
Multimeric RGD-Based Strategies for Selective Drug Delivery to Tumor Tissues

Abstract
:1. Introduction

2. Design of Multimeric RGD Compounds for Drug Delivery
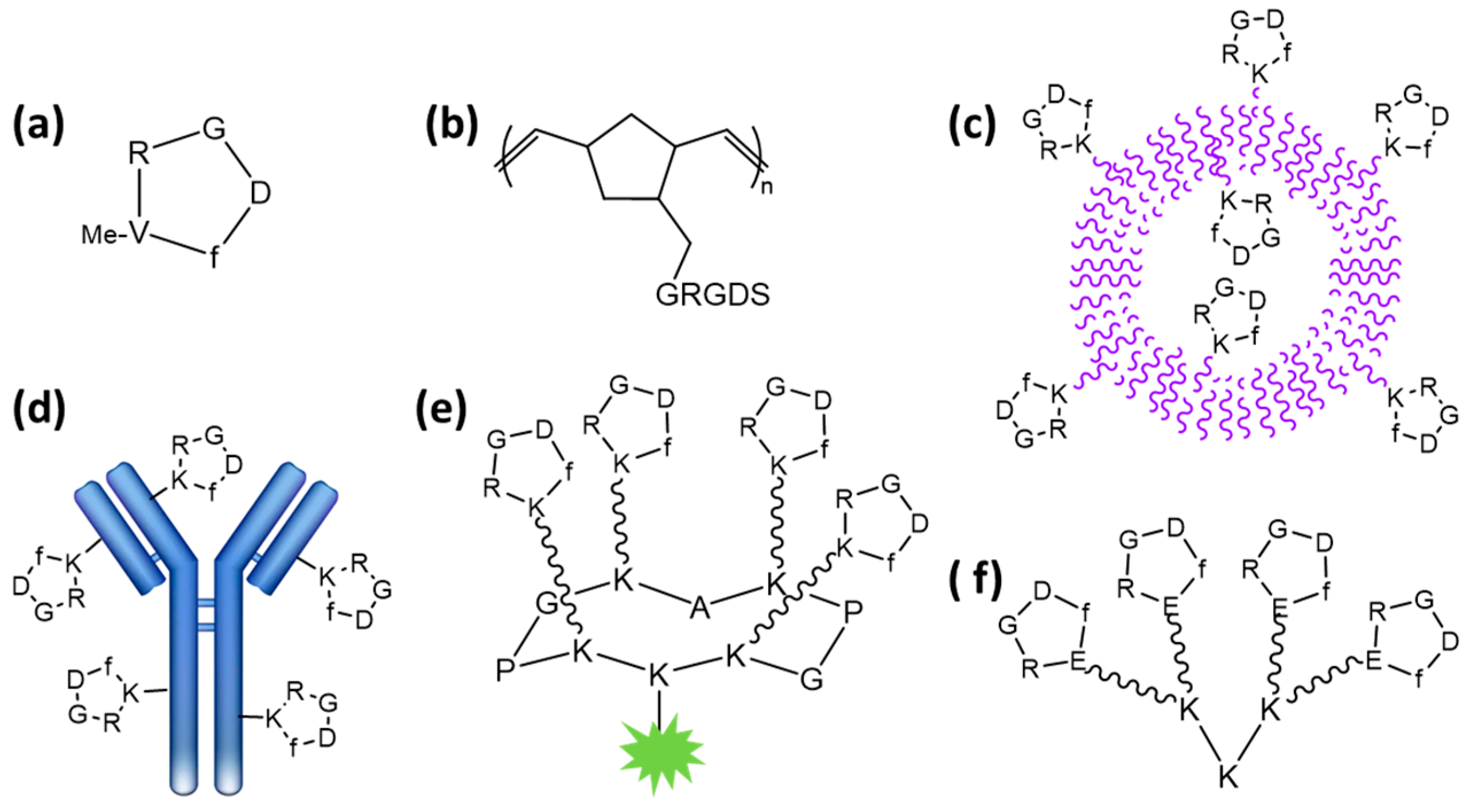
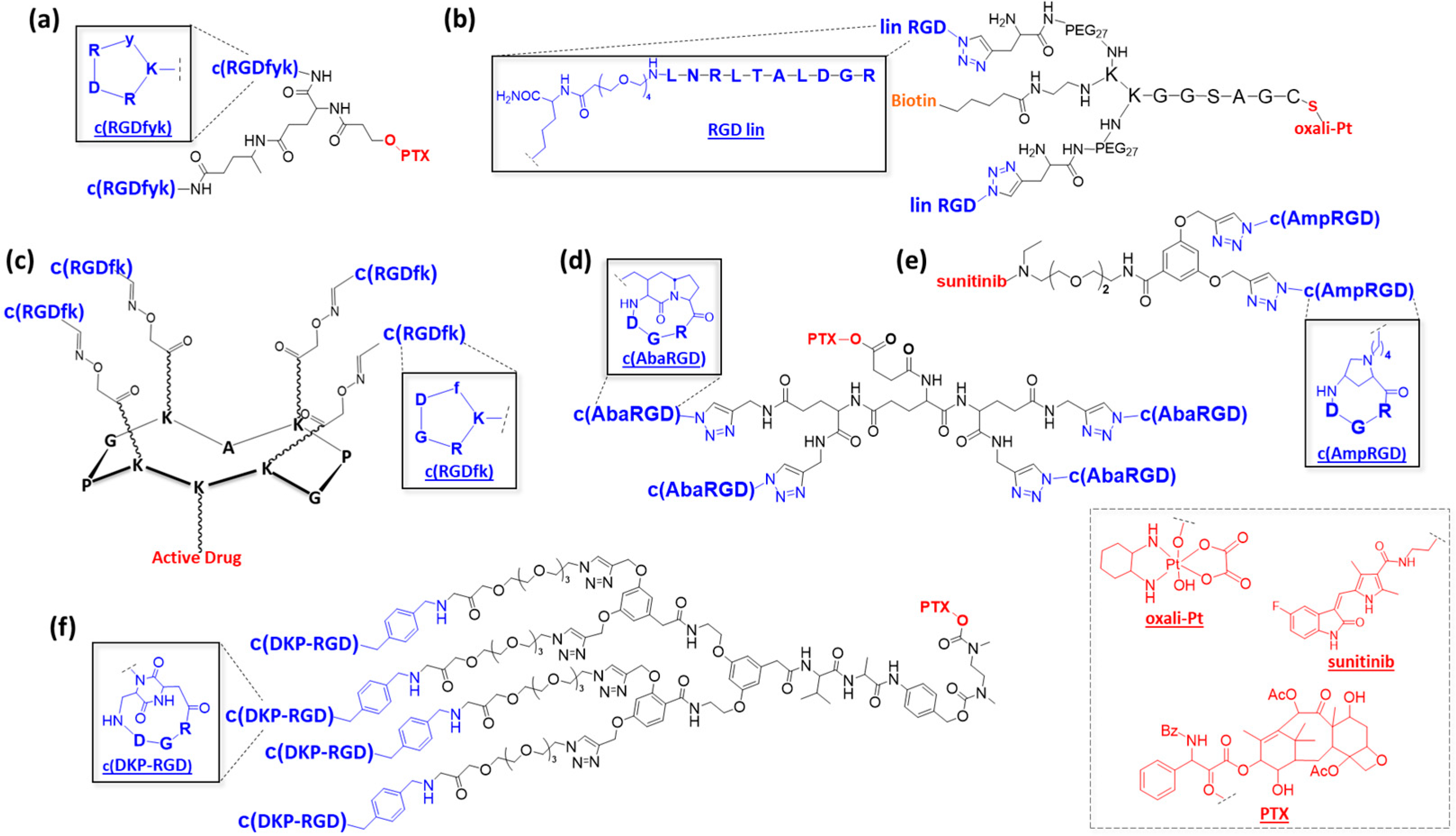
2.1. Using Low-Molecular-Weight Scaffolds

2.2. Using Polymers and Nanoparticles

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
3. Use of Cleavable Linker for Multimeric RGD-Based Drug Delivery
3.1. General Overview: Enzymatically Cleavable Linkers
3.2. General Overview: Physically and Chemically Cleavable Linkers
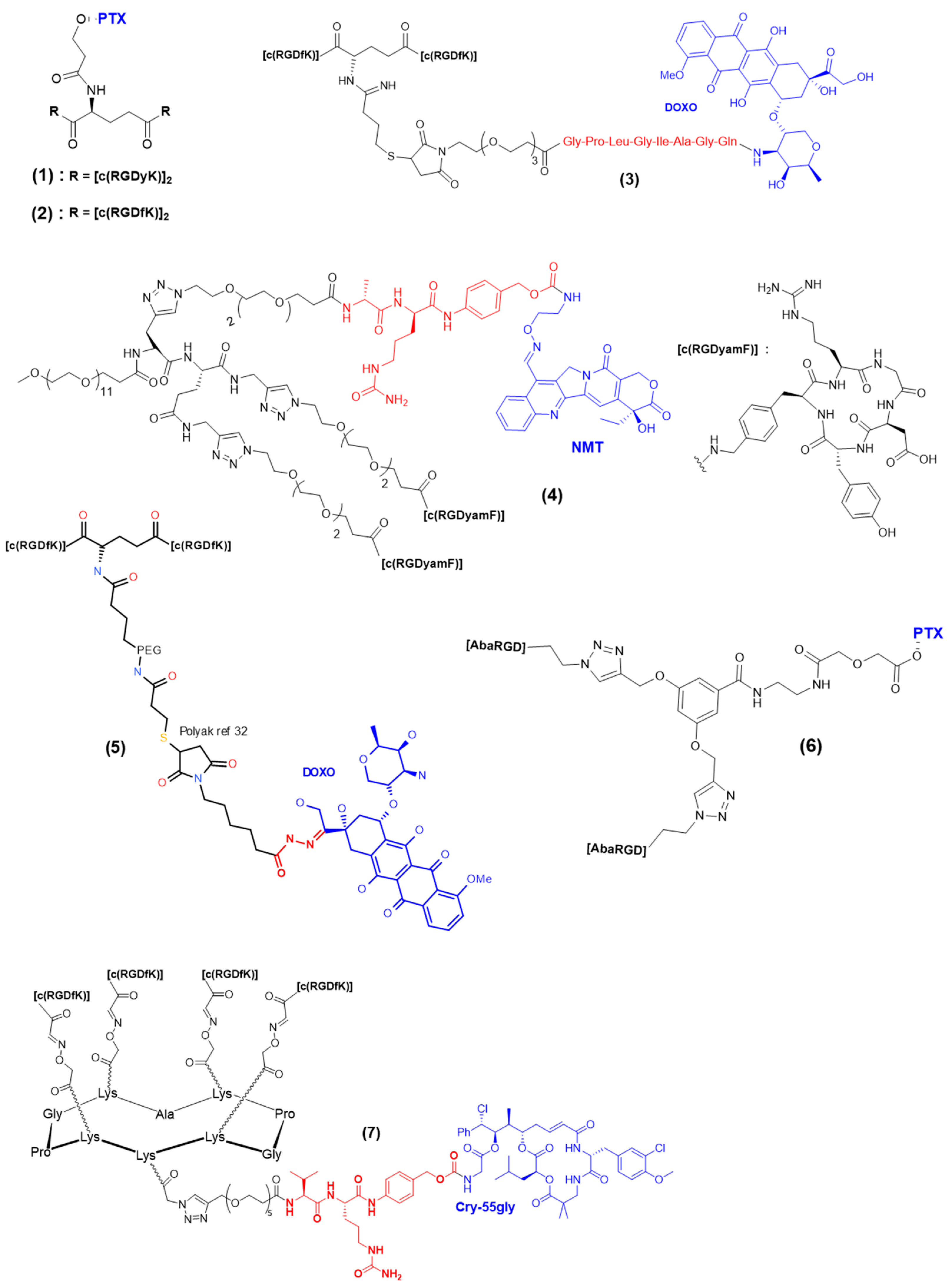
3.3. Cleavable Linkers in Multimeric RGD-Based Drug Delivery
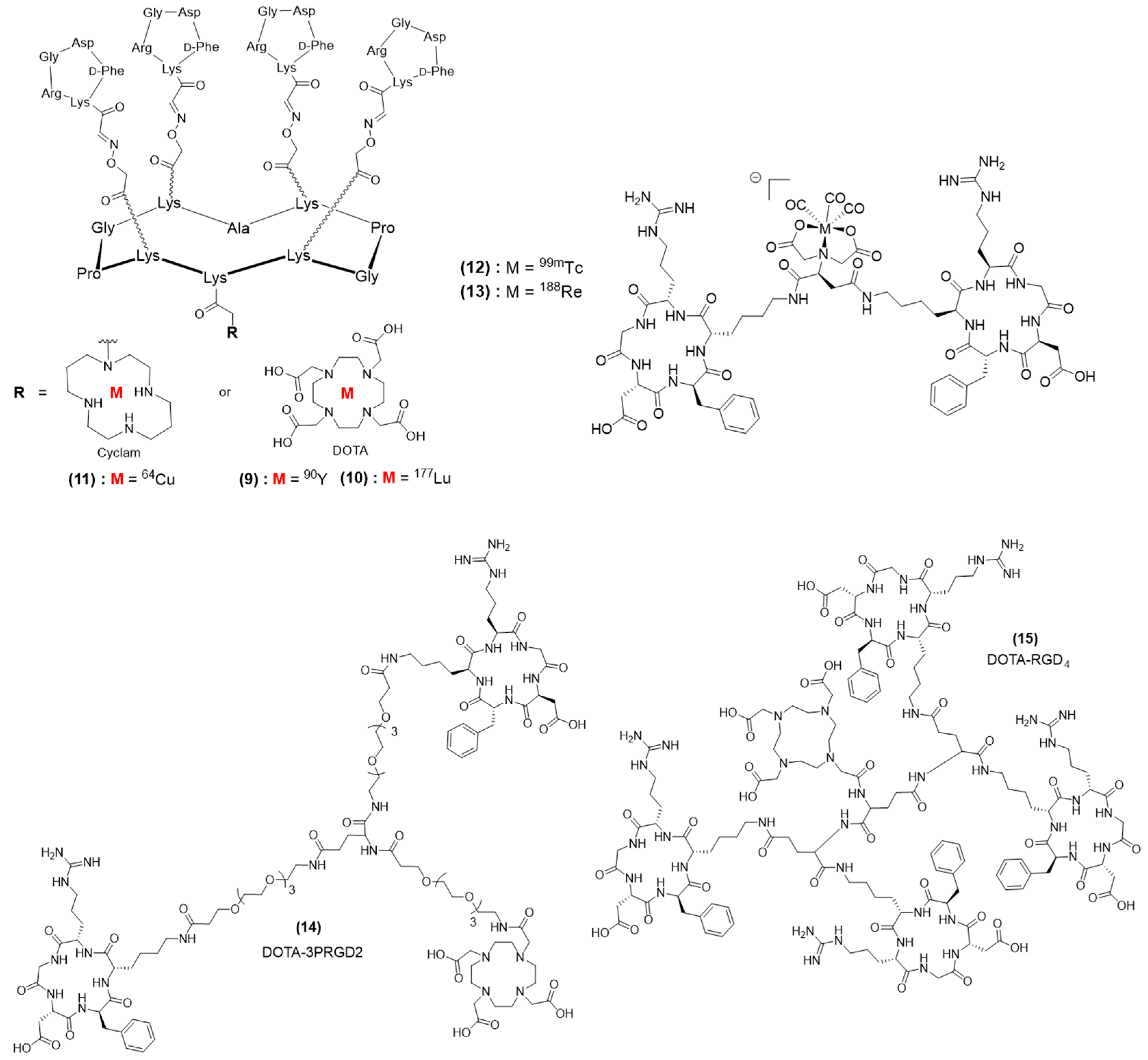
4. Multimeric RGD-Based Theranostic Systems
4.1. Multimeric RGD Theranostic Systems Combining Multiple Effectors
4.2. Multimeric RGD Systems with a Single Theranostic Effector
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody therapy of cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef]
- Chari, R.V.J.; Miller, M.L.; Widdison, W.C. Antibody-Drug Conjugates: An Emerging Concept in Cancer Therapy. Angew. Chem. Int. Ed. 2014, 53, 3796–3827. [Google Scholar] [CrossRef] [PubMed]
- Elgundi, Z.; Reslan, M.; Cruz, E.; Sifniotis, V.; Kayser, V. The state-of-play and future of antibody therapeutics. Adv. Drug Deliv. Rev. 2017, 122, 2–19. [Google Scholar] [CrossRef] [PubMed]
- Vivès, E.; Schmidt, J.; Pèlegrin, A. Cell-penetrating and cell-targeting peptides in drug delivery. Biochim. Biophys. Acta (BBA) Rev. Cancer 2008, 1786, 126–138. [Google Scholar] [CrossRef] [PubMed]
- Pierschbacher, M.D.; Ruoslahti, E. Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule. Nature 1984, 309, 30–33. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Oldberg, A.; Hayman, E.G.; Pierschbacher, M.D.; Ruoslahti, E. Complete amino acid sequence of human vitronectin deduced from cDNA. Similarity of cell attachment sites in vitronectin and fibronectin. EMBO J. 1985, 4, 2519–2524. [Google Scholar] [CrossRef]
- Grant, D.S.; Tashiro, K.-I.; Segui-Real, B.; Yamada, Y.; Martin, G.R.; Kleinman, H.K. Two different laminin domains mediate the differentiation of human endothelial cells into capillary-like structures in vitro. Cell 1989, 58, 933–943. [Google Scholar] [CrossRef]
- Pierschbacher, M.D.; Ruoslahti, E. Influence of stereochemistry of the sequence Arg-Gly-Asp-Xaa on binding specificity in cell adhesion. J. Biol. Chem. 1987, 262, 17294–17298. [Google Scholar] [CrossRef] [PubMed]
- Aumailley, M.; Gurrath, M.; Müller, G.; Calvete, J.; Timpl, R.; Kessler, H. Arg-Gly-Asp constrained within cyclic pentapoptides Strong and selective inhibitors of cell adhesion to vitronectin and laminin fragment P1. FEBS Lett. 1991, 291, 50–54. [Google Scholar] [CrossRef] [PubMed]
- Kessler, H. Conformation and Biological Activity of Cyclic Peptides. Angew. Chem. Int. Ed. 1982, 21, 512–523. [Google Scholar] [CrossRef]
- Dechantsreiter, M.A.; Planker, E.; Mathä, B.; Lohof, E.; Hölzemann, G.; Jonczyk, A.; Goodman, S.L.; Kessler, H. N-Methylated Cyclic RGD Peptides as Highly Active and Selective αVβ3 Integrin Antagonists. J. Med. Chem. 1999, 42, 3033–3040. [Google Scholar] [CrossRef] [PubMed]
- Mason, W.P. End of the road: Confounding results of the CORE trial terminate the arduous journey of cilengitide for glioblastoma. Neuro-Oncology 2015, 17, 634–635. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chen, X.; Yu, J.; Yuan, S. Preliminary Clinical Application of RGD-Containing Peptides as PET Radiotracers for Imaging Tumors. Front. Oncol. 2022, 12, 837952. [Google Scholar] [CrossRef]
- Battistini, L.; Bugatti, K.; Sartori, A.; Curti, C.; Zanardi, F. RGD Peptide-Drug Conjugates as Effective Dual Targeting Platforms: Recent Advances. Eur. J. Org. Chem. 2021, 2021, 2506–2528. [Google Scholar] [CrossRef]
- Mammen, M.; Choi, S.K.; Whitesides, G.M. Polyvalent interactions in biological systems: Implications for design and use of multivalent ligands and inhibitors. Angew. Chem. Int. Ed. 1998, 37, 2754–2794. [Google Scholar] [CrossRef]
- Maynard, H.D.; Okada, S.Y.; Grubbs, R.H. Inhibition of Cell Adhesion to Fibronectin by Oligopeptide-Substituted Polynorbornenes. J. Am. Chem. Soc. 2001, 123, 1275–1279. [Google Scholar] [CrossRef] [PubMed]
- Marchi-Artzner, V.; Lorz, B.; Hellerer, U.; Kantlehner, M.; Kessler, H.; Sackmann, E. Selective Adhesion of Endothelial Cells to Artificial Membranes with a Synthetic RGD-Lipopeptide. Chem. A Eur. J. 2001, 7, 1095–1101. [Google Scholar] [CrossRef]
- Lin, W.; Ma, G.; Kampf, N.; Yuan, Z.; Chen, S. Development of Long-Circulating Zwitterionic Cross-Linked Micelles for Active-Targeted Drug Delivery. Biomacromolecules 2016, 17, 2010–2018. [Google Scholar] [CrossRef]
- Kok, R.J.; Schraa, A.J.; Bos, E.J.; Moorlag, H.E.; Ásgeirsdóttir, S.A.; Everts, M.; Meijer, D.K.F.; Molema, G. Preparation and Functional Evaluation of RGD-Modified Proteins as αvβ3 Integrin Directed Therapeutics. Bioconjugate Chem. 2001, 13, 128–135. [Google Scholar] [CrossRef]
- Thumshirn, G.; Hersel, U.; Goodman, S.L.; Kessler, H. Multimeric Cyclic RGD Peptides as Potential Tools for Tumor Targeting: Solid-Phase Peptide Synthesis and Chemoselective Oxime Ligation. Chem. Eur. J. 2003, 9, 2717–2725. [Google Scholar] [CrossRef]
- Boturyn, D.; Coll, J.-L.; Garanger, E.; Favrot, M.-C.; Dumy, P. Template Assembled Cyclopeptides as Multimeric System for Integrin Targeting and Endocytosis. J. Am. Chem. Soc. 2004, 126, 5730–5739. [Google Scholar] [CrossRef]
- Sancey, L.; Garanger, E.; Foillard, S.; Schoehn, G.; Hurbin, A.; Albiges-Rizo, C.; Boturyn, D.; Souchier, C.; Grichine, A.; Dumy, P.; et al. Clustering and internalization of integrin αVβ3 with a tetrameric RGD-synthetic peptide. Mol. Ther. 2009, 17, 837–843. [Google Scholar] [CrossRef]
- Gestwicki, J.E.; Cairo, C.W.; Strong, L.E.; Oetjen, K.A.; Kiessling, L.L. Influencing Receptor−Ligand Binding Mechanisms with Multivalent Ligand Architecture. J. Am. Chem. Soc. 2002, 124, 14922–14933. [Google Scholar] [CrossRef] [PubMed]
- Cairo, C.W.; Gestwicki, J.E.; Kanai, M.; Kiessling, L.L. Control of Multivalent Interactions by Binding Epitope Density. J. Am. Chem. Soc. 2002, 124, 1615–1619. [Google Scholar] [CrossRef]
- Casi, G.; Neri, D. Antibody–Drug Conjugates and Small Molecule–Drug Conjugates: Opportunities and Challenges for the Development of Selective Anticancer Cytotoxic Agents. J. Med. Chem. 2015, 58, 8751–8761. [Google Scholar] [CrossRef] [PubMed]
- Boturyn, D.; Dumy, P. Tumor Targeting with RGD Peptide Ligands-Design of New Molecular Conjugates for Imaging and Therapy of Cancers. Anti-Cancer Agents Med. Chem. 2007, 7, 552–558. [Google Scholar] [CrossRef]
- Liu, S. Radiolabeled Multimeric Cyclic RGD Peptides as Integrin αvβ3 Targeted Radiotracers for Tumor Imaging. Mol. Pharm. 2006, 3, 472–487. [Google Scholar] [CrossRef] [PubMed]
- Meyer, A.; Auernheimer, J.; Modlinger, A.; Kessler, H. Targeting RGD Recognizing Integrins: Drug Development, Biomaterial Research, Tumor Imaging and Targeting. Curr. Pharm. Des. 2006, 12, 2723–2747. [Google Scholar] [CrossRef]
- Chen, X.; Plasencia, C.; Hou, Y.; Neamati, N. Synthesis and Biological Evaluation of Dimeric RGD Peptide−Paclitaxel Conjugate as a Model for Integrin-Targeted Drug Delivery. J. Med. Chem. 2005, 48, 1098–1106. [Google Scholar] [CrossRef]
- Conibear, A.C.; Hager, S.; Mayr, J.; Klose, M.H.M.; Keppler, B.K.; Kowol, C.R.; Heffeter, P.; Becker, C.F.W. Multifunctional αvβ6 Integrin-Specific Peptide–Pt(IV) Conjugates for Cancer Cell Targeting. Bioconjugate Chem. 2017, 28, 2429–2439. [Google Scholar] [CrossRef]
- Garanger, E.; Boturyn, D.; Jin, Z.; Dumy, P.; Favrot, M.-C.; Coll, J.-L. New Multifunctional Molecular Conjugate Vector for Targeting, Imaging, and Therapy of Tumors. Mol. Ther. 2005, 12, 1168–1175. [Google Scholar] [CrossRef]
- Galibert, M.; Sancey, L.; Renaudet, O.; Coll, J.-L.; Dumy, P.; Boturyn, D. Application of click–click chemistry to the synthesis of new multivalent RGD conjugates. Org. Biomol. Chem. 2010, 8, 5133–5138. [Google Scholar] [CrossRef]
- Dufort, S.; Sancey, L.; Hurbin, A.; Foillard, S.; Boturyn, D.; Dumy, P.; Coll, J.-L. Targeted delivery of a proapoptotic peptide to tumors in vivo. J. Drug Target. 2011, 19, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Karageorgis, A.; Claron, M.; Jugé, R.; Aspord, C.; Thoreau, F.; Leloup, C.; Kucharczak, J.; Plumas, J.; Henry, M.; Hurbin, A.; et al. Systemic Delivery of Tumor-Targeted Bax-Derived Membrane-Active Peptides for the Treatment of Melanoma Tumors in a Humanized SCID Mouse Model. Mol. Ther. 2017, 25, 534–546. [Google Scholar] [CrossRef]
- Borbély, A.; Thoreau, F.; Figueras, E.; Kadri, M.; Coll, J.-L.; Boturyn, D.; Sewald, N. Synthesis and Biological Characterization of Monomeric and Tetrameric RGD-Cryptophycin Conjugates. Chem. A Eur. J. 2020, 26, 2602–2605. [Google Scholar] [CrossRef] [PubMed]
- Pilkington-Miksa, M.; Arosio, D.; Battistini, L.; Belvisi, L.; De Matteo, M.; Vasile, F.; Burreddu, P.; Carta, P.; Rassu, G.; Perego, P.; et al. Design, synthesis and biological evaluation of novel cRGD-paclitaxel conjugates for integrin-assisted drug delivery. Org. Biomol. Chem. 2012, 23, 1610–1622. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, A.; Arosio, D.; Perego, P.; De Cesare, M.; Carenini, N.; Zaffaroni, N.; De Matteo, M.; Manzoni, L. Design, synthesis and biological evaluation of novel dimeric and tetrameric cRGD-paclitaxel conjugates for integrin-assisted drug delivery. Org. Biomol. Chem 2015, 13, 7530–7541. [Google Scholar] [CrossRef]
- Sartori, A.; Portioli, E.; Battistini, L.; Calorini, L.; Pupi, A.; Vacondio, F.; Arosio, D.; Bianchini, F.; Zanardi, F. Synthesis of Novel c(AmpRGD)–Sunitinib Dual Conjugates as Molecular Tools Targeting the αvβ3 Integrin/VEGFR2 Couple and Impairing Tumor-Associated Angiogenesis. J. Med. Chem. 2017, 60, 248–262. [Google Scholar] [CrossRef]
- Dias, A.R.M.; Pina, A.; Corso, A.D.; Arosio, D.; Belvisi, L.; Pignataro, L.; Caruso, M.; Gennari, C. Multivalency Increases the Binding Strength of RGD Peptidomimetic-Paclitaxel Conjugates to Integrin αV β3. Chem. A Eur. J. 2017, 23, 14410–14415. [Google Scholar] [CrossRef]
- Albanese, A.; Tang, P.S.; Chan, W.C. The effect of nanoparticle size, shape, and surface chemistry on biological systems. Annu. Rev. Biomed. Eng. 2012, 14, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Gratton, S.E.; Ropp, P.A.; Pohlhaus, P.D.; Luft, J.C.; Madden, V.J.; Napier, M.E.; DeSimone, J.M. The effect of particle design on cellular internalization pathways. Proc. Natl. Acad. Sci. USA 2008, 105, 11613–11618. [Google Scholar] [CrossRef]
- Chithrani, B.D.; Ghazani, A.A.; Chan, W.C.W. Determining the Size and Shape Dependence of Gold Nanoparticle Uptake into Mammalian Cells. Nano Lett. 2006, 6, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Huo, S.; Ma, H.; Huang, K.; Liu, J.; Wei, T.; Jin, S.; Zhang, J.; He, S.; Liang, X.-J. Superior Penetration and Retention Behavior of 50 nm Gold Nanoparticles in Tumors. Cancer Res 2013, 73, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Lao, J.; Madani, J.; Puértolas, T.; Álvarez, M.; Hernández, A.; Pazo-Cid, R.; Artal, Á.; Torres, A.A. Liposomal Doxorubicin in the Treatment of Breast Cancer Patients: A Review. J. Drug Deliv. 2013, 2013, 4564091. [Google Scholar] [CrossRef] [PubMed]
- Kalyane, D.; Raval, N.; Maheshwari, R.; Tambe, V.; Kalia, K.; Tekade, R.K. Employment of enhanced permeability and retention effect (EPR): Nanoparticle-based precision tools for targeting of therapeutic and diagnostic agent in cancer. Mater. Sci. Eng. C 2019, 98, 1252–1276. [Google Scholar] [CrossRef]
- Schiffelers, R.; Ansari, A.; Xu, J.; Zhou, Q.; Tang, Q.; Storm, G.; Molema, G.; Lu, P.Y.; Scaria, P.V.; Woodle, M.C. Cancer siRNA therapy by tumor selective delivery with ligand-targeted sterically stabilized nanoparticle. Nucleic Acids Res. 2004, 32, e149. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Wu, J.; Liu, Y.; Yu, M.; Zhao, L.; Zhu, X.; Bhasin, S.; Li, Q.; Ha, E.; Shi, J.; et al. Ultra-pH-Responsive and Tumor-Penetrating Nanoplatform for Targeted siRNA Delivery with Robust Anti-Cancer Efficacy. Angew. Chem. Int. Ed. 2016, 55, 7091–7094. [Google Scholar] [CrossRef]
- Xiong, X.-B.; Mahmud, A.; Uludag, H.; Lavasanifar, A. Multifunctional Polymeric Micelles for Enhanced Intracellular Delivery of Doxorubicin to Metastatic Cancer Cells. Pharm. Res. 2008, 25, 2555–2566. [Google Scholar] [CrossRef]
- Zhang, L.; Zhu, S.; Qian, L.; Pei, Y.; Qiu, Y.; Jiang, Y. RGD-modified PEG–PAMAM–DOX conjugates: In vitro and in vivo studies for glioma. Eur. J. Pharm. Biopharm. 2011, 79, 232–240. [Google Scholar] [CrossRef]
- Bahadur, K.C.R.; Thapa, B.; Xu, P. pH and redox dual responsive nanoparticle for nuclear targeted drug delivery. Mol. Pharm. 2012, 9, 2719–2729. [Google Scholar] [CrossRef]
- Chen, W.; Zou, Y.; Zhong, Z.; Haag, R. Cyclo(RGD)-Decorated Reduction-Responsive Nanogels Mediate Targeted Chemotherapy of Integrin Overexpressing Human Glioblastoma In Vivo. Small 2017, 13. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhang, X.; Liu, Y.; Liu, C.; Jiang, B.; Jiang, Y. Tumor penetrability and anti-angiogenesis using iRGD-mediated delivery of doxorubicin-polymer conjugates. Biomaterials 2014, 35, 8735–8747. [Google Scholar] [CrossRef] [PubMed]
- Eldar-Boock, A.; Miller, K.; Sanchis, J.; Lupu, R.; Vicent, M.J.; Satchi-Fainaro, R. Integrin-assisted drug delivery of nano-scaled polymer therapeutics bearing paclitaxel. Biomaterials 2011, 32, 3862–3874. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Liu, Y.; Su, S.; Li, W.; Chen, C.; Wu, Y. Anti-tumor activity of paclitaxel through dual-targeting carrier of cyclic RGD and transferrin conjugated hyperbranched copolymer nanoparticles. Biomaterials 2012, 33, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Graf, N.; Bielenberg, D.R.; Kolishetti, N.; Muus, C.; Banyard, J.; Farokhzad, O.C.; Lippard, S.J. αVβ3 Integrin-Targeted PLGA-PEG Nanoparticles for Enhanced Anti-tumor Efficacy of a Pt(IV) Prodrug. ACS Nano 2012, 6, 4530–4539. [Google Scholar] [CrossRef]
- Yadav, A.S.; Radharani, N.N.V.; Gorain, M.; Bulbule, A.; Shetti, D.; Roy, G.; Baby, T.; Kundu, G.C. RGD functionalized chitosan nanoparticle mediated targeted delivery of raloxifene selectively suppresses angiogenesis and tumor growth in breast cancer. Nanoscale 2020, 12, 10664–10684. [Google Scholar] [CrossRef]
- Song, S.; Mao, G.; Du, J.; Zhu, X. Novel RGD containing, temozolomide-loading nanostructured lipid carriers for glioblastoma multiforme chemotherapy. Drug Deliv. 2016, 23, 1404–1408. [Google Scholar] [CrossRef]
- Murphy, E.A.; Majeti, B.K.; Barnes, L.A.; Makale, M.; Weis, S.M.; Lutu-Fuga, K.; Wrasidlo, W.; Cheresh, D.A. Nanoparticle-mediated drug delivery to tumor vasculature suppresses metastasis. Proc. Natl. Acad. Sci. USA 2008, 105, 9343–9348. [Google Scholar] [CrossRef]
- Cao, Y.; Zhou, Y.; Zhuang, Q.; Cui, L.; Xu, X.; Xu, R.; He, X. Anti-tumor effect of RGD modified PTX loaded liposome on prostatic cancer. Int. J. Clin. Exp. Med. 2015, 8, 12182–12191. [Google Scholar]
- Zhang, J.; Yuan, Z.-F.; Wang, Y.; Chen, W.-H.; Luo, G.-F.; Cheng, S.-X.; Zhuo, R.-X.; Zhang, X.-Z. Multifunctional Envelope-Type Mesoporous Silica Nanoparticles for Tumor-Triggered Targeting Drug Delivery. J. Am. Chem. Soc. 2013, 135, 5068–5073. [Google Scholar] [CrossRef]
- Xu, H.; Wang, Z.; Li, Y.; Guo, Y.; Zhou, H.; Li, Y.; Wu, F.; Zhang, L.; Yang, X.; Lu, B.; et al. Preparation and characterization of a dual-receptor mesoporous silica nanoparticle–hyaluronic acid–RGD peptide targeting drug delivery system. RSC Adv. 2016, 6, 40427–40435. [Google Scholar] [CrossRef]
- Lei, Q.; Qiu, W.-X.; Hu, J.-J.; Cao, P.-X.; Zhu, C.-H.; Cheng, H.; Zhang, X.-Z. Multifunctional Mesoporous Silica Nanoparticles with Thermal-Responsive Gatekeeper for NIR Light-Triggered Chemo/Photothermal-Therapy. Small 2016, 12, 4286–4298. [Google Scholar] [CrossRef]
- Robinson, J.T.; Tabakman, S.M.; Liang, Y.; Wang, H.; Casalongue, H.S.; Vinh, D.; Dai, H. Ultrasmall Reduced Graphene Oxide with High Near-Infrared Absorbance for Photothermal Therapy. J. Am. Chem. Soc. 2011, 133, 6825–6831. [Google Scholar] [CrossRef] [PubMed]
- Paris, J.L.; Villaverde, G.; Gómez-Graña, S.; Vallet-Regí, M. Nanoparticles for multimodal antivascular therapeutics: Dual drug release, photothermal and photodynamic therapy. Acta Biomater. 2020, 101, 459–468. [Google Scholar] [CrossRef]
- Schleich, N.; Po, C.; Jacobs, D.; Ucakar, B.; Gallez, B.; Danhier, F.; Préat, V. Comparison of active, passive and magnetic targeting to tumors of multifunctional paclitaxel/SPIO-loaded nanoparticles for tumor imaging and therapy. J. Control. Release 2014, 194, 82–91. [Google Scholar] [CrossRef]
- Yang, K.; Liu, Y.; Liu, Y.; Zhang, Q.; Kong, C.; Yi, C.; Zhou, Z.; Wang, Z.; Zhang, G.; Zhang, Y.; et al. Cooperative Assembly of Magneto-Nanovesicles with Tunable Wall Thickness and Permeability for MRI-Guided Drug Delivery. J. Am. Chem. Soc. 2018, 140, 4666–4677. [Google Scholar] [CrossRef]
- Zhang, P.; Wang, C.; Zhao, J.; Xiao, A.; Shen, Q.; Li, L.; Li, J.; Zhang, J.; Min, Q.; Chen, J.; et al. Near Infrared-Guided Smart Nanocarriers for MicroRNA-Controlled Release of Doxorubicin/siRNA with Intracellular ATP as Fuel. ACS Nano 2016, 10, 3637–3647. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Valdepérez, D.; Jin, Q.; Yang, B.; Li, Z.; Wu, Y.; Pelaz, B.; Parak, W.J.; Ji, J. Dual Enzymatic Reaction-Assisted Gemcitabine Delivery Systems for Programmed Pancreatic Cancer Therapy. ACS Nano 2017, 11, 1281–1291. [Google Scholar] [CrossRef] [PubMed]
- Kunjachan, S.; Pola, R.; Gremse, F.; Theek, B.; Ehling, J.; Moeckel, D.; Hermanns-Sachweh, B.; Pechar, M.; Ulbrich, K.; Hennink, W.E.; et al. Passive versus Active Tumor Targeting Using RGD- and NGR-Modified Polymeric Nanomedicines. Nano Lett. 2014, 14, 972–981. [Google Scholar] [CrossRef]
- Xu, X.; Saw, P.E.; Tao, W.; Li, Y.; Ji, X.; Bhasin, S.; Liu, Y.; Ayyash, D.; Rasmussen, J.; Huo, M.; et al. ROS-Responsive Polyprodrug Nanoparticles for Triggered Drug Delivery and Effective Cancer Therapy. Adv. Mater. 2017, 29, 1700141. [Google Scholar] [CrossRef]
- Francisco, J.A.; Cerveny, C.G.; Meyer, D.L.; Mixan, B.J.; Klussman, K.; Chace, D.F.; Rejniak, S.X.; Gordon, K.A.; DeBlanc, R.; Toki, B.E.; et al. cAC10-vcMMAE, an anti-CD30–monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood 2003, 102, 1458–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borbély, A.; Figueras, E.; Martins, A.; Esposito, S.; Auciello, G.; Monteagudo, E.; Di Marco, A.; Summa, V.; Cordella, P.; Perego, R.; et al. Synthesis and Biological Evaluation of RGD⁻Cryptophycin Conjugates for Targeted Drug Delivery. Pharmaceutics 2019, 11, 151. [Google Scholar] [CrossRef] [PubMed]
- Battersby, J.E.; Hancock, W.S.; Canova-Davis, E.; Oeswein, J.; O’onnor, B. Diketopiperazine Formation and N-Terminal Degradation in Recombinant Human Growth Hormone. Int. J. Pept. Protein Res. 1994, 44, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Caculitan, N.G.; Chuh, J.D.C.; Ma, Y.; Zhang, D.; Kozak, K.R.; Liu, Y.; Pillow, T.H.; Sadowsky, J.; Cheung, T.K.; Phung, Q.; et al. Cathepsin B Is Dispensable for Cellular Processing of Cathepsin B-Cleavable Antibody–Drug Conjugates. Cancer Res 2017, 77, 7027–7037. [Google Scholar] [CrossRef]
- Borbély, A.; Figueras, E.; Martins, A.; Bodero, L.; Dias, A.R.M.; Rivas, P.L.; Pina, A.; Arosio, D.; Gallinari, P.; Frese, M.; et al. Conjugates of Cryptophycin and RGD or iso DGR Peptidomimetics for Targeted Drug Delivery. Chemistryopen 2019, 8, 737–742. [Google Scholar] [CrossRef] [PubMed]
- Dal Corso, A.; Caruso, M.; Belvisi, L.; Arosio, D.; Piarulli, U.; Albanese, C.; Gasparri, F.; Marsiglio, A.; Sola, F.; Troiani, S.; et al. Synthesis and Biological Evaluation of RGD Peptidomimetic-Paclitaxel Conjugates Bearing Lysosomally Cleavable Linkers. Chem. A Eur. J. 2015, 21, 6921–6929. [Google Scholar] [CrossRef] [PubMed]
- Dias, A.R.M.; Bodero, L.; Martins, A.; Arosio, D.; Gazzola, S.; Belvisi, L.; Pignataro, L.; Steinkühler, C.; Corso, A.D.; Gennari, C.; et al. Synthesis and Biological Evaluation of RGD and iso DGR–Monomethyl Auristatin Conjugates Targeting Integrin α V β 3. ChemMedChem 2019, 14, 938–942. [Google Scholar] [CrossRef]
- Bodero, L.; Rivas, P.L.; Korsak, B.; Hechler, T.; Pahl, A.; Muller, C.; Arosio, D.; Pignataro, L.; Gennari, C.; Piarulli, U. Synthesis and biological evaluation of RGD and isoDGR peptidomimetic-α-amanitin conjugates for tumor-targeting. Beilstein J. Org. Chem. 2018, 14, 407–415. [Google Scholar] [CrossRef]
- Rivas, P.L.; Ranđelović, I.; Dias, A.R.M.; Pina, A.; Arosio, D.; Tóvári, J.; Mező, G.; Corso, A.D.; Pignataro, L.; Gennari, C.; et al. Synthesis and Biological Evaluation of Paclitaxel Conjugates Involving Linkers Cleavable by Lysosomal Enzymes and αV β3 -Integrin Ligands for Tumor Targeting. Eur. J. Org. Chem. 2018, 2018, 2902–2909. [Google Scholar] [CrossRef]
- Dias, A.R.M.; Pina, A.; Dean, A.; Lerchen, H.-G.; Caruso, M.; Gasparri, F.; Fraietta, I.; Troiani, S.; Arosio, D.; Belvisi, L.; et al. Neutrophil Elastase Promotes Linker Cleavage and Paclitaxel Release from an Integrin-Targeted Conjugate. Chem. A Eur. J. 2019, 25, 1696–1700. [Google Scholar] [CrossRef]
- Ren, H.; Zeng, X.-Z.; Zhao, X.-X.; Hou, D.-Y.; Yao, H.; Yaseen, M.; Zhao, L.; Xu, W.-H.; Wang, H.; Li, L.-L. A bioactivated in vivo assembly nanotechnology fabricated NIR probe for small pancreatic tumor intraoperative imaging. Nat. Commun. 2022, 13, 1–13. [Google Scholar] [CrossRef]
- Han, H.; Jin, Q.; Wang, Y.; Chen, Y.; Ji, J. The rational design of a gemcitabine prodrug with AIE-based intracellular light-up characteristics for selective suppression of pancreatic cancer cells. Chem. Commun. 2015, 51, 17435–17438. [Google Scholar] [CrossRef] [PubMed]
- Pina, A.; Corso, A.D.; Caruso, M.; Belvisi, L.; Arosio, D.; Zanella, S.; Gasparri, F.; Albanese, C.; Cucchi, U.; Fraietta, I.; et al. Targeting Integrin αV β3 with Theranostic RGD-Camptothecin Conjugates Bearing a Disulfide Linker: Biological Evaluation Reveals a Complex Scenario. ChemistrySelect 2017, 2, 4759–4766. [Google Scholar] [CrossRef]
- Nam, E.J.; Kang, J.H.; Sung, S.; Sa, K.H.; Kim, K.H.; Seo, J.S.; Kim, J.-H.; Han, S.W.; Kim, I.S.; Kang, Y.M. A Matrix Metalloproteinase 1-Cleavable Composite Peptide Derived from Transforming Growth Factor β-Inducible Gene h3 Potently Inhibits Collagen-Induced Arthritis. Arthritis Rheum. 2013, 65, 1753–1763. [Google Scholar] [CrossRef] [PubMed]
- Wenk, C.H.F.; Josserand, V.; Dumy, P.; Coll, J.-L.; Boturyn, D. Integrin and matrix metalloprotease dual-targeting with an MMP substrate–RGD conjugate. Org. Biomol. Chem. 2012, 11, 448–452. [Google Scholar] [CrossRef]
- Rivas, P.L.; Müller, C.; Breunig, C.; Hechler, T.; Pahl, A.; Arosio, D.; Belvisi, L.; Pignataro, L.; Corso, A.D.; Gennari, C. β-Glucuronidase triggers extracellular MMAE release from an integrin-targeted conjugate. Org. Biomol. Chem. 2019, 17, 4705–4710. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Diao, Y.; Li, W.; Yang, Z.; Zhang, L.; Chen, Z.; Wu, Y. RGD peptide conjugation results in enhanced antitumor activity of PD0325901 against glioblastoma by both tumor-targeting delivery and combination therapy. Int. J. Pharm. 2016, 505, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-Y.; Liu, L.-H.; Rong, L.; Qiu, W.-X.; Jia, H.-Z.; Li, B.; Li, F.; Zhang, X.-Z. A Dual-FRET-Based Versatile Prodrug for Real-Time Drug Release Monitoring and In Situ Therapeutic Efficacy Evaluation. Adv. Funct. Mater. 2015, 25, 7317–7326. [Google Scholar] [CrossRef]
- Gilad, Y.; Noy, E.; Senderowitz, H.; Albeck, A.; Firer, M.A.; Gellerman, G. Synthesis, biological studies and molecular dynamics of new anticancer RGD-based peptide conjugates for targeted drug delivery. Bioorg. Med. Chem. 2016, 24, 294–303. [Google Scholar] [CrossRef]
- Luciano, M.P.; Nourian, S.; Gorka, A.P.; Nani, R.R.; Nagaya, T.; Kobayashi, H.; Schnermann, M.J. A near-infrared light-mediated cleavable linker strategy using the heptamethine cyanine chromophore. Methods Enzymol. 2020, 641, 245–275. [Google Scholar] [CrossRef] [PubMed]
- Klausen, M.; Blanchard-Desce, M. Two-photon uncaging of bioactive compounds: Starter guide to an efficient IR light switch. J. Photochem. Photobiol. C: Photochem. Rev. 2021, 48. [Google Scholar] [CrossRef]
- Bernard, S.; Audisio, D.; Riomet, M.; Bregant, S.; Sallustrau, A.; Plougastel, L.; Decuypere, E.; Gabillet, S.; Kumar, R.A.; Elyian, J.; et al. Bioorthogonal Click and Release Reaction of Iminosydnones with Cycloalkynes. Angew. Chem. Int. Ed. 2017, 56, 15612–15616. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Pan, Z.; Yu, B.; De La Cruz, L.K.; Zheng, Y.; Ke, B.; Wang, B. Click and release: Bioorthogonal approaches to “on-demand” activation of prodrugs. Chem. Soc. Rev. 2019, 48, 1077–1094. [Google Scholar] [CrossRef]
- Versteegen, R.M.; Rossin, R.; Hoeve, W.T.; Janssen, H.M.; Robillard, M.S. Click to Release: Instantaneous Doxorubicin Elimination upon Tetrazine Ligation. Angew. Chem. Int. Ed. 2013, 52, 14112–14116. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Li, Z.; Chen, K.; Wu, Z.; He, L.; Neamati, N.; Chen, X. Evaluation of biodistribution and anti-tumor effect of a dimeric RGD peptide–paclitaxel conjugate in mice with breast cancer. Eur. J. Nucl. Med. 2008, 35, 1489–1498. [Google Scholar] [CrossRef]
- Ryppa, C.; Mann-Steinberg, H.; Biniossek, M.L.; Satchi-Fainaro, R.; Kratz, F. In vitro and in vivo evaluation of a paclitaxel conjugate with the divalent peptide E-[c(RGDfK)2] that targets integrin αvβ3. Int. J. Pharm. 2009, 368, 89–97. [Google Scholar] [CrossRef]
- Ryppa, C.; Mann-Steinberg, H.; Fichtner, I.; Weber, H.; Satchi-Fainaro, R.; Biniossek, M.L.; Kratz, F. In Vitro and in Vivo Evaluation of Doxorubicin Conjugates with the Divalent Peptide E-[c(RGDfK)2] that Targets Integrin αvβ3. Bioconjugate Chem. 2008, 19, 1414–1422. [Google Scholar] [CrossRef]
- Pozzo, A.D.; Esposito, E.; Ni, M.; Muzi, L.; Pisano, C.; Bucci, F.; Vesci, L.; Castorina, M.; Penco, S. Conjugates of a Novel 7-Substituted Camptothecin with RGD-Peptides as αvβ3 Integrin Ligands: An Approach to Tumor-Targeted Therapy. Bioconjugate Chem. 2010, 21, 1956–1967. [Google Scholar] [CrossRef]
- Polyak, D.; Ryppa, C.; Eldar-Boock, A.; Ofek, P.; Many, A.; Licha, K.; Kratz, F.; Satchi-Fainaro, R. Development of PEGylated doxorubicin-E-[c(RGDfK)2] conjugate for integrin-targeted cancer therapy. Polym. Adv. Technol. 2011, 22, 103–113. [Google Scholar] [CrossRef]
- Lu, J.; Jiang, F.; Lu, A.; Zhang, G. Linkers Having a Crucial Role in Antibody–Drug Conjugates. Int. J. Mol. Sci. 2016, 17, 561. [Google Scholar] [CrossRef]
- Bandara, N.; Reynolds, T.J.S.; Schehr, R.; Bandari, R.P.; Diebolder, P.; Krieger, S.; Xu, J.; Miao, Y.; Rogers, B.E.; Smith, C.J. Matched-pair, 86Y/90Y-labeled, bivalent RGD/bombesin antagonist, [RGD-Glu-[DO3A]-6-Ahx-RM2], as a potential theranostic agent for prostate cancer. Nucl. Med. Biol. 2018, 62–63, 71–77. [Google Scholar] [CrossRef]
- Lee, M.H.; Kim, J.Y.; Han, J.H.; Bhuniya, S.; Sessler, J.L.; Kang, C.; Kim, J.S. Direct Fluorescence Monitoring of the Delivery and Cellular Uptake of a Cancer-Targeted RGD Peptide-Appended Naphthalimide Theragnostic Prodrug. J. Am. Chem. Soc. 2012, 134, 12668–12674. [Google Scholar] [CrossRef] [PubMed]
- Nahrwold, M.; Weiß, C.; Bogner, T.; Mertink, F.; Conradi, J.; Sammet, B.; Palmisano, R.; Gracia, S.R.; Preuße, T.; Sewald, N. Conjugates of Modified Cryptophycins and RGD-Peptides Enter Target Cells by Endocytosis. J. Med. Chem. 2013, 56, 1853–1864. [Google Scholar] [CrossRef]
- Yu, G.; Yang, Z.; Fu, X.; Yung, B.C.; Yang, J.; Mao, Z.; Shao, L.; Hua, B.; Liu, Y.; Zhang, F.; et al. Polyrotaxane-based supramolecular theranostics. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Wei, Y.; Sun, Y.; Bao, J.; Yao, F.; Li, Z.; Meng, F.; Hu, C.; Storm, G.; Zhong, Z. Cyclic RGD-Functionalized and Disulfide-Crosslinked Iodine-Rich Polymersomes as a Robust and Smart Theranostic Agent for Targeted CT Imaging and Chemotherapy of Tumor. Theranostics 2019, 9, 8061–8072. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Wang, K.; Sun, L.; Sun, B.; Yang, M.; Chen, H.; Wang, Y.; Sun, J.; Dong, L. Application of graphene quantum dots for simultaneous fluorescence imaging and tumor-targeted drug delivery. Sens. Actuators B Chem. 2018, 256, 616–623. [Google Scholar] [CrossRef]
- Bozon-Petitprin, A.; Bacot, S.; Gauchez, A.-S.; Ahmadi, M.; Bourre, J.C.; Marti-Batlle, D.; Perret, P.; Broisat, A.; Riou, L.M.; Claron, M.; et al. Targeted radionuclide therapy with RAFT-RGD radiolabelled with 90Y or 177Lu in a mouse model of αvβ3-expressing tumours. Eur. J. Nucl. Med. 2015, 42, 252–263. [Google Scholar] [CrossRef]
- Jin, Z.-H.; Furukawa, T.; Degardin, M.; Sugyo, A.; Tsuji, A.B.; Yamasaki, T.; Kawamura, K.; Fujibayashi, Y.; Zhang, M.-R.; Boturyn, D.; et al. αVβ3 Integrin-Targeted Radionuclide Therapy with 64Cu-cyclam-RAFT-c(-RGDfK-)4. Mol. Cancer Ther. 2016, 15, 2076–2085. [Google Scholar] [CrossRef]
- Jin, Z.-H.; Tsuji, A.B.; Degardin, M.; Sugyo, A.; Yoshii, Y.; Nagatsu, K.; Zhang, M.-R.; Fujibayashi, Y.; Dumy, P.; Boturyn, D.; et al. Uniform intratumoral distribution of radioactivity produced using two different radioagents, 64Cu-cyclam-RAFT-c(-RGDfK-)4 and 64Cu-ATSM, improves therapeutic efficacy in a small animal tumor model. EJNMMI Res. 2018, 8, 54. [Google Scholar] [CrossRef]
- Jin, Z.-H.; Tsuji, A.B.; Degardin, M.; Sugyo, A.; Obara, S.; Wakizaka, H.; Nagatsu, K.; Hu, K.; Zhang, M.-R.; Dumy, P.; et al. Radiotheranostic Agent 64Cu-cyclam-RAFT-c(-RGDfK-)4 for Management of Peritoneal Metastasis in Ovarian Cancer. Clin. Cancer Res. 2020, 26, 6230–6241. [Google Scholar] [CrossRef]
- Luna-Gutiérrez, M.; Ferro-Flores, G.; Ocampo-García, B.; Jiménez-Mancilla, N.; Morales-Avila, E.; De León-Rodríguez, L.; Isaac-Olivé, K. 177Lu-labeled monomeric, dimeric and multimeric RGD peptides for the therapy of tumors expressing α(ν)β(3) integrins. J. Label. Compd. Radiopharm. 2012, 55, 140–148. [Google Scholar] [CrossRef]
- Shi, J.; Fan, D.; Dong, C.; Liu, H.; Jia, B.; Zhao, H.; Jin, X.; Liu, Z.; Li, F.; Wang, F. Anti-tumor Effect of Integrin Targeted 177Lu-3PRGD2 and Combined Therapy with Endostar. Theranostics 2014, 4, 256–266. [Google Scholar] [CrossRef]
- Lee, B.C.; Moon, B.S.; Kim, J.S.; Jung, J.H.; Park, H.S.; Katzenellenbogen, J.A.; Kim, S.E. Synthesis and biological evaluation of RGD peptides with the99mTc/188Re chelated iminodiacetate core: Highly enhanced uptake and excretion kinetics of theranostics against tumor angiogenesis. RSC Adv. 2012, 3, 782–792. [Google Scholar] [CrossRef]
- Liu, Z.; Shi, J.; Jia, B.; Yu, Z.; Liu, Y.; Zhao, H.; Li, F.; Tian, J.; Chen, X.; Liu, S.; et al. Two 90Y-Labeled Multimeric RGD Peptides RGD4 and 3PRGD2 for Integrin Targeted Radionuclide Therapy. Mol. Pharm. 2011, 8, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Li, S.; Jin, Y.; Wang, J.Y.; Li, W.; Wu, W.; Hong, Z. Multimerization Increases Tumor Enrichment of Peptide–Photosensitizer Conjugates. Molecules 2019, 24, 817. [Google Scholar] [CrossRef]
- Liu, S.; Vorobyova, I.; Park, R.; Conti, P.S. Biodistribution and Radiation Dosimetry of the Integrin Marker 64Cu-BaBaSar-RGD2 Determined from Whole-Body PET/CT in a Non-Human Primate. Front. Phys. 2017, 5. [Google Scholar] [CrossRef]
- Lopez-Rodriguez, V.; Gaspar-Carcamo, R.; Pedraza-Lopez, M.; Rojas-Calderon, E.; de Murphy, C.A.; Ferro-Flores, G.; Avila-Rodriguez, M. Preparation and preclinical evaluation of 66Ga-DOTA-E(c(RGDfK))2 as a potential theranostic radiopharmaceutical. Nucl. Med. Biol. 2015, 42, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Li, R.; Deng, H.; Li, Y.; Cui, Y.; Zhang, H.; Dai, W.; He, B.; Zheng, Y.; Wang, X.; et al. Receptor mediated transcytosis in biological barrier: The influence of receptor character and their ligand density on the transmembrane pathway of active-targeting nanocarriers. Biomaterials 2018, 180, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.; Ullah, I.; Kim, N.; Lim, J.; Shin, J.; Lee, S.C.; Jeon, S.; Kim, S.H.; Kumar, P.; Lee, S.-K. Intranasal delivery of cancer-targeting doxorubicin-loaded PLGA nanoparticles arrests glioblastoma growth. J. Drug Target. 2020, 28, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Liet, B.; Laigre, E.; Goyard, D.; Todaro, B.; Tiertant, C.; Boturyn, D.; Berthet, N.; Renaudet, O. Multifunctional Glycoconjugates for Recruiting Natural Antibodies against Cancer Cells. Chem. A Eur. J. 2019, 25, 15508–15515. [Google Scholar] [CrossRef] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cossu, J.; Thoreau, F.; Boturyn, D. Multimeric RGD-Based Strategies for Selective Drug Delivery to Tumor Tissues. Pharmaceutics 2023, 15, 525. https://doi.org/10.3390/pharmaceutics15020525
Cossu J, Thoreau F, Boturyn D. Multimeric RGD-Based Strategies for Selective Drug Delivery to Tumor Tissues. Pharmaceutics. 2023; 15(2):525. https://doi.org/10.3390/pharmaceutics15020525
Chicago/Turabian StyleCossu, Jordan, Fabien Thoreau, and Didier Boturyn. 2023. "Multimeric RGD-Based Strategies for Selective Drug Delivery to Tumor Tissues" Pharmaceutics 15, no. 2: 525. https://doi.org/10.3390/pharmaceutics15020525