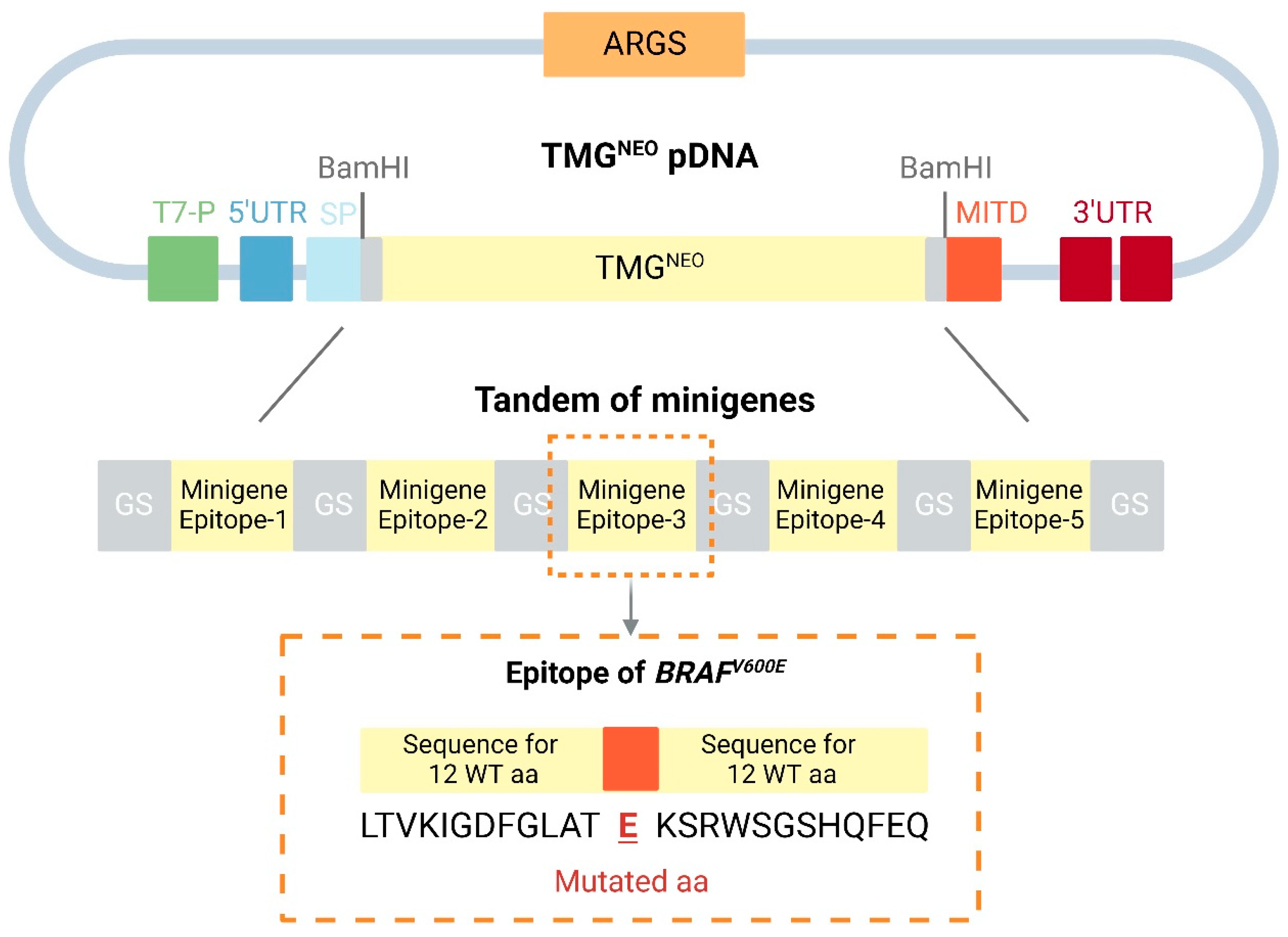
4.1. Neoantigen mRNA Vaccines
4.2. TAA mRNA Vaccines
4.3. mRNA Encoding Ab
4.4. Immunomodulator mRNA Vaccines
4.5. Protein Replacement Therapy
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brenner, S.; Jacob, F.; Meselson, M. An unstable intermediate carrying information from genes to ribosomes for protein synthesis. Nature 1961, 190, 576–581. [Google Scholar] [CrossRef]
- Wolff, J.A.; Malone, R.W.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P.L. Direct gene transfer into mouse muscle in vivo. Science 1990, 247, 1465–1468. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Pogocki, D.; Schoneich, C. Chemical stability of nucleic acid-derived drugs. J. Pharm. Sci. 2000, 89, 443–456. [Google Scholar] [CrossRef]
- Gross, J.D.; Moerke, N.J.; von der Haar, T.; Lugovskoy, A.A.; Sachs, A.B.; McCarthy, J.E.G.; Wagner, G. Ribosome loading onto the mRNA cap is driven by conformational coupling between eIF4G and eIF4E. Cell 2003, 115, 739–750. [Google Scholar] [CrossRef]
- Flaherty, S.M.; Fortes, P.; Izaurralde, E.; Mattaj, I.W.; Gilmartin, G.M. Participation of the nuclear cap binding complex in pre-mRNA 3′ processing. Proc. Natl. Acad. Sci. USA 1997, 94, 11893–11898. [Google Scholar] [CrossRef]
- Gilmartin, G.M.; McDevitt, M.A.; Nevins, J.R. Multiple factors are required for specific RNA cleavage at a poly(A) addition site. Genes Dev. 1988, 2, 578–587. [Google Scholar] [CrossRef]
- Cooke, C.; Alwine, J.C. The cap and the 3′ splice site similarly affect polyadenylation efficiency. Mol. Cell. Biol. 1996, 16, 2579–2584. [Google Scholar] [CrossRef]
- Konarska, M.M.; Padgett, R.A.; Sharp, P.A. Recognition of cap structure in splicing in vitro of mRNA precursors. Cell 1984, 38, 731–736. [Google Scholar] [CrossRef]
- Lewis, J.D.; Izaurralde, E. The role of the cap structure in RNA processing and nuclear export. Eur. J. Biochem. 1997, 247, 461–469. [Google Scholar] [CrossRef]
- Visa, N.; Izaurralde, E.; Ferreira, J.; Daneholt, B.; Mattaj, I.W. A nuclear cap-binding complex binds Balbiani ring pre-mRNA cotranscriptionally and accompanies the ribonucleoprotein particle during nuclear export. J. Cell Biol. 1996, 133, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Zust, R.; Cervantes-Barragan, L.; Habjan, M.; Maier, R.; Neuman, B.W.; Ziebuhr, J.; Szretter, K.J.; Baker, S.C.; Barchet, W.; Diamond, M.S.; et al. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat. Immunol. 2011, 12, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Jemielity, J.; Fowler, T.; Zuberek, J.; Stepinski, J.; Lewdorowicz, M.; Niedzwiecka, A.; Stolarski, R.; Darzynkiewicz, E.; Rhoads, R.E. Novel “anti-reverse” cap analogs with superior translational properties. RNA 2003, 9, 1108–1122. [Google Scholar] [CrossRef] [PubMed]
- Grudzien-Nogalska, E.; Jemielity, J.; Kowalska, J.; Darzynkiewicz, E.; Rhoads, R.E. Phosphorothioate cap analogs stabilize mRNA and increase translational efficiency in mammalian cells. RNA 2007, 13, 1745–1755. [Google Scholar] [CrossRef]
- Kuhn, A.N.; Diken, M.; Kreiter, S.; Selmi, A.; Kowalska, J.; Jemielity, J.; Darzynkiewicz, E.; Huber, C.; Tureci, O.; Sahin, U. Phosphorothioate cap analogs increase stability and translational efficiency of RNA vaccines in immature dendritic cells and induce superior immune responses in vivo. Gene Ther. 2010, 17, 961–971. [Google Scholar] [CrossRef]
- Wojtczak, B.A.; Sikorski, P.J.; Fac-Dabrowska, K.; Nowicka, A.; Warminski, M.; Kubacka, D.; Nowak, E.; Nowotny, M.; Kowalska, J.; Jemielity, J. 5′-Phosphorothiolate Dinucleotide Cap Analogues: Reagents for Messenger RNA Modification and Potent Small-Molecular Inhibitors of Decapping Enzymes. J. Am. Chem. Soc. 2018, 140, 5987–5999. [Google Scholar] [CrossRef]
- Rydzik, A.M.; Kulis, M.; Lukaszewicz, M.; Kowalska, J.; Zuberek, J.; Darzynkiewicz, Z.M.; Darzynkiewicz, E.; Jemielity, J. Synthesis and properties of mRNA cap analogs containing imidodiphosphate moiety—Fairly mimicking natural cap structure, yet resistant to enzymatic hydrolysis. Bioorg. Med. Chem. 2012, 20, 1699–1710. [Google Scholar] [CrossRef]
- Kowalska, J.; Wypijewska del Nogal, A.; Darzynkiewicz, Z.M.; Buck, J.; Nicola, C.; Kuhn, A.N.; Lukaszewicz, M.; Zuberek, J.; Strenkowska, M.; Ziemniak, M.; et al. Synthesis, properties, and biological activity of boranophosphate analogs of the mRNA cap: Versatile tools for manipulation of therapeutically relevant cap-dependent processes. Nucleic Acids Res. 2014, 42, 10245–10264. [Google Scholar] [CrossRef]
- Henderson, J.M.; Ujita, A.; Hill, E.; Yousif-Rosales, S.; Smith, C.; Ko, N.; McReynolds, T.; Cabral, C.R.; Escamilla-Powers, J.R.; Houston, M.E. Cap 1 Messenger RNA Synthesis with Co-transcriptional CleanCap((R)) Analog by In Vitro Transcription. Curr. Protoc. 2021, 1, e39. [Google Scholar] [CrossRef]
- Lockless, S.W.; Cheng, H.T.; Hodel, A.E.; Quiocho, F.A.; Gershon, P.D. Recognition of capped RNA substrates by VP39, the vaccinia virus-encoded mRNA cap-specific 2′-O-methyltransferase. Biochemistry 1998, 37, 8564–8574. [Google Scholar] [CrossRef]
- Sutton, G.; Grimes, J.M.; Stuart, D.I.; Roy, P. Bluetongue virus VP4 is an RNA-capping assembly line. Nat. Struct. Mol. Biol. 2007, 14, 449–451. [Google Scholar] [CrossRef] [PubMed]
- Malone, R.W.; Felgner, P.L.; Verma, I.M. Cationic liposome-mediated RNA transfection. Proc. Natl. Acad. Sci. USA 1989, 86, 6077–6081. [Google Scholar] [CrossRef]
- Zarghampoor, F.; Azarpira, N.; Khatami, S.R.; Behzad-Behbahani, A.; Foroughmand, A.M. Improved translation efficiency of therapeutic mRNA. Gene 2019, 707, 231–238. [Google Scholar] [CrossRef]
- Ding, Y.; Tang, Y.; Kwok, C.K.; Zhang, Y.; Bevilacqua, P.C.; Assmann, S.M. In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature 2014, 505, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Kozak, M. An analysis of 5′-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res. 1987, 15, 8125–8148. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.P.; Overton, K.W.; Wang, C.L. Tuning gene expression with synthetic upstream open reading frames. Proc. Natl. Acad. Sci. USA 2013, 110, 11284–11289. [Google Scholar] [CrossRef]
- Oliveira, C.C.; McCarthy, J.E. The relationship between eukaryotic translation and mRNA stability. A short upstream open reading frame strongly inhibits translational initiation and greatly accelerates mRNA degradation in the yeast Saccharomyces cerevisiae. J. Biol. Chem. 1995, 270, 8936–8943. [Google Scholar] [CrossRef]
- Sobczak, K.; Krzyzosiak, W.J. Structural determinants of BRCA1 translational regulation. J. Biol. Chem. 2002, 277, 17349–17358. [Google Scholar] [CrossRef]
- Jia, L.; Mao, Y.; Ji, Q.; Dersh, D.; Yewdell, J.W.; Qian, S.B. Decoding mRNA translatability and stability from the 5′ UTR. Nat. Struct. Mol. Biol. 2020, 27, 814–821. [Google Scholar] [CrossRef]
- Komar, A.A.; Hatzoglou, M. Cellular IRES-mediated translation: The war of ITAFs in pathophysiological states. Cell Cycle 2011, 10, 229–240. [Google Scholar] [CrossRef]
- Vivinus, S.; Baulande, S.; van Zanten, M.; Campbell, F.; Topley, P.; Ellis, J.H.; Dessen, P.; Coste, H. An element within the 5′ untranslated region of human Hsp70 mRNA which acts as a general enhancer of mRNA translation. Eur. J. Biochem. 2001, 268, 1908–1917. [Google Scholar] [CrossRef] [PubMed]
- Orlandini von Niessen, A.G.; Poleganov, M.A.; Rechner, C.; Plaschke, A.; Kranz, L.M.; Fesser, S.; Diken, M.; Lower, M.; Vallazza, B.; Beissert, T.; et al. Improving mRNA-Based Therapeutic Gene Delivery by Expression-Augmenting 3′ UTRs Identified by Cellular Library Screening. Mol. Ther. 2019, 27, 824–836. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Zhang, G.; Makeyev, E.V. RNA-binding protein HuR autoregulates its expression by promoting alternative polyadenylation site usage. Nucleic Acids Res. 2012, 40, 787–800. [Google Scholar] [CrossRef] [PubMed]
- Tiedje, C.; Kotlyarov, A.; Gaestel, M. Molecular mechanisms of phosphorylation-regulated TTP (tristetraprolin) action and screening for further TTP-interacting proteins. Biochem. Soc. Trans. 2010, 38, 1632–1637. [Google Scholar] [CrossRef]
- Vlasova, I.A.; Bohjanen, P.R. Posttranscriptional regulation of gene networks by GU-rich elements and CELF proteins. RNA Biol. 2008, 5, 201–207. [Google Scholar] [CrossRef]
- Chaudhuri, A.; Das, S.; Das, B. Localization elements and zip codes in the intracellular transport and localization of messenger RNAs in Saccharomyces cerevisiae. Wiley Interdiscip. Rev. RNA 2020, 11, e1591. [Google Scholar] [CrossRef]
- Castillo-Hair, S.M.; Seelig, G. Machine Learning for Designing Next-Generation mRNA Therapeutics. Acc. Chem. Res. 2022, 55, 24–34. [Google Scholar] [CrossRef]
- Leppek, K.; Byeon, G.W.; Kladwang, W.; Wayment-Steele, H.K.; Kerr, C.H.; Xu, A.F.; Kim, D.S.; Topkar, V.V.; Choe, C.; Rothschild, D.; et al. Combinatorial optimization of mRNA structure, stability, and translation for RNA-based therapeutics. Nat. Commun. 2022, 13, 1536. [Google Scholar] [CrossRef]
- Sample, P.J.; Wang, B.; Reid, D.W.; Presnyak, V.; McFadyen, I.J.; Morris, D.R.; Seelig, G. Human 5′ UTR design and variant effect prediction from a massively parallel translation assay. Nat. Biotechnol. 2019, 37, 803–809. [Google Scholar] [CrossRef]
- Cao, J.; Novoa, E.M.; Zhang, Z.; Chen, W.C.W.; Liu, D.; Choi, G.C.G.; Wong, A.S.L.; Wehrspaun, C.; Kellis, M.; Lu, T.K. High-throughput 5′ UTR engineering for enhanced protein production in non-viral gene therapies. Nat. Commun. 2021, 12, 4138. [Google Scholar] [CrossRef]
- Asrani, K.H.; Farelli, J.D.; Stahley, M.R.; Miller, R.L.; Cheng, C.J.; Subramanian, R.R.; Brown, J.M. Optimization of mRNA untranslated regions for improved expression of therapeutic mRNA. RNA Biol. 2018, 15, 756–762. [Google Scholar] [CrossRef] [PubMed]
- Schoenberg, D.R. Mechanisms of endonuclease-mediated mRNA decay. Wiley Interdiscip. Rev. RNA 2011, 2, 582–600. [Google Scholar] [CrossRef]
- Presnyak, V.; Alhusaini, N.; Chen, Y.H.; Martin, S.; Morris, N.; Kline, N.; Olson, S.; Weinberg, D.; Baker, K.E.; Graveley, B.R.; et al. Codon optimality is a major determinant of mRNA stability. Cell 2015, 160, 1111–1124. [Google Scholar] [CrossRef]
- Gustafsson, C.; Govindarajan, S.; Minshull, J. Codon bias and heterologous protein expression. Trends Biotechnol. 2004, 22, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Alexaki, A.; Kames, J.; Holcomb, D.D.; Athey, J.; Santana-Quintero, L.V.; Lam, P.V.N.; Hamasaki-Katagiri, N.; Osipova, E.; Simonyan, V.; Bar, H.; et al. Codon and Codon-Pair Usage Tables (CoCoPUTs): Facilitating Genetic Variation Analyses and Recombinant Gene Design. J. Mol. Biol. 2019, 431, 2434–2441. [Google Scholar] [CrossRef] [PubMed]
- Tats, A.; Tenson, T.; Remm, M. Preferred and avoided codon pairs in three domains of life. BMC Genom. 2008, 9, 463. [Google Scholar] [CrossRef] [PubMed]
- Tulloch, F.; Atkinson, N.J.; Evans, D.J.; Ryan, M.D.; Simmonds, P. RNA virus attenuation by codon pair deoptimisation is an artefact of increases in CpG/UpA dinucleotide frequencies. Elife 2014, 3, e04531. [Google Scholar] [CrossRef]
- Hia, F.; Yang, S.F.; Shichino, Y.; Yoshinaga, M.; Murakawa, Y.; Vandenbon, A.; Fukao, A.; Fujiwara, T.; Landthaler, M.; Natsume, T.; et al. Codon bias confers stability to human mRNAs. EMBO Rep. 2019, 20, e48220. [Google Scholar] [CrossRef]
- Vaidyanathan, S.; Azizian, K.T.; Haque, A.; Henderson, J.M.; Hendel, A.; Shore, S.; Antony, J.S.; Hogrefe, R.I.; Kormann, M.S.D.; Porteus, M.H.; et al. Uridine Depletion and Chemical Modification Increase Cas9 mRNA Activity and Reduce Immunogenicity without HPLC Purification. Mol. Ther. Nucleic Acids 2018, 12, 530–542. [Google Scholar] [CrossRef]
- Diebold, S.S.; Massacrier, C.; Akira, S.; Paturel, C.; Morel, Y.; Reis e Sousa, C. Nucleic acid agonists for Toll-like receptor 7 are defined by the presence of uridine ribonucleotides. Eur. J. Immunol. 2006, 36, 3256–3267. [Google Scholar] [CrossRef]
- Uzri, D.; Gehrke, L. Nucleotide sequences and modifications that determine RIG-I/RNA binding and signaling activities. J. Virol. 2009, 83, 4174–4184. [Google Scholar] [CrossRef]
- Saito, T.; Owen, D.M.; Jiang, F.; Marcotrigiano, J.; Gale, M., Jr. Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature 2008, 454, 523–527. [Google Scholar] [CrossRef]
- Runge, S.; Sparrer, K.M.J.; Lässig, C.; Hembach, K.; Baum, A.; García-Sastre, A.; Söding, J.; Conzelmann, K.-K.; Hopfner, K.-P. In vivo ligands of MDA5 and RIG-I in measles virus-infected cells. PLoS Pathog. 2014, 10, e1004081. [Google Scholar] [CrossRef]
- Chiang, C.; Beljanski, V.; Yin, K.; Olagnier, D.; Ben Yebdri, F.; Steel, C.; Goulet, M.-L.; DeFilippis, V.R.; Streblow, D.N.; Haddad, E.K.; et al. Sequence-Specific Modifications Enhance the Broad-Spectrum Antiviral Response Activated by RIG-I Agonists. J. Virol. 2015, 89, 8011–8025. [Google Scholar] [CrossRef] [PubMed]
- Brule, C.E.; Grayhack, E.J. Synonymous Codons: Choose Wisely for Expression. Trends Genet. 2017, 33, 283–297. [Google Scholar] [CrossRef]
- Yu, C.H.; Dang, Y.; Zhou, Z.; Wu, C.; Zhao, F.; Sachs, M.S.; Liu, Y. Codon Usage Influences the Local Rate of Translation Elongation to Regulate Co-translational Protein Folding. Mol. Cell 2015, 59, 744–754. [Google Scholar] [CrossRef] [PubMed]
- Walsh, I.M.; Bowman, M.A.; Soto Santarriaga, I.F.; Rodriguez, A.; Clark, P.L. Synonymous codon substitutions perturb cotranslational protein folding in vivo and impair cell fitness. Proc. Natl. Acad. Sci. USA 2020, 117, 3528–3534. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, L.; Lin, A.; Xu, C.; Li, Z.; Liu, K.; Liu, B.; Ma, X.; Zhao, F.; Yao, W. Lineardesign: Efficient algorithms for optimized mrna sequence design. arXiv 2020, arXiv:2004.10177. [Google Scholar]
- Vinciguerra, P.; Stutz, F. mRNA export: An assembly line from genes to nuclear pores. Curr. Opin. Cell Biol. 2004, 16, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Kahvejian, A.; Svitkin, Y.V.; Sukarieh, R.; M’Boutchou, M.-N.; Sonenberg, N. Mammalian poly(A)-binding protein is a eukaryotic translation initiation factor, which acts via multiple mechanisms. Genes Dev. 2005, 19, 104–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, N.K.; Coller, J.M.; Dickson, K.S.; Wickens, M. Multiple portions of poly(A)-binding protein stimulate translation in vivo. EMBO J. 2000, 19, 4723–4733. [Google Scholar] [CrossRef]
- Siddiqui, N.; Mangus, D.A.; Chang, T.-C.; Palermino, J.-M.; Shyu, A.-B.; Gehring, K. Poly(A) Nuclease Interacts with the C-terminal Domain of Polyadenylate-binding Protein Domain from Poly(A)-binding Protein *. J. Biol. Chem. 2007, 282, 25067–25075. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-Y.A.; Shyu, A.-B. Mechanisms of deadenylation-dependent decay. Wiley Interdiscip. Rev. RNA 2011, 2, 167–183. [Google Scholar] [CrossRef] [PubMed]
- Nanjappa, D.P.; Babu, N.; Khanna-Gupta, A.; O’Donohue, M.-F.; Sips, P.; Chakraborty, A. Poly (A)-specific ribonuclease (PARN): More than just “mRNA stock clearing”. Life Sci. 2021, 285, 119953. [Google Scholar] [CrossRef]
- Meijer, H.A.; Bushell, M.; Hill, K.; Gant, T.W.; Willis, A.E.; Jones, P.; de Moor, C.H. A novel method for poly(A) fractionation reveals a large population of mRNAs with a short poly(A) tail in mammalian cells. Nucleic Acids Res. 2007, 35, e132. [Google Scholar] [CrossRef]
- Lima, S.A.; Chipman, L.B.; Nicholson, A.L.; Chen, Y.H.; Yee, B.A.; Yeo, G.W.; Coller, J.; Pasquinelli, A.E. Short poly(A) tails are a conserved feature of highly expressed genes. Nat. Struct. Mol. Biol. 2017, 24, 1057–1063. [Google Scholar] [CrossRef]
- Munroe, D.; Jacobson, A. mRNA poly(A) tail, a 3′ enhancer of translational initiation. Mol. Cell. Biol. 1990, 10, 3441–3455. [Google Scholar] [CrossRef]
- Holtkamp, S.; Kreiter, S.; Selmi, A.; Simon, P.; Koslowski, M.; Huber, C.; Türeci, O.; Sahin, U. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood 2006, 108, 4009–4017. [Google Scholar] [CrossRef]
- Mockey, M.; Goncalves, C.; Dupuy, F.P.; Lemoine, F.M.; Pichon, C.; Midoux, P. mRNA transfection of dendritic cells: Synergistic effect of ARCA mRNA capping with Poly(A) chains in cis and in trans for a high protein expression level. Biochem. Biophys. Res. Commun. 2006, 340, 1062–1068. [Google Scholar] [CrossRef]
- Jalkanen, A.L.; Coleman, S.J.; Wilusz, J. Determinants and implications of mRNA poly(A) tail size--does this protein make my tail look big? Semin. Cell Dev. Biol. 2014, 34, 24–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strzelecka, D.; Smietanski, M.; Sikorski, P.J.; Warminski, M.; Kowalska, J.; Jemielity, J. Phosphodiester modifications in mRNA poly(A) tail prevent deadenylation without compromising protein expression. RNA 2020, 26, 1815–1837. [Google Scholar] [CrossRef]
- Sànchez, R.; Marzluff, W.F. The Stem-Loop Binding Protein Is Required for Efficient Translation of Histone mRNA In Vivo and In Vitro. Mol. Cell. Biol. 2002, 22, 7093–7104. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Slevin, M.K.; Marzluff, W.F.; Rhoads, R.E. Synthetic mRNA with Superior Properties that Mimics the Intracellular Fates of Natural Histone mRNA. Methods Mol. Biol. 2016, 1428, 93–114. [Google Scholar] [CrossRef] [PubMed]
- Thess, A.; Grund, S.; Mui, B.L.; Hope, M.J.; Baumhof, P.; Fotin-Mleczek, M.; Schlake, T. Sequence-engineered mRNA Without Chemical Nucleoside Modifications Enables an Effective Protein Therapy in Large Animals. Mol. Ther. 2015, 23, 1456–1464. [Google Scholar] [CrossRef]
- Boo, S.H.; Kim, Y.K. The emerging role of RNA modifications in the regulation of mRNA stability. Exp. Mol. Med. 2020, 52, 400–408. [Google Scholar] [CrossRef]
- Nance, K.D.; Meier, J.L. Modifications in an Emergency: The Role of N1-Methylpseudouridine in COVID-19 Vaccines. ACS Cent. Sci. 2021, 7, 748–756. [Google Scholar] [CrossRef]
- Mauger, D.M.; Cabral, B.J.; Presnyak, V.; Su, S.V.; Reid, D.W.; Goodman, B.; Link, K.; Khatwani, N.; Reynders, J.; Moore, M.J.; et al. mRNA structure regulates protein expression through changes in functional half-life. Proc. Natl. Acad. Sci. USA 2019, 116, 24075–24083. [Google Scholar] [CrossRef]
- Nelson, J.; Sorensen, E.W.; Mintri, S.; Rabideau, A.E.; Zheng, W.; Besin, G.; Khatwani, N.; Su, S.V.; Miracco, E.J.; Issa, W.J.; et al. Impact of mRNA chemistry and manufacturing process on innate immune activation. Sci. Adv. 2020, 6, eaaz6893. [Google Scholar] [CrossRef]
- Bialkowski, L.; Van der Jeught, K.; Renmans, D.; van Weijnen, A.; Heirman, C.; Keyaerts, M.; Breckpot, K.; Thielemans, K. Adjuvant-Enhanced mRNA Vaccines. Methods Mol. Biol. 2017, 1499, 179–191. [Google Scholar] [CrossRef]
- Karikó, K.; Muramatsu, H.; Ludwig, J.; Weissman, D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res. 2011, 39, e142. [Google Scholar] [CrossRef] [Green Version]
- Karikó, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA recognition by Toll-like receptors: The impact of nucleoside modification and the evolutionary origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef]
- Karikó, K.; Muramatsu, H.; Welsh, F.A.; Ludwig, J.; Kato, H.; Akira, S.; Weissman, D. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol. Ther. 2008, 16, 1833–1840. [Google Scholar] [CrossRef]
- Kormann, M.S.; Hasenpusch, G.; Aneja, M.K.; Nica, G.; Flemmer, A.W.; Herber-Jonat, S.; Huppmann, M.; Mays, L.E.; Illenyi, M.; Schams, A.; et al. Expression of therapeutic proteins after delivery of chemically modified mRNA in mice. Nat. Biotechnol. 2011, 29, 154–157. [Google Scholar] [CrossRef]
- Svitkin, Y.V.; Cheng, Y.M.; Chakraborty, T.; Presnyak, V.; John, M.; Sonenberg, N. N1-methyl-pseudouridine in mRNA enhances translation through eIF2α-dependent and independent mechanisms by increasing ribosome density. Nucleic Acids Res. 2017, 45, 6023–6036. [Google Scholar] [CrossRef]
- Mays, L.E.; Ammon-Treiber, S.; Mothes, B.; Alkhaled, M.; Rottenberger, J.; Müller-Hermelink, E.S.; Grimm, M.; Mezger, M.; Beer-Hammer, S.; von Stebut, E.; et al. Modified Foxp3 mRNA protects against asthma through an IL-10-dependent mechanism. J. Clin. Investig. 2013, 123, 1216–1228. [Google Scholar] [CrossRef]
- Arango, D.; Sturgill, D.; Alhusaini, N.; Dillman, A.A.; Sweet, T.J.; Hanson, G.; Hosogane, M.; Sinclair, W.R.; Nanan, K.K.; Mandler, M.D.; et al. Acetylation of Cytidine in mRNA Promotes Translation Efficiency. Cell 2018, 175, 1872–1886.e24. [Google Scholar] [CrossRef]
- Hou, X.; Zaks, T.; Langer, R.; Dong, Y. Lipid nanoparticles for mRNA delivery. Nat. Rev. Mater. 2021, 6, 1078–1094. [Google Scholar] [CrossRef]
- Phua, K.K.; Leong, K.W.; Nair, S.K. Transfection efficiency and transgene expression kinetics of mRNA delivered in naked and nanoparticle format. J. Control. Release 2013, 166, 227–233. [Google Scholar] [CrossRef]
- Diken, M.; Kreiter, S.; Selmi, A.; Britten, C.M.; Huber, C.; Türeci, Ö.; Sahin, U. Selective uptake of naked vaccine RNA by dendritic cells is driven by macropinocytosis and abrogated upon DC maturation. Gene Ther. 2011, 18, 702–708. [Google Scholar] [CrossRef]
- Selmi, A.; Vascotto, F.; Kautz-Neu, K.; Türeci, Ö.; Sahin, U.; von Stebut, E.; Diken, M.; Kreiter, S. Uptake of synthetic naked RNA by skin-resident dendritic cells via macropinocytosis allows antigen expression and induction of T-cell responses in mice. Cancer Immunol. Immunother. 2016, 65, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D.K.; Jasny, E.; Yoon, H.; Horscroft, N.; Schanen, B.; Geter, T.; Fotin-Mleczek, M.; Petsch, B.; Wittman, V. Adjuvant effects of a sequence-engineered mRNA vaccine: Translational profiling demonstrates similar human and murine innate response. J. Transl. Med. 2017, 15, 1. [Google Scholar] [CrossRef] [PubMed]
- Carralot, J.P.; Probst, J.; Hoerr, I.; Scheel, B.; Teufel, R.; Jung, G.; Rammensee, H.G.; Pascolo, S. Polarization of immunity induced by direct injection of naked sequence-stabilized mRNA vaccines. Cell. Mol. Life Sci. 2004, 61, 2418–2424. [Google Scholar] [CrossRef] [PubMed]
- Joe, P.T.; Christopoulou, I.; van Hoecke, L.; Schepens, B.; Ysenbaert, T.; Heirman, C.; Thielemans, K.; Saelens, X.; Aerts, J.L. Intranodal administration of mRNA encoding nucleoprotein provides cross-strain immunity against influenza in mice. J. Transl. Med. 2019, 17, 242. [Google Scholar] [CrossRef] [PubMed]
- Kreiter, S.; Selmi, A.; Diken, M.; Koslowski, M.; Britten, C.M.; Huber, C.; Türeci, O.; Sahin, U. Intranodal vaccination with naked antigen-encoding RNA elicits potent prophylactic and therapeutic antitumoral immunity. Cancer Res. 2010, 70, 9031–9040. [Google Scholar] [CrossRef] [PubMed]
- Kreiter, S.; Diken, M.; Selmi, A.; Diekmann, J.; Attig, S.; Hüsemann, Y.; Koslowski, M.; Huber, C.; Türeci, Ö.; Sahin, U. FLT3 ligand enhances the cancer therapeutic potency of naked RNA vaccines. Cancer Res. 2011, 71, 6132–6142. [Google Scholar] [CrossRef]
- Petsch, B.; Schnee, M.; Vogel, A.B.; Lange, E.; Hoffmann, B.; Voss, D.; Schlake, T.; Thess, A.; Kallen, K.J.; Stitz, L.; et al. Protective efficacy of in vitro synthesized, specific mRNA vaccines against influenza A virus infection. Nat. Biotechnol. 2012, 30, 1210–1216. [Google Scholar] [CrossRef]
- Van Tendeloo, V.F.; Ponsaerts, P.; Lardon, F.; Nijs, G.; Lenjou, M.; Van Broeckhoven, C.; Van Bockstaele, D.R.; Berneman, Z.N. Highly efficient gene delivery by mRNA electroporation in human hematopoietic cells: Superiority to lipofection and passive pulsing of mRNA and to electroporation of plasmid cDNA for tumor antigen loading of dendritic cells. Blood 2001, 98, 49–56. [Google Scholar] [CrossRef]
- Morgan, R.A.; Yang, J.C.; Kitano, M.; Dudley, M.E.; Laurencot, C.M.; Rosenberg, S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010, 18, 843–851. [Google Scholar] [CrossRef]
- Ang, W.X.; Ng, Y.Y.; Xiao, L.; Chen, C.; Li, Z.; Chi, Z.; Tay, J.C.-K.; Tan, W.K.; Zeng, J.; Toh, H.C.; et al. Electroporation of NKG2D RNA CAR Improves Vγ9Vδ2 T Cell Responses against Human Solid Tumor Xenografts. Mol. Ther.-Oncolytics 2020, 17, 421–430. [Google Scholar] [CrossRef]
- Cu, Y.; Broderick, K.E.; Banerjee, K.; Hickman, J.; Otten, G.; Barnett, S.; Kichaev, G.; Sardesai, N.Y.; Ulmer, J.B.; Geall, A. Enhanced Delivery and Potency of Self-Amplifying mRNA Vaccines by Electroporation in Situ. Vaccines 2013, 1, 367–383. [Google Scholar] [CrossRef] [Green Version]
- Johansson, D.X.; Ljungberg, K.; Kakoulidou, M.; Liljeström, P. Intradermal electroporation of naked replicon RNA elicits strong immune responses. PLoS ONE 2012, 7, e29732. [Google Scholar] [CrossRef]
- Stamatatos, L.; Leventis, R.; Zuckermann, M.J.; Silvius, J.R. Interactions of cationic lipid vesicles with negatively charged phospholipid vesicles and biological membranes. Biochemistry 1988, 27, 3917–3925. [Google Scholar] [CrossRef] [PubMed]
- Felgner, P.L.; Gadek, T.R.; Holm, M.; Roman, R.; Chan, H.W.; Wenz, M.; Northrop, J.P.; Ringold, G.M.; Danielsen, M. Lipofection: A highly efficient, lipid-mediated DNA-transfection procedure. Proc. Natl. Acad. Sci. USA 1987, 84, 7413–7417. [Google Scholar] [CrossRef]
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Krienke, C.; Kolb, L.; Diken, E.; Streuber, M.; Kirchhoff, S.; Bukur, T.; Akilli-Öztürk, Ö.; Kranz, L.M.; Berger, H.; Petschenka, J.; et al. A noninflammatory mRNA vaccine for treatment of experimental autoimmune encephalomyelitis. Science 2021, 371, 145–153. [Google Scholar] [CrossRef]
- Sahin, U.; Oehm, P.; Derhovanessian, E.; Jabulowsky, R.A.; Vormehr, M.; Gold, M.; Maurus, D.; Schwarck-Kokarakis, D.; Kuhn, A.N.; Omokoko, T.; et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature 2020, 585, 107–112. [Google Scholar] [CrossRef]
- Qureischi, M.; Mohr, J.; Arellano-Viera, E.; Knudsen, S.E.; Vohidov, F.; Garitano-Trojaola, A. Chapter One—mRNA-based therapies: Preclinical and clinical applications. Int. Rev. Cell Mol. Biol. 2022, 372, 1–54. [Google Scholar] [PubMed]
- Leung, A.K.; Tam, Y.Y.; Chen, S.; Hafez, I.M.; Cullis, P.R. Microfluidic Mixing: A General Method for Encapsulating Macromolecules in Lipid Nanoparticle Systems. J. Phys. Chem. B 2015, 119, 8698–8706. [Google Scholar] [CrossRef]
- Jahn, A.; Stavis, S.M.; Hong, J.S.; Vreeland, W.N.; DeVoe, D.L.; Gaitan, M. Microfluidic mixing and the formation of nanoscale lipid vesicles. ACS Nano 2010, 4, 2077–2087. [Google Scholar] [CrossRef] [PubMed]
- Granot, Y.; Peer, D. Delivering the right message: Challenges and opportunities in lipid nanoparticles-mediated modified mRNA therapeutics-An innate immune system standpoint. Semin. Immunol. 2017, 34, 68–77. [Google Scholar] [CrossRef]
- Huotari, J.; Helenius, A. Endosome maturation. EMBO J. 2011, 30, 3481–3500. [Google Scholar] [CrossRef]
- Walsh, C.L.; Nguyen, J.; Tiffany, M.R.; Szoka, F.C. Synthesis, characterization, and evaluation of ionizable lysine-based lipids for siRNA delivery. Bioconjug. Chem. 2013, 24, 36–43. [Google Scholar] [CrossRef]
- Heyes, J.; Palmer, L.; Bremner, K.; MacLachlan, I. Cationic lipid saturation influences intracellular delivery of encapsulated nucleic acids. J. Control. Release 2005, 107, 276–287. [Google Scholar] [CrossRef]
- Fenton, O.S.; Kauffman, K.J.; McClellan, R.L.; Appel, E.A.; Dorkin, J.R.; Tibbitt, M.W.; Heartlein, M.W.; DeRosa, F.; Langer, R.; Anderson, D.G. Bioinspired Alkenyl Amino Alcohol Ionizable Lipid Materials for Highly Potent In Vivo mRNA Delivery. Adv. Mater. 2016, 28, 2939–2943. [Google Scholar] [CrossRef]
- Miao, L.; Lin, J.; Huang, Y.; Li, L.; Delcassian, D.; Ge, Y.; Shi, Y.; Anderson, D.G. Synergistic lipid compositions for albumin receptor mediated delivery of mRNA to the liver. Nat. Commun. 2020, 11, 2424. [Google Scholar] [CrossRef]
- Sabnis, S.; Kumarasinghe, E.S.; Salerno, T.; Mihai, C.; Ketova, T.; Senn, J.J.; Lynn, A.; Bulychev, A.; McFadyen, I.; Chan, J.; et al. A Novel Amino Lipid Series for mRNA Delivery: Improved Endosomal Escape and Sustained Pharmacology and Safety in Non-human Primates. Mol. Ther. 2018, 26, 1509–1519. [Google Scholar] [CrossRef] [PubMed]
- Hassett, K.J.; Benenato, K.E.; Jacquinet, E.; Lee, A.; Woods, A.; Yuzhakov, O.; Himansu, S.; Deterling, J.; Geilich, B.M.; Ketova, T.; et al. Optimization of Lipid Nanoparticles for Intramuscular Administration of mRNA Vaccines. Mol. Ther. Nucleic Acids 2019, 15, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, K.J.; Dorkin, J.R.; Yang, J.H.; Heartlein, M.W.; DeRosa, F.; Mir, F.F.; Fenton, O.S.; Anderson, D.G. Optimization of Lipid Nanoparticle Formulations for mRNA Delivery in Vivo with Fractional Factorial and Definitive Screening Designs. Nano Lett. 2015, 15, 7300–7306. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, W.; Nguyen, G.N.; Zhang, C.; Zeng, C.; Yan, J.; Du, S.; Hou, X.; Li, W.; Jiang, J.; et al. Functionalized lipid-like nanoparticles for in vivo mRNA delivery and base editing. Sci. Adv. 2020, 6, eabc2315. [Google Scholar] [CrossRef]
- Semple, S.C.; Akinc, A.; Chen, J.; Sandhu, A.P.; Mui, B.L.; Cho, C.K.; Sah, D.W.; Stebbing, D.; Crosley, E.J.; Yaworski, E.; et al. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 2010, 28, 172–176. [Google Scholar] [CrossRef]
- Akinc, A.; Maier, M.A.; Manoharan, M.; Fitzgerald, K.; Jayaraman, M.; Barros, S.; Ansell, S.; Du, X.; Hope, M.J.; Madden, T.D.; et al. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. NanoTechnol. 2019, 14, 1084–1087. [Google Scholar] [CrossRef]
- Veiga, N.; Goldsmith, M.; Granot, Y.; Rosenblum, D.; Dammes, N.; Kedmi, R.; Ramishetti, S.; Peer, D. Cell specific delivery of modified mRNA expressing therapeutic proteins to leukocytes. Nat. Commun. 2018, 9, 4493. [Google Scholar] [CrossRef]
- Patel, S.; Ryals, R.C.; Weller, K.K.; Pennesi, M.E.; Sahay, G. Lipid nanoparticles for delivery of messenger RNA to the back of the eye. J. Control. Release 2019, 303, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, S.; Tonnu, N.; Tachikawa, K.; Limphong, P.; Vega, J.B.; Karmali, P.P.; Chivukula, P.; Verma, I.M. Systemic delivery of factor IX messenger RNA for protein replacement therapy. Proc. Natl. Acad. Sci. USA 2017, 114, E1941–E1950. [Google Scholar] [CrossRef]
- Schoenmaker, L.; Witzigmann, D.; Kulkarni, J.A.; Verbeke, R.; Kersten, G.; Jiskoot, W.; Crommelin, D.J.A. mRNA-lipid nanoparticle COVID-19 vaccines: Structure and stability. Int. J. Pharm. 2021, 601, 120586. [Google Scholar] [CrossRef] [PubMed]
- Finn, J.D.; Smith, A.R.; Patel, M.C.; Shaw, L.; Youniss, M.R.; van Heteren, J.; Dirstine, T.; Ciullo, C.; Lescarbeau, R.; Seitzer, J.; et al. A Single Administration of CRISPR/Cas9 Lipid Nanoparticles Achieves Robust and Persistent In Vivo Genome Editing. Cell Rep. 2018, 22, 2227–2235. [Google Scholar] [CrossRef]
- Han, X.; Zhang, H.; Butowska, K.; Swingle, K.L.; Alameh, M.G.; Weissman, D.; Mitchell, M.J. An ionizable lipid toolbox for RNA delivery. Nat. Commun. 2021, 12, 7233. [Google Scholar] [CrossRef] [PubMed]
- Witzigmann, D.; Kulkarni, J.A.; Leung, J.; Chen, S.; Cullis, P.R.; van der Meel, R. Lipid nanoparticle technology for therapeutic gene regulation in the liver. Adv. Drug Deliv. Rev. 2020, 159, 344–363. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, J.A.; Witzigmann, D.; Leung, J.; Tam, Y.Y.C.; Cullis, P.R. On the role of helper lipids in lipid nanoparticle formulations of siRNA. Nanoscale 2019, 11, 21733–21739. [Google Scholar] [CrossRef]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA COVID-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Lee, R.J. The role of helper lipids in lipid nanoparticles (LNPs) designed for oligonucleotide delivery. Adv. Drug Deliv. Rev. 2016, 99, 129–137. [Google Scholar] [CrossRef]
- Miao, L.; Zhang, Y.; Huang, L. mRNA vaccine for cancer immunotherapy. Mol. Cancer 2021, 20, 41. [Google Scholar] [CrossRef]
- Ambegia, E.; Ansell, S.; Cullis, P.; Heyes, J.; Palmer, L.; MacLachlan, I. Stabilized plasmid-lipid particles containing PEG-diacylglycerols exhibit extended circulation lifetimes and tumor selective gene expression. Biochim. Biophys. Acta 2005, 1669, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Tao, W.; Liu, D.; Wu, J.; Guo, Z.; Ji, X.; Bharwani, Z.; Zhao, L.; Zhao, X.; Farokhzad, O.C.; et al. Surface De-PEGylation Controls Nanoparticle-Mediated siRNA Delivery In Vitro and In Vivo. Theranostics 2017, 7, 1990–2002. [Google Scholar] [CrossRef]
- Heyes, J.; Hall, K.; Tailor, V.; Lenz, R.; MacLachlan, I. Synthesis and characterization of novel poly(ethylene glycol)-lipid conjugates suitable for use in drug delivery. J. Control. Release 2006, 112, 280–290. [Google Scholar] [CrossRef]
- Cheng, Q.; Wei, T.; Farbiak, L.; Johnson, L.T.; Dilliard, S.A.; Siegwart, D.J. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR-Cas gene editing. Nat. Nanotechnol. 2020, 15, 313–320. [Google Scholar] [CrossRef]
- Chen, J.; Ye, Z.; Huang, C.; Qiu, M.; Song, D.; Li, Y.; Xu, Q. Lipid nanoparticle-mediated lymph node-targeting delivery of mRNA cancer vaccine elicits robust CD8(+) T cell response. Proc. Natl. Acad. Sci. USA 2022, 119, e2207841119. [Google Scholar] [CrossRef]
- Dilliard, S.A.; Cheng, Q.; Siegwart, D.J. On the mechanism of tissue-specific mRNA delivery by selective organ targeting nanoparticles. Proc. Natl. Acad. Sci. USA 2021, 118, e2109256118. [Google Scholar] [CrossRef]
- Liu, S.; Cheng, Q.; Wei, T.; Yu, X.; Johnson, L.T.; Farbiak, L.; Siegwart, D.J. Membrane-destabilizing ionizable phospholipids for organ-selective mRNA delivery and CRISPR-Cas gene editing. Nat. Mater. 2021, 20, 701–710. [Google Scholar] [CrossRef]
- Parhiz, H.; Shuvaev, V.V.; Pardi, N.; Khoshnejad, M.; Kiseleva, R.Y.; Brenner, J.S.; Uhler, T.; Tuyishime, S.; Mui, B.L.; Tam, Y.K.; et al. PECAM-1 directed re-targeting of exogenous mRNA providing two orders of magnitude enhancement of vascular delivery and expression in lungs independent of apolipoprotein E-mediated uptake. J. Control. Release 2018, 291, 106–115. [Google Scholar] [CrossRef]
- Tombácz, I.; Laczkó, D.; Shahnawaz, H.; Muramatsu, H.; Natesan, A.; Yadegari, A.; Papp, T.E.; Alameh, M.G.; Shuvaev, V.; Mui, B.L.; et al. Highly efficient CD4+ T cell targeting and genetic recombination using engineered CD4+ cell-homing mRNA-LNPs. Mol. Ther. 2021, 29, 3293–3304. [Google Scholar] [CrossRef] [PubMed]
- Rurik, J.G.; Tombácz, I.; Yadegari, A.; Méndez Fernández, P.O.; Shewale, S.V.; Li, L.; Kimura, T.; Soliman, O.Y.; Papp, T.E.; Tam, Y.K.; et al. CAR T cells produced in vivo to treat cardiac injury. Science 2022, 375, 91–96. [Google Scholar] [CrossRef]
- Kong, N.; Tao, W.; Ling, X.; Wang, J.; Xiao, Y.; Shi, S.; Ji, X.; Shajii, A.; Gan, S.T.; Kim, N.Y.; et al. Synthetic mRNA nanoparticle-mediated restoration of p53 tumor suppressor sensitizes p53-deficient cancers to mTOR inhibition. Sci. Transl. Med. 2019, 11, eaaw1565. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhao, L.; Xu, X.; Bertrand, N.; Choi, W.I.; Yameen, B.; Shi, J.; Shah, V.; Mulvale, M.; MacLean, J.L.; et al. Hydrophobic Cysteine Poly(disulfide)-based Redox-Hypersensitive Nanoparticle Platform for Cancer Theranostics. Angew. Chem. Int. Ed. Engl. 2015, 54, 9218–9223. [Google Scholar] [CrossRef] [PubMed]
- Gilleron, J.; Querbes, W.; Zeigerer, A.; Borodovsky, A.; Marsico, G.; Schubert, U.; Manygoats, K.; Seifert, S.; Andree, C.; Stöter, M.; et al. Image-based analysis of lipid nanoparticle-mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat. Biotechnol. 2013, 31, 638–646. [Google Scholar] [CrossRef]
- Rosenblum, D.; Gutkin, A.; Kedmi, R.; Ramishetti, S.; Veiga, N.; Jacobi, A.M.; Schubert, M.S.; Friedmann-Morvinski, D.; Cohen, Z.R.; Behlke, M.A.; et al. CRISPR-Cas9 genome editing using targeted lipid nanoparticles for cancer therapy. Sci. Adv. 2020, 6, eabc9450. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Shen, R.; Vuong, I.; Reynolds, R.A.; Shears, M.J.; Yao, Z.C.; Hu, Y.; Cho, W.J.; Kong, J.; Reddy, S.K.; et al. Multi-step screening of DNA/lipid nanoparticles and co-delivery with siRNA to enhance and prolong gene expression. Nat. Commun. 2022, 13, 4282. [Google Scholar] [CrossRef]
- Zhao, P.; Hou, X.; Yan, J.; Du, S.; Xue, Y.; Li, W.; Xiang, G.; Dong, Y. Long-term storage of lipid-like nanoparticles for mRNA delivery. Bioact. Mater. 2020, 5, 358–363. [Google Scholar] [CrossRef]
- Li, S.; Hu, Y.; Li, A.; Lin, J.; Hsieh, K.; Schneiderman, Z.; Zhang, P.; Zhu, Y.; Qiu, C.; Kokkoli, E.; et al. Payload distribution and capacity of mRNA lipid nanoparticles. Nat. Commun. 2022, 13, 5561. [Google Scholar] [CrossRef]
- Patel, A.K.; Kaczmarek, J.C.; Bose, S.; Kauffman, K.J.; Mir, F.; Heartlein, M.W.; DeRosa, F.; Langer, R.; Anderson, D.G. Inhaled Nanoformulated mRNA Polyplexes for Protein Production in Lung Epithelium. Adv. Mater. 2019, 31, e1805116. [Google Scholar] [CrossRef]
- Kaczmarek, J.C.; Patel, A.K.; Kauffman, K.J.; Fenton, O.S.; Webber, M.J.; Heartlein, M.W.; DeRosa, F.; Anderson, D.G. Polymer-Lipid Nanoparticles for Systemic Delivery of mRNA to the Lungs. Angew. Chem. Int. Ed. Engl. 2016, 55, 13808–13812. [Google Scholar] [CrossRef]
- Rejman, J.; Tavernier, G.; Bavarsad, N.; Demeester, J.; De Smedt, S.C. mRNA transfection of cervical carcinoma and mesenchymal stem cells mediated by cationic carriers. J. Control. Release 2010, 147, 385–391. [Google Scholar] [CrossRef]
- Kim, Y.H.; Park, J.H.; Lee, M.; Kim, Y.H.; Park, T.G.; Kim, S.W. Polyethylenimine with acid-labile linkages as a biodegradable gene carrier. J. Control. Release 2005, 103, 209–219. [Google Scholar] [CrossRef]
- Lv, H.; Zhang, S.; Wang, B.; Cui, S.; Yan, J. Toxicity of cationic lipids and cationic polymers in gene delivery. J. Control. Release 2006, 114, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Piotrowski-Daspit, A.S.; Kauffman, A.C.; Bracaglia, L.G.; Saltzman, W.M. Polymeric vehicles for nucleic acid delivery. Adv. Drug Deliv. Rev. 2020, 156, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Kamaly, N.; Yameen, B.; Wu, J.; Farokhzad, O.C. Degradable Controlled-Release Polymers and Polymeric Nanoparticles: Mechanisms of Controlling Drug Release. Chem. Rev. 2016, 116, 2602–2663. [Google Scholar] [CrossRef]
- Islam, M.A.; Xu, Y.; Tao, W.; Ubellacker, J.M.; Lim, M.; Aum, D.; Lee, G.Y.; Zhou, K.; Zope, H.; Yu, M.; et al. Restoration of tumour-growth suppression in vivo via systemic nanoparticle-mediated delivery of PTEN mRNA. Nat. Biomed. Eng. 2018, 2, 850–864. [Google Scholar] [CrossRef] [PubMed]
- Parayath, N.N.; Stephan, S.B.; Koehne, A.L.; Nelson, P.S.; Stephan, M.T. In vitro-transcribed antigen receptor mRNA nanocarriers for transient expression in circulating T cells in vivo. Nat. Commun. 2020, 11, 6080. [Google Scholar] [CrossRef]
- Teixeira, H.F.; Bruxel, F.; Fraga, M.; Schuh, R.S.; Zorzi, G.K.; Matte, U.; Fattal, E. Cationic nanoemulsions as nucleic acids delivery systems. Int. J. Pharm. 2017, 534, 356–367. [Google Scholar] [CrossRef]
- Blakney, A.K.; Ip, S.; Geall, A.J. An Update on Self-Amplifying mRNA Vaccine Development. Vaccines 2021, 9, 97. [Google Scholar] [CrossRef]
- Udhayakumar, V.K.; De Beuckelaer, A.; McCaffrey, J.; McCrudden, C.M.; Kirschman, J.L.; Vanover, D.; Van Hoecke, L.; Roose, K.; Deswarte, K.; De Geest, B.G.; et al. Arginine-Rich Peptide-Based mRNA Nanocomplexes Efficiently Instigate Cytotoxic T Cell Immunity Dependent on the Amphipathic Organization of the Peptide. Adv. Healthc. Mater. 2017, 6, 1601412. [Google Scholar] [CrossRef]
- Hoerr, I.; Obst, R.; Rammensee, H.G.; Jung, G. In vivo application of RNA leads to induction of specific cytotoxic T lymphocytes and antibodies. Eur. J. Immunol. 2000, 30, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Scheel, B.; Teufel, R.; Probst, J.; Carralot, J.P.; Geginat, J.; Radsak, M.; Jarrossay, D.; Wagner, H.; Jung, G.; Rammensee, H.G.; et al. Toll-like receptor-dependent activation of several human blood cell types by protamine-condensed mRNA. Eur. J. Immunol. 2005, 35, 1557–1566. [Google Scholar] [CrossRef] [PubMed]
- Scheel, B.; Braedel, S.; Probst, J.; Carralot, J.P.; Wagner, H.; Schild, H.; Jung, G.; Rammensee, H.G.; Pascolo, S. Immunostimulating capacities of stabilized RNA molecules. Eur. J. Immunol. 2004, 34, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Fotin-Mleczek, M.; Duchardt, K.M.; Lorenz, C.; Pfeiffer, R.; Ojkić-Zrna, S.; Probst, J.; Kallen, K.J. Messenger RNA-based vaccines with dual activity induce balanced TLR-7 dependent adaptive immune responses and provide antitumor activity. J. Immunother. 2011, 34, 1–15. [Google Scholar] [CrossRef]
- Weide, B.; Pascolo, S.; Scheel, B.; Derhovanessian, E.; Pflugfelder, A.; Eigentler, T.K.; Pawelec, G.; Hoerr, I.; Rammensee, H.G.; Garbe, C. Direct injection of protamine-protected mRNA: Results of a phase 1/2 vaccination trial in metastatic melanoma patients. J. Immunother. 2009, 32, 498–507. [Google Scholar] [CrossRef]
- Sebastian, M.; Papachristofilou, A.; Weiss, C.; Früh, M.; Cathomas, R.; Hilbe, W.; Wehler, T.; Rippin, G.; Koch, S.D.; Scheel, B.; et al. Phase Ib study evaluating a self-adjuvanted mRNA cancer vaccine (RNActive®) combined with local radiation as consolidation and maintenance treatment for patients with stage IV non-small cell lung cancer. BMC Cancer 2014, 14, 748. [Google Scholar] [CrossRef]
- Fu, C.; Jiang, A. Dendritic Cells and CD8 T Cell Immunity in Tumor Microenvironment. Front. Immunol. 2018, 9, 3059. [Google Scholar] [CrossRef]
- Garrido, F.; Aptsiauri, N. Cancer immune escape: MHC expression in primary tumours versus metastases. Immunology 2019, 158, 255–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhang, Z.; Luo, J.; Han, X.; Wei, Y.; Wei, X. mRNA vaccine: A potential therapeutic strategy. Mol. Cancer 2021, 20, 33. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Johnson, B.A., 3rd; Lutz, E.R.; Laheru, D.A.; Jaffee, E.M. Targeting neoantigens to augment antitumour immunity. Nat. Rev. Cancer 2017, 17, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Zhang, X.; Qi, Y.; Zhang, Q.; Liu, W. Application of mass spectrometry-based MHC immunopeptidome profiling in neoantigen identification for tumor immunotherapy. Biomed. Pharm. 2019, 120, 109542. [Google Scholar] [CrossRef]
- Lennerz, V.; Fatho, M.; Gentilini, C.; Frye, R.A.; Lifke, A.; Ferel, D.; Wölfel, C.; Huber, C.; Wölfel, T. The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc. Natl. Acad. Sci. USA 2005, 102, 16013–16018. [Google Scholar] [CrossRef]
- Esprit, A.; de Mey, W.; Bahadur Shahi, R.; Thielemans, K.; Franceschini, L.; Breckpot, K. Neo-Antigen mRNA Vaccines. Vaccines 2020, 8, 776. [Google Scholar] [CrossRef]
- Pritchard, A.L.; Burel, J.G.; Neller, M.A.; Hayward, N.K.; Lopez, J.A.; Fatho, M.; Lennerz, V.; Wölfel, T.; Schmidt, C.W. Exome Sequencing to Predict Neoantigens in Melanoma. Cancer Immunol. Res. 2015, 3, 992–998. [Google Scholar] [CrossRef]
- Chang, T.C.; Carter, R.A.; Li, Y.; Li, Y.; Wang, H.; Edmonson, M.N.; Chen, X.; Arnold, P.; Geiger, T.L.; Wu, G.; et al. The neoepitope landscape in pediatric cancers. Genome Med. 2017, 9, 78. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Scheper, W.; Kvistborg, P. Cancer Neoantigens. Annu. Rev. Immunol. 2019, 37, 173–200. [Google Scholar] [CrossRef]
- Rock, K.L.; Goldberg, A.L. Degradation of cell proteins and the generation of MHC class I-presented peptides. Annu. Rev. Immunol. 1999, 17, 739–779. [Google Scholar] [CrossRef]
- Vigneron, N.; Van den Eynde, B.J. Insights into the processing of MHC class I ligands gained from the study of human tumor epitopes. Cell Mol. Life Sci. 2011, 68, 1503–1520. [Google Scholar] [CrossRef]
- Raghavan, M.; Del Cid, N.; Rizvi, S.M.; Peters, L.R. MHC class I assembly: Out and about. Trends Immunol. 2008, 29, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Strønen, E.; Toebes, M.; Kelderman, S.; van Buuren, M.M.; Yang, W.; van Rooij, N.; Donia, M.; Böschen, M.-L.; Lund-Johansen, F.; Olweus, J.; et al. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science 2016, 352, 1337–1341. [Google Scholar] [CrossRef]
- Zhao, W.; Wu, J.; Chen, S.; Zhou, Z. Shared neoantigens: Ideal targets for off-the-shelf cancer immunotherapy. Pharmacogenomics 2020, 21, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Bear, A.S.; Blanchard, T.; Cesare, J.; Ford, M.J.; Richman, L.P.; Xu, C.; Baroja, M.L.; McCuaig, S.; Costeas, C.; Gabunia, K.; et al. Biochemical and functional characterization of mutant KRAS epitopes validates this oncoprotein for immunological targeting. Nat. Commun. 2021, 12, 4365. [Google Scholar] [CrossRef]
- Smalley Rumfield, C.; Roller, N.; Pellom, S.T.; Schlom, J.; Jochems, C. Therapeutic Vaccines for HPV-Associated Malignancies. Immunotargets Ther. 2020, 9, 167–200. [Google Scholar] [CrossRef] [PubMed]
- Grunwitz, C.; Salomon, N.; Vascotto, F.; Selmi, A.; Bukur, T.; Diken, M.; Kreiter, S.; Türeci, Ö.; Sahin, U. HPV16 RNA-LPX vaccine mediates complete regression of aggressively growing HPV-positive mouse tumors and establishes protective T cell memory. Oncoimmunology 2019, 8, e1629259. [Google Scholar] [CrossRef]
- Wells, D.K.; van Buuren, M.M.; Dang, K.K.; Hubbard-Lucey, V.M.; Sheehan, K.C.F.; Campbell, K.M.; Lamb, A.; Ward, J.P.; Sidney, J.; Blazquez, A.B.; et al. Key Parameters of Tumor Epitope Immunogenicity Revealed Through a Consortium Approach Improve Neoantigen Prediction. Cell 2020, 183, 818–834.e13. [Google Scholar] [CrossRef]
- Stone, J.D.; Chervin, A.S.; Kranz, D.M. T-cell receptor binding affinities and kinetics: Impact on T-cell activity and specificity. Immunology 2009, 126, 165–176. [Google Scholar] [CrossRef]
- Yadav, M.; Jhunjhunwala, S.; Phung, Q.T.; Lupardus, P.; Tanguay, J.; Bumbaca, S.; Franci, C.; Cheung, T.K.; Fritsche, J.; Weinschenk, T.; et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 2014, 515, 572–576. [Google Scholar] [CrossRef] [PubMed]
- Vita, R.; Overton, J.A.; Greenbaum, J.A.; Ponomarenko, J.; Clark, J.D.; Cantrell, J.R.; Wheeler, D.K.; Gabbard, J.L.; Hix, D.; Sette, A.; et al. The immune epitope database (IEDB) 3.0. Nucleic Acids Res. 2015, 43, D405–D412. [Google Scholar] [CrossRef] [PubMed]
- Shao, W.; Pedrioli, P.G.A.; Wolski, W.; Scurtescu, C.; Schmid, E.; Vizcaíno, J.A.; Courcelles, M.; Schuster, H.; Kowalewski, D.; Marino, F.; et al. The SysteMHC Atlas project. Nucleic Acids Res. 2018, 46, D1237–D1247. [Google Scholar] [CrossRef]
- Vizcaíno, J.A.; Csordas, A.; del-Toro, N.; Dianes, J.A.; Griss, J.; Lavidas, I.; Mayer, G.; Perez-Riverol, Y.; Reisinger, F.; Ternent, T.; et al. 2016 update of the PRIDE database and its related tools. Nucleic Acids Res. 2016, 44, D447–D456. [Google Scholar] [CrossRef] [PubMed]
- Lundegaard, C.; Lamberth, K.; Harndahl, M.; Buus, S.; Lund, O.; Nielsen, M. NetMHC-3.0: Accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8-11. Nucleic Acids Res. 2008, 36, W509–W512. [Google Scholar] [CrossRef]
- Fotakis, G.; Trajanoski, Z.; Rieder, D. Computational cancer neoantigen prediction: Current status and recent advances. Immunooncol. Technol. 2021, 12, 100052. [Google Scholar] [CrossRef]
- Hoof, I.; Peters, B.; Sidney, J.; Pedersen, L.E.; Sette, A.; Lund, O.; Buus, S.; Nielsen, M. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics 2009, 61, 1–13. [Google Scholar] [CrossRef]
- Richman, L.P.; Vonderheide, R.H.; Rech, A.J. Neoantigen Dissimilarity to the Self-Proteome Predicts Immunogenicity and Response to Immune Checkpoint Blockade. Cell Syst. 2019, 9, 375–382.e4. [Google Scholar] [CrossRef]
- Hundal, J.; Carreno, B.M.; Petti, A.A.; Linette, G.P.; Griffith, O.L.; Mardis, E.R.; Griffith, M. pVAC-Seq: A genome-guided in silico approach to identifying tumor neoantigens. Genome Med. 2016, 8, 11. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef]
- Sarkizova, S.; Klaeger, S.; Le, P.M.; Li, L.W.; Oliveira, G.; Keshishian, H.; Hartigan, C.R.; Zhang, W.; Braun, D.A.; Ligon, K.L.; et al. A large peptidome dataset improves HLA class I epitope prediction across most of the human population. Nat. Biotechnol. 2020, 38, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Yossef, R.; Tran, E.; Deniger, D.C.; Gros, A.; Pasetto, A.; Parkhurst, M.R.; Gartner, J.J.; Prickett, T.D.; Cafri, G.; Robbins, P.F.; et al. Enhanced detection of neoantigen-reactive T cells targeting unique and shared oncogenes for personalized cancer immunotherapy. JCI Insight 2018, 3, e122467. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.; Ahmadzadeh, M.; Lu, Y.C.; Gros, A.; Turcotte, S.; Robbins, P.F.; Gartner, J.J.; Zheng, Z.; Li, Y.F.; Ray, S.; et al. Immunogenicity of somatic mutations in human gastrointestinal cancers. Science 2015, 350, 1387–1390. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Zou, Z.; Du, J.; Su, S.; Shao, J.; Meng, F.; Yang, J.; Xu, Q.; Ding, N.; Yang, Y.; et al. Neoantigen identification strategies enable personalized immunotherapy in refractory solid tumors. J. Clin. Investig. 2019, 129, 2056–2070. [Google Scholar] [CrossRef] [PubMed]
- Kreiter, S.; Selmi, A.; Diken, M.; Sebastian, M.; Osterloh, P.; Schild, H.; Huber, C.; Türeci, O.; Sahin, U. Increased antigen presentation efficiency by coupling antigens to MHC class I trafficking signals. J. Immunol. 2008, 180, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zaro, J.L.; Shen, W.C. Fusion protein linkers: Property, design and functionality. Adv. Drug Deliv. Rev. 2013, 65, 1357–1369. [Google Scholar] [CrossRef]
- van Rosmalen, M.; Krom, M.; Merkx, M. Tuning the Flexibility of Glycine-Serine Linkers To Allow Rational Design of Multidomain Proteins. Biochemistry 2017, 56, 6565–6574. [Google Scholar] [CrossRef]
- Ingels, J.; De Cock, L.; Mayer, R.L.; Devreker, P.; Weening, K.; Heyns, K.; Lootens, N.; De Smet, S.; Brusseel, M.; De Munter, S.; et al. Small-scale manufacturing of neoantigen-encoding messenger RNA for early-phase clinical trials. Cytotherapy 2022, 24, 213–222. [Google Scholar] [CrossRef]
- Cafri, G.; Gartner, J.J.; Zaks, T.; Hopson, K.; Levin, N.; Paria, B.C.; Parkhurst, M.R.; Yossef, R.; Lowery, F.J.; Jafferji, M.S.; et al. mRNA vaccine-induced neoantigen-specific T cell immunity in patients with gastrointestinal cancer. J. Clin. Investig. 2020, 130, 5976–5988. [Google Scholar] [CrossRef]
- Hollingsworth, R.E.; Jansen, K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines 2019, 4, 7. [Google Scholar] [CrossRef]
- Faghfuri, E.; Pourfarzi, F.; Faghfouri, A.H.; Abdoli Shadbad, M.; Hajiasgharzadeh, K.; Baradaran, B. Recent developments of RNA-based vaccines in cancer immunotherapy. Expert Opin. Biol. Ther. 2021, 21, 201–218. [Google Scholar] [CrossRef] [PubMed]
- Coulie, P.G.; Van den Eynde, B.J.; van der Bruggen, P.; Boon, T. Tumour antigens recognized by T lymphocytes: At the core of cancer immunotherapy. Nat. Rev. Cancer 2014, 14, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Ott, P.A.; Wu, C.J. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat. Rev. Immunol. 2018, 18, 168–182. [Google Scholar] [CrossRef]
- Chen, Y.T.; Scanlan, M.J.; Sahin, U.; Türeci, O.; Gure, A.O.; Tsang, S.; Williamson, B.; Stockert, E.; Pfreundschuh, M.; Old, L.J. A testicular antigen aberrantly expressed in human cancers detected by autologous antibody screening. Proc. Natl. Acad. Sci. USA 1997, 94, 1914–1918. [Google Scholar] [CrossRef] [PubMed]
- Buonaguro, L.; Petrizzo, A.; Tornesello, M.L.; Buonaguro, F.M. Translating tumor antigens into cancer vaccines. Clin. Vaccine Immunol. 2011, 18, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Novellino, L.; Castelli, C.; Parmiani, G. A listing of human tumor antigens recognized by T cells: March 2004 update. Cancer Immunol. Immunother. 2005, 54, 187–207. [Google Scholar] [CrossRef]
- Simon, P.; Omokoko, T.A.; Breitkreuz, A.; Hebich, L.; Kreiter, S.; Attig, S.; Konur, A.; Britten, C.M.; Paret, C.; Dhaene, K.; et al. Functional TCR retrieval from single antigen-specific human T cells reveals multiple novel epitopes. Cancer Immunol. Res. 2014, 2, 1230–1244. [Google Scholar] [CrossRef] [PubMed]
- Cheever, M.A.; Allison, J.P.; Ferris, A.S.; Finn, O.J.; Hastings, B.M.; Hecht, T.T.; Mellman, I.; Prindiville, S.A.; Viner, J.L.; Weiner, L.M.; et al. The prioritization of cancer antigens: A national cancer institute pilot project for the acceleration of translational research. Clin. Cancer Res. 2009, 15, 5323–5337. [Google Scholar] [CrossRef]
- Aikins, M.E.; Xu, C.; Moon, J.J. Engineered Nanoparticles for Cancer Vaccination and Immunotherapy. Acc Chem. Res. 2020, 53, 2094–2105. [Google Scholar] [CrossRef]
- Conry, R.M.; LoBuglio, A.F.; Wright, M.; Sumerel, L.; Pike, M.J.; Johanning, F.; Benjamin, R.; Lu, D.; Curiel, D.T. Characterization of a messenger RNA polynucleotide vaccine vector. Cancer Res. 1995, 55, 1397–1400. [Google Scholar]
- Zhou, W.Z.; Hoon, D.S.; Huang, S.K.; Fujii, S.; Hashimoto, K.; Morishita, R.; Kaneda, Y. RNA melanoma vaccine: Induction of antitumor immunity by human glycoprotein 100 mRNA immunization. Hum. Gene Ther. 1999, 10, 2719–2724. [Google Scholar] [CrossRef] [PubMed]
- Perche, F.; Benvegnu, T.; Berchel, M.; Lebegue, L.; Pichon, C.; Jaffrès, P.A.; Midoux, P. Enhancement of dendritic cells transfection in vivo and of vaccination against B16F10 melanoma with mannosylated histidylated lipopolyplexes loaded with tumor antigen messenger RNA. Nanomedicine 2011, 7, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, L.; Xu, Z.; Miao, L.; Huang, L. mRNA Vaccine with Antigen-Specific Checkpoint Blockade Induces an Enhanced Immune Response against Established Melanoma. Mol. Ther. 2018, 26, 420–434. [Google Scholar] [CrossRef] [PubMed]
- Oberli, M.A.; Reichmuth, A.M.; Dorkin, J.R.; Mitchell, M.J.; Fenton, O.S.; Jaklenec, A.; Anderson, D.G.; Langer, R.; Blankschtein, D. Lipid Nanoparticle Assisted mRNA Delivery for Potent Cancer Immunotherapy. Nano Lett. 2017, 17, 1326–1335. [Google Scholar] [CrossRef]
- Mai, Y.; Guo, J.; Zhao, Y.; Ma, S.; Hou, Y.; Yang, J. Intranasal delivery of cationic liposome-protamine complex mRNA vaccine elicits effective anti-tumor immunity. Cell Immunol. 2020, 354, 104143. [Google Scholar] [CrossRef]
- Do, A.S.S.; Amano, T.; Edwards, L.A.; Zhang, L.; De Peralta-Venturina, M.; Yu, J.S. CD133 mRNA-Loaded Dendritic Cell Vaccination Abrogates Glioma Stem Cell Propagation in Humanized Glioblastoma Mouse Model. Mol. Ther. Oncolytics 2020, 18, 295–303. [Google Scholar] [CrossRef]
- He, Q.; Gao, H.; Tan, D.; Zhang, H.; Wang, J.Z. mRNA cancer vaccines: Advances, trends and challenges. Acta Pharm. Sin. B 2022, 12, 2969–2989. [Google Scholar] [CrossRef]
- Lichtenegger, F.S.; Schnorfeil, F.M.; Rothe, M.; Deiser, K.; Altmann, T.; Bücklein, V.L.; Köhnke, T.; Augsberger, C.; Konstandin, N.P.; Spiekermann, K.; et al. Toll-like receptor 7/8-matured RNA-transduced dendritic cells as post-remission therapy in acute myeloid leukaemia: Results of a phase I trial. Clin. Transl. Immunol. 2020, 9, e1117. [Google Scholar] [CrossRef]
- Van Tendeloo, V.F.; Van de Velde, A.; Van Driessche, A.; Cools, N.; Anguille, S.; Ladell, K.; Gostick, E.; Vermeulen, K.; Pieters, K.; Nijs, G.; et al. Induction of complete and molecular remissions in acute myeloid leukemia by Wilms’ tumor 1 antigen-targeted dendritic cell vaccination. Proc. Natl. Acad. Sci. USA 2010, 107, 13824–13829. [Google Scholar] [CrossRef]
- Anguille, S.; Van de Velde, A.L.; Smits, E.L.; Van Tendeloo, V.F.; Juliusson, G.; Cools, N.; Nijs, G.; Stein, B.; Lion, E.; Van Driessche, A.; et al. Dendritic cell vaccination as postremission treatment to prevent or delay relapse in acute myeloid leukemia. Blood 2017, 130, 1713–1721. [Google Scholar] [CrossRef]
- Kübler, H.; Scheel, B.; Gnad-Vogt, U.; Miller, K.; Schultze-Seemann, W.; Vom Dorp, F.; Parmiani, G.; Hampel, C.; Wedel, S.; Trojan, L.; et al. Self-adjuvanted mRNA vaccination in advanced prostate cancer patients: A first-in-man phase I/IIa study. J. Immunother. Cancer 2015, 3, 26. [Google Scholar] [CrossRef]
- Sebastian, M.; Schröder, A.; Scheel, B.; Hong, H.S.; Muth, A.; von Boehmer, L.; Zippelius, A.; Mayer, F.; Reck, M.; Atanackovic, D.; et al. A phase I/IIa study of the mRNA-based cancer immunotherapy CV9201 in patients with stage IIIB/IV non-small cell lung cancer. Cancer Immunol. Immunother. 2019, 68, 799–812. [Google Scholar] [CrossRef]
- Linch, M.; Papai, Z.; Takacs, I.; Imedio, E.R.; Kühnle, M.-C.; Derhovanessian, E.; Vogler, I.; Renken, S.; Graham, P.; Sahin, U.; et al. 421 A first-in-human (FIH) phase I/IIa clinical trial assessing a ribonucleic acid lipoplex (RNA-LPX) encoding shared tumor antigens for immunotherapy of prostate cancer; preliminary analysis of PRO-MERIT. J. Immunother. Cancer 2021, 9, A451. [Google Scholar] [CrossRef]
- Chung, D.J.; Sharma, S.; Rangesa, M.; DeWolf, S.; Elhanati, Y.; Perica, K.; Young, J.W. Langerhans dendritic cell vaccine bearing mRNA-encoded tumor antigens induces antimyeloma immunity after autotransplant. Blood Adv. 2022, 6, 1547–1558. [Google Scholar] [CrossRef] [PubMed]
- Papachristofilou, A.; Hipp, M.M.; Klinkhardt, U.; Früh, M.; Sebastian, M.; Weiss, C.; Pless, M.; Cathomas, R.; Hilbe, W.; Pall, G.; et al. Phase Ib evaluation of a self-adjuvanted protamine formulated mRNA-based active cancer immunotherapy, BI1361849 (CV9202), combined with local radiation treatment in patients with stage IV non-small cell lung cancer. J. Immunother. Cancer 2019, 7, 38. [Google Scholar] [CrossRef] [PubMed]
- Reinhard, K.; Rengstl, B.; Oehm, P.; Michel, K.; Billmeier, A.; Hayduk, N.; Klein, O.; Kuna, K.; Ouchan, Y.; Wöll, S.; et al. An RNA vaccine drives expansion and efficacy of claudin-CAR-T cells against solid tumors. Science 2020, 367, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Strebhardt, K.; Ullrich, A. Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat. Rev. Cancer 2008, 8, 473–480. [Google Scholar] [CrossRef]
- Chen, W.; Yuan, Y.; Jiang, X. Antibody and antibody fragments for cancer immunotherapy. J. Control. Release 2020, 328, 395–406. [Google Scholar] [CrossRef]
- Baas, P.; Scherpereel, A.; Nowak, A.K.; Fujimoto, N.; Peters, S.; Tsao, A.S.; Mansfield, A.S.; Popat, S.; Jahan, T.; Antonia, S.; et al. First-line nivolumab plus ipilimumab in unresectable malignant pleural mesothelioma (CheckMate 743): A multicentre, randomised, open-label, phase 3 trial. Lancet 2021, 397, 375–386. [Google Scholar] [CrossRef]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Yokoi, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. Durvalumab after Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 1919–1929. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P.W.H.I. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef]
- Van Hoecke, L.; Roose, K. How mRNA therapeutics are entering the monoclonal antibody field. J. Transl. Med. 2019, 17, 54. [Google Scholar] [CrossRef] [PubMed]
- Schlake, T.; Thran, M.; Fiedler, K.; Heidenreich, R.; Petsch, B.; Fotin-Mleczek, M. mRNA: A Novel Avenue to Antibody Therapy? Mol. Ther. 2019, 27, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Thran, M.; Mukherjee, J.; Pönisch, M.; Fiedler, K.; Thess, A.; Mui, B.L.; Hope, M.J.; Tam, Y.K.; Horscroft, N.; Heidenreich, R.; et al. mRNA mediates passive vaccination against infectious agents, toxins, and tumors. EMBO Mol. Med. 2017, 9, 1434–1447. [Google Scholar] [CrossRef]
- Rybakova, Y.; Kowalski, P.S.; Huang, Y.; Gonzalez, J.T.; Heartlein, M.W.; DeRosa, F.; Delcassian, D.; Anderson, D.G. mRNA Delivery for Therapeutic Anti-HER2 Antibody Expression In Vivo. Mol. Ther. 2019, 27, 1415–1423. [Google Scholar] [CrossRef]
- Wang, Y.; Tiruthani, K.; Li, S.; Hu, M.; Zhong, G.; Tang, Y.; Roy, S.; Zhang, L.; Tan, J.; Liao, C.; et al. mRNA Delivery of a Bispecific Single-Domain Antibody to Polarize Tumor-Associated Macrophages and Synergize Immunotherapy against Liver Malignancies. Adv. Mater. 2021, 33, e2007603. [Google Scholar] [CrossRef]
- Klinger, M.; Benjamin, J.; Kischel, R.; Stienen, S.; Zugmaier, G. Harnessing T cells to fight cancer with BiTE® antibody constructs--past developments and future directions. Immunol. Rev. 2016, 270, 193–208. [Google Scholar] [CrossRef]
- Stadler, C.R.; Bähr-Mahmud, H.; Celik, L.; Hebich, B.; Roth, A.S.; Roth, R.P.; Karikó, K.; Türeci, Ö.; Sahin, U. Elimination of large tumors in mice by mRNA-encoded bispecific antibodies. Nat. Med. 2017, 23, 815–817. [Google Scholar] [CrossRef]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef] [PubMed]
- Demaria, O.; Cornen, S.; Daëron, M.; Morel, Y.; Medzhitov, R.; Vivier, E. Harnessing innate immunity in cancer therapy. Nature 2019, 574, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Aricò, E.; Castiello, L.; Capone, I.; Gabriele, L.; Belardelli, F. Type I Interferons and Cancer: An Evolving Story Demanding Novel Clinical Applications. Cancers 2019, 11, 1943. [Google Scholar] [CrossRef] [Green Version]
- Jeong, S.; Park, S.H. Co-Stimulatory Receptors in Cancers and Their Implications for Cancer Immunotherapy. Immune Netw. 2020, 20, e3. [Google Scholar] [CrossRef] [PubMed]
- Ong, G.H.; Lian, B.S.X.; Kawasaki, T.; Kawai, T. Exploration of Pattern Recognition Receptor Agonists as Candidate Adjuvants. Front. Cell Infect Microbiol. 2021, 11, 745016. [Google Scholar] [CrossRef]
- Lai, I.; Swaminathan, S.; Baylot, V.; Mosley, A.; Dhanasekaran, R.; Gabay, M.; Felsher, D.W. Lipid nanoparticles that deliver IL-12 messenger RNA suppress tumorigenesis in MYC oncogene-driven hepatocellular carcinoma. J. Immunother. Cancer 2018, 6, 125. [Google Scholar] [CrossRef]
- Liu, J.Q.; Zhang, C.; Zhang, X.; Yan, J.; Zeng, C.; Talebian, F.; Lynch, K.; Zhao, W.; Hou, X.; Du, S.; et al. Intratumoral delivery of IL-12 and IL-27 mRNA using lipid nanoparticles for cancer immunotherapy. J. Control. Release 2022, 345, 306–313. [Google Scholar] [CrossRef]
- Hotz, C.; Wagenaar, T.R.; Gieseke, F.; Bangari, D.S.; Callahan, M.; Cao, H.; Diekmann, J.; Diken, M.; Grunwitz, C.; Hebert, A.; et al. Local delivery of mRNA-encoded cytokines promotes antitumor immunity and tumor eradication across multiple preclinical tumor models. Sci. Transl. Med. 2021, 13, eabc7804. [Google Scholar] [CrossRef] [PubMed]
- Haabeth, O.A.W.; Blake, T.R.; McKinlay, C.J.; Tveita, A.A.; Sallets, A.; Waymouth, R.M.; Wender, P.A.; Levy, R. Local Delivery of Ox40l, Cd80, and Cd86 mRNA Kindles Global Anticancer Immunity. Cancer Res. 2019, 79, 1624–1634. [Google Scholar] [CrossRef]
- Hess, P.R.; Boczkowski, D.; Nair, S.K.; Snyder, D.; Gilboa, E. Vaccination with mRNAs encoding tumor-associated antigens and granulocyte-macrophage colony-stimulating factor efficiently primes CTL responses, but is insufficient to overcome tolerance to a model tumor/self antigen. Cancer Immunol. Immunother. 2006, 55, 672–683. [Google Scholar] [CrossRef]
- Kreiter, S.; Diken, M.; Selmi, A.; Petschenka, J.; Türeci, Ö.; Sahin, U. FLT3 Ligand as a Molecular Adjuvant for Naked RNA Vaccines. Methods Mol. Biol. 2016, 1428, 163–175. [Google Scholar] [CrossRef]
- De Keersmaecker, B.; Claerhout, S.; Carrasco, J.; Bar, I.; Corthals, J.; Wilgenhof, S.; Neyns, B.; Thielemans, K. TriMix and tumor antigen mRNA electroporated dendritic cell vaccination plus ipilimumab: Link between T-cell activation and clinical responses in advanced melanoma. J. Immunother. Cancer 2020, 8, e000329. [Google Scholar] [CrossRef] [PubMed]
- Jansen, Y.; Kruse, V.; Corthals, J.; Schats, K.; van Dam, P.J.; Seremet, T.; Heirman, C.; Brochez, L.; Kockx, M.; Thielemans, K.; et al. A randomized controlled phase II clinical trial on mRNA electroporated autologous monocyte-derived dendritic cells (TriMixDC-MEL) as adjuvant treatment for stage III/IV melanoma patients who are disease-free following the resection of macrometastases. Cancer Immunol. Immunother. 2020, 69, 2589–2598. [Google Scholar] [CrossRef]
- Wilgenhof, S.; Van Nuffel, A.M.T.; Benteyn, D.; Corthals, J.; Aerts, C.; Heirman, C.; Van Riet, I.; Bonehill, A.; Thielemans, K.; Neyns, B. A phase IB study on intravenous synthetic mRNA electroporated dendritic cell immunotherapy in pretreated advanced melanoma patients. Ann. Oncol. 2013, 24, 2686–2693. [Google Scholar] [CrossRef] [PubMed]
- Arance Fernandez, A.N.A.M.; Baurain, J.-F.; Vulsteke, C.; Rutten, A.; Soria, A.; Carrasco, J.; Neyns, B.; De Keersmaecker, B.; Van Assche, T.; Lindmark, B. A phase I study (E011-MEL) of a TriMix-based mRNA immunotherapy (ECI-006) in resected melanoma patients: Analysis of safety and immunogenicity. J. Clin. Oncol. 2019, 37 (Suppl. 15), 2641. [Google Scholar] [CrossRef]
- Bialkowski, L.; van Weijnen, A.; Van der Jeught, K.; Renmans, D.; Daszkiewicz, L.; Heirman, C.; Stangé, G.; Breckpot, K.; Aerts, J.L.; Thielemans, K. Intralymphatic mRNA vaccine induces CD8 T-cell responses that inhibit the growth of mucosally located tumours. Sci. Rep. 2016, 6, 22509. [Google Scholar] [CrossRef]
- Hewitt, S.L.; Bai, A.; Bailey, D.; Ichikawa, K.; Zielinski, J.; Karp, R.; Apte, A.; Arnold, K.; Zacharek, S.J.; Iliou, M.S.; et al. Durable anticancer immunity from intratumoral administration of IL-23, IL-36γ, and OX40L mRNAs. Sci. Transl. Med. 2019, 11, eaat9143. [Google Scholar] [CrossRef] [PubMed]
- Eigentler, T.; Bauernfeind, F.G.; Becker, J.C.; Brossart, P.; Fluck, M.; Heinzerling, L.; Krauss, J.; Mohr, P.; Ochsenreither, S.; Schreiber, J.S.; et al. A phase I dose-escalation and expansion study of intratumoral CV8102 as single-agent or in combination with anti-PD-1 antibodies in patients with advanced solid tumors. J. Clin. Oncol. 2020, 38 (Suppl. 15), 3096. [Google Scholar] [CrossRef]
- Löffler, M.W.; Gori, S.; Izzo, F.; Mayer-Mokler, A.; Ascierto, P.A.; Königsrainer, A.; Ma, Y.T.; Sangro, B.; Francque, S.; Vonghia, L.; et al. Phase I/II Multicenter Trial of a Novel Therapeutic Cancer Vaccine, HepaVac-101, for Hepatocellular Carcinoma. Clin. Cancer Res. 2022, 28, 2555–2566. [Google Scholar] [CrossRef]
- Patel, M.R.; Bauer, T.M.; Jimeno, A.; Wang, D.; LoRusso, P.; Do, K.T.; Stemmer, S.M.; Maurice-Dror, C.; Geva, R.; Zacharek, S.; et al. A phase I study of mRNA-2752, a lipid nanoparticle encapsulating mRNAs encoding human OX40L, IL-23, and IL-36γ, for intratumoral (iTu) injection alone and in combination with durvalumab. J. Clin. Oncol. 2020, 38 (Suppl. 15), 3092. [Google Scholar] [CrossRef]
- Gao, L.; Wu, Z.X.; Assaraf, Y.G.; Chen, Z.S.; Wang, L. Overcoming anti-cancer drug resistance via restoration of tumor suppressor gene function. Drug Resist. Updat 2021, 57, 100770. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Tamborero, D.; Gonzalez-Perez, A.; Perez-Llamas, C.; Deu-Pons, J.; Kandoth, C.; Reimand, J.; Lawrence, M.S.; Getz, G.; Bader, G.D.; Ding, L.; et al. Comprehensive identification of mutational cancer driver genes across 12 tumor types. Sci. Rep. 2013, 3, 2650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.X.; Wang, Y.; Ding, J.; Jiang, A.; Wang, J.; Yu, M.; Blake, S.; Liu, S.; Bieberich, C.J.; Farokhzad, O.C.; et al. Reactivation of the tumor suppressor PTEN by mRNA nanoparticles enhances antitumor immunity in preclinical models. Sci. Transl. Med. 2021, 13, eaba9772. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Chen, J.; Zhou, H.; Zeng, X.; Ruan, Z.; Pu, Z.; Jiang, X.; Matsui, A.; Zhu, L.; Amoozgar, Z.; et al. Combining p53 mRNA nanotherapy with immune checkpoint blockade reprograms the immune microenvironment for effective cancer therapy. Nat. Commun. 2022, 13, 758. [Google Scholar] [CrossRef] [PubMed]
- Qiu, M.; Tang, Y.; Chen, J.; Muriph, R.; Ye, Z.; Huang, C.; Evans, J.; Henske, E.P.; Xu, Q. Lung-selective mRNA delivery of synthetic lipid nanoparticles for the treatment of pulmonary lymphangioleiomyomatosis. Proc. Natl. Acad. Sci. USA 2022, 119, e2116271119. [Google Scholar] [CrossRef]
- Segel, M.; Lash, B.; Song, J.; Ladha, A.; Liu, C.C.; Jin, X.; Mekhedov, S.L.; Macrae, R.K.; Koonin, E.V.; Zhang, F. Mammalian retrovirus-like protein PEG10 packages its own mRNA and can be pseudotyped for mRNA delivery. Science 2021, 373, 882–889. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Period | Product | Type | Study | Phase | Sponsor | Formulation | Route | Other Therapy | Response |
|---|---|---|---|---|---|---|---|---|---|
| 2020–2023 | IVAC_W_bre1_uID and IVAC_M_uID | TNBC | NCT02316457 | Phase I | BioNTech SE | LPX | i.v. | / | ongoing |
| 2017–2019 | IVAC MUTANOME, RBL001/RBL002 (BNT121) | melanoma | NCT02035956 | Phase I | BioNTech SE | naked mRNA | i.n. | / | not published |
| 2017–2024 | Autogene cevumeran (RO7198457, BNT122) | solid tumors | NCT03289962 | Phase I | Genentech, Inc. | naked mRNA | i.v. | Atezolizumab | ongoing |
| 2017–2025 | mRNA-4157 | solid tumors | NCT03313778 | Phase I | ModernaTX, Inc. | LNP | i.m. | Pembrolizumab | ongoing |
| 2018–2020 | NCI-4650 | solid tumors | NCT03480152 | Phase I/II | National Cancer Institute (NCI) | / | i.m. | / | safe with a slight adverse event [209] |
| 2018–2021 | personalized mRNA tumor vaccine | solid tumors in digestive system | NCT03468244 | NA | Changhai Hospital | LPP | s.c. | / | not published |
| 2019–2024 | RO7198457 | advanced melanoma | NCT03815058 | Phase II | Genentech, Inc. | LPX | i.v. | Pembrolizumab | ongoing |
| 2019–2024 | mRNA-4157 | high risk of recurrence melanoma | NCT03897881 | Phase II | ModernaTX, Inc. | naked mRNA | / | Pembrolizumab | ongoing |
| 2019–2022 | personalized mRNA tumor vaccine | esophageal cancer, NSCLC | NCT03908671 | / | Stemirna Therapeutics | LPP | s.c. | / | not published |
| 2019–2023 | RO7198457 | pancreatic cancer | NCT04161755 | Phase I | Memorial Sloan Kettering Cancer Center | LPX | / | Atezolizumab, chemotherapy (mFOLFIRINOX) | ongoing |
| 2020–2025 | RO7198457 | NSCLC | NCT04267237 | Phase II | Hoffmann-La Roche | LPX | i.v. | Atezolizumab | withdrawn |
| 2022–2023 | SW1115C3 | solid tumor | NCT05198752 | Phase I | Stemirna Therapeutics | LPP | / | / | ongoing |
| 2022–2025 | neoantigen tumor vaccine | gastric cancer, esophageal cancer, and liver cancer | NCT05192460 | / | Jianming Xu | / | / | PD-1/L1 drugs | ongoing |
| 2022–2026 | GRT-C901/GRT-R902 | colonic neoplasms and colorectal neoplasms | NCT05456165 | Phase II | Gritstone bio, Inc. | chimpanzee adenovirus | i.m | Atezolizumab, Ipilimumab, chemotherapy | ongoing |
| 2019–2025 | RO7198457 | NSCLC | NCT04267237 | Phase II | Hoffmann-La Roche | LPX | i.v. | Atezolizumab | ongoing |
| 2019–2024 | RO7198457 | melanoma | NCT03815058 | Phase II | Genentech, Inc. | LPX | i.v. | Pembrolizumab | ongoing |
| Period | Product/TAA | Type | Study | Phase | Sponsor | Formulation | Route | Other Therapy | Response |
|---|---|---|---|---|---|---|---|---|---|
| 2022–2027 | mRNA-4359 (mRNA encoding IDO and PD-L1) | advanced solid tumors | NCT05533697 | Phase I/II | ModernaTX, Inc. | / | i.m. | Pembrolizumab | ongoing |
| 2009–2013 | CV9103 (mRNA encoding 4 PSAs, PSCA, PSMA, and STEAP1) | hormonal refractory prostate cancer | NCT00831467 | Phase I/II | CureVac AG | protamine-stabilized mRNA | i.d. | / | well tolerated, prolonged patient survival [231] |
| 2013–2017 | CV9104 (mRNA encoding PSA, PSMA, PSCA, STEAP1, PAP, and MUC1) | PCa | NCT01817738 | Phase I/II | CureVac AG | protamine-stabilized mRNA | i.d. | / | not published |
| 2007–2009 | mRNA in AML cell lysate | AML | NCT00514189 | Phase I | M.D. Anderson Cancer Center | DCs loaded | i.v. | / | not published |
| 2010–2024 | tumor mRNA | PCa | NCT01197625 | Phase I/II | Oslo University Hospital | DCs loaded | i.v. | / | not published |
| 2007–2014 | GRNVAC1 (mRNA encoding hTERT, LAMP) | AML | NCT00510133 | Phase II | Asterias Biotherapeutics | DCs loaded | i.v. | / | not published |
| 2011–2013 | DC-006 vaccine (mRNA encoding hTERT, survivin) | recurrent epithelial OC | NCT01334047 | Phase I/II | Steinar Aamdal | DCs loaded | i.d. | / | not published |
| 2009–2012 | mRNA encoding hTERT, survivin, and tumor mRNA | metastatic malignant melanoma | NCT00961844 | Phase I/II | Steinar Aamdal | DCs loaded and ex vivo T cell expansion and reinfusion | i.v. | Temozolomide | not published |
| 2009–2014 | CV9201 (mRNA encoding NY-ESO-1, MAGE-C1/C2, survivin, and 5T4) | NSCLC | NCT00923312 | Phase I/II | CureVac AG | protamine-stabilized mRNA | / | / | well tolerated and therapeutic [232] |
| 2011–2023 | tumor mRNA | melanoma | NCT01456104 | Phase I | Memorial Sloan Kettering Cancer Center | DCs loaded | i.d. | / | ongoing |
| 2020–2025 | BNT111 (mRNA encoding NY-ESO-1, MAGE-A3, tyrosinase, and TPTE) | unresectable/stage III/stage IV melanoma | NCT04526899 | Phase II | BioNTech SE | LPX | i.v. | Cemiplimab | ongoing |
| 2019–2023 | W_ova1 Vaccine (3 OC TAA mRNAs) | OC | NCT04163094 | Phase I | University Medical Center Groningen | LPX | i.v. | adjuvant chemotherapy | ongoing |
| 2020–2023 | BNT112 (mRNA encoding kallikrein-2/3, acid phosphatase prostate, HOXB13, and NK3 homeobox 1) | PCa | NCT04382898 | Phase I/II | BioNTech SE | LPX | i.v. | Cemiplimab | an acceptable safety profile [233] |
| 2020–2025 | BNT113 (mRNA encoding E6/E7) | unresectable/metastatic/recurrent head and neck cancer | NCT04534205 | Phase II | BioNTech SE | LPX | i.v. | Pembrolizumab | ongoing |
| 2013–2022 | mRNA encoding CT7, MAGE-A3, and WT1 | multiple myeloma | NCT01995708 | Phase I | Memorial Sloan Kettering Cancer Center | LCs loaded | s.c. | / | safe and therapeutic with a slight adverse event [234] |
| 2015–2019 | mRNA encoding WT1 and PRAME | AML | NCT02405338 | Phase I/II | Medigene AG | DCs loaded | i.d. | / | not published |
| 2012–2018 | mRNA encoding WT1, PRAME, and CMVpp65 | AML | NCT01734304 | Phase I/II | Ludwig-Maximilians—University of Munich | TLR7/8-matured DCs loaded | i.v. | / | feasible and safe with a slight adverse event [228] |
| 2009–2014 | mRNA encoding hTERT, survivin, and p53 | breast cancer and malignant melanoma | NCT00978913 | Phase I | Inge Marie Svane | DCs loaded | i.d. | Cyclophosphamide | not published |
| 2017–2021 | CV9202 (BI 1361849, mRNA encoding MUC1, survivin, NY-ESO-1, 5T4, MAGE-C1/C2) | NSCLC | NCT03164772 | Phase I/II | Ludwig Institute for Cancer Research | LNP | i.d. | Durvalumab, Tremelumumab | with an adverse event |
| 2013–2016 | CV9202 (BI 1361849) | NSCLC | NCT01915524 | Phase I | CureVac AG | LNP | i.d. | Radiotherapy, an EGFR tyrosine kinase inhibitor | well tolerated with an adverse event [169,235] |
| Target | mAb | Type |
|---|---|---|
| anti-CD20 antibody | rituximab | lymphoma, chronic lymphocytic leukemia |
| anti-EGFR antibody | cetuximab | head and neck cancer and colorectal cancer |
| anti-HER2 antibody | trastuzumab | HER2-positive metastatic breast cancer |
| Immune Checkpoint Inhibitors | Location | Ligand | mAb | Product |
|---|---|---|---|---|
| PD-1 inhibitors | T cells | PD-L1 | Pembrolizumab | Keytruda |
| Nivolumab | Opdivo | |||
| Cemiplimab | Libtayo | |||
| PD-L1 inhibitors | Cancer cells | PD-1 | Atezolizumab | Tecentriq |
| Avelumab | Bavencio | |||
| Durvalumab | Imfinzi | |||
| CTLA-4 inhibitors | T cells | CD80, CD86 | Ipilimumab | Yervoy |
| Tremelimumab | Imjudo |
| Period | Product | Type | Study | Phase | Sponsor | Formulation | Route | Other Therapy | Response |
|---|---|---|---|---|---|---|---|---|---|
| 2020–2024 | BNT141 (mRNA encoding anti-Claudin18.2 monoclonal antibody) | unresectable or metastatic CLDN18.2-positive gastric, pancreatic, ovarian, and biliary tract tumors | NCT04683939 | Phase I/II | BioNTech SE | LNP | i.v. | nab-paclitaxel, gemcitabine | ongoing |
| 2022–2026 | BNT142 (mRNA encoding antibodies targeting CD3 × CLDN6) | solid tumor | NCT05262530 | Phase I/II | BioNTech SE | LNP | i.v. | / | ongoing |
| Period | Product | Type | Study | Phase | Sponsor | Formulation | Route | Other Therapy | Response |
|---|---|---|---|---|---|---|---|---|---|
| 2017–2023 | CV8102 (mRNA encoding TLR7/8/RIG-I agonist) | advanced solid tumors | NCT03291002 | Phase I | CureVac | non-coding, non-capped RNA | i.t. | anti-PD-1 Ab | well tolerated without dose-limiting toxicities [267] |
| 2017–2019 | CV8102 | HCC | NCT03203005 | Phase I/II | National Cancer Institute, Naples | non-coding, non-capped RNA | i.d. | Cyclophosphamide, IMA970A (multipeptide-based HCC vaccine) | safe with a side effect [268] |
| 2016–2023 | mRNA-2752 (mRNA encoding OX40L, IL-23, and IL-36γ) | high-risk DCIS | NCT02872025 | Phase I | Laura Esserman | LNP | Intralesional injection | Pembrolizumab | well tolerated with slight dose-limiting toxicities [269] |
| 2018–2023 | mRNA-2752 (mRNA encoding OX40L, IL-23, and IL-36γ) | advanced malignancies | NCT03739931 | Phase I | ModernaTX, Inc. | LNP | i.t. | Durvalumab | ongoing |
| 2017–2022 | mRNA-2416 (mRNA encoding OX40L) | relapsed/refractory solid tumor malignancies or lymphoma and OC | NCT03323398 | Phase I/II | ModernaTX, Inc. | LNP | i.t. | Durvalumab | not published |
| 2019–2024 | BNT131 (SAR441000, mRNA encoding IL-12sc, IFNα-2b, GM-CSF, and IL-15sushi) | metastatic neoplasm | NCT03871348 | Phase I | Sanofi | saline-formulated mixture | i.t. | Cemiplimab REGN2810 | ongoing |
| 2019–2027 | MEDI1191 (mRNA encoding IL-12) | advanced solid tumors | NCT03946800 | Phase I | MedImmune LLC | LNP | i.t. | Durvalumab | ongoing |
| 2020–2026 | BNT151 (mRNA encoding IL-2) | solid tumors | NCT04455620 | Phase I/II | BioNTech SE | LPX | i.v. | / | ongoing |
| 2021–2023 | BNT152 (mRNA encoding IL-7) plus BNT153 (mRNA encoding IL-2) | solid tumor | NCT04710043 | Phase I | BioNTech SE | LPX | i.v. | / | ongoing |
| 2022–2027 | ABOD2011 (mRNA encoding IL-12) | advanced solid tumors | NCT05392699 | Phase I | Cancer Institute and Hospital, Chinese Academy of Medical Sciences | naked mRNA | i.t. | / | ongoing |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, H.; Zhang, Y.; Wang, G.; Yang, W.; Xu, Y. mRNA-Based Therapeutics in Cancer Treatment. Pharmaceutics 2023, 15, 622. https://doi.org/10.3390/pharmaceutics15020622
Sun H, Zhang Y, Wang G, Yang W, Xu Y. mRNA-Based Therapeutics in Cancer Treatment. Pharmaceutics. 2023; 15(2):622. https://doi.org/10.3390/pharmaceutics15020622
Chicago/Turabian StyleSun, Han, Yu Zhang, Ge Wang, Wen Yang, and Yingjie Xu. 2023. "mRNA-Based Therapeutics in Cancer Treatment" Pharmaceutics 15, no. 2: 622. https://doi.org/10.3390/pharmaceutics15020622