
Mechanistic Insight into the Early Stages of Toroidal Pore Formation by the Antimicrobial Peptide Smp24

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Patch Clamp
2.2. Molecular Dynamics Simulations
2.2.1. Single Peptide Bilayer Models
2.2.2. Multi-Peptide Bilayer Models
2.2.3. Peptide-Pore Models
2.2.4. Statistical Analysis
3. Results
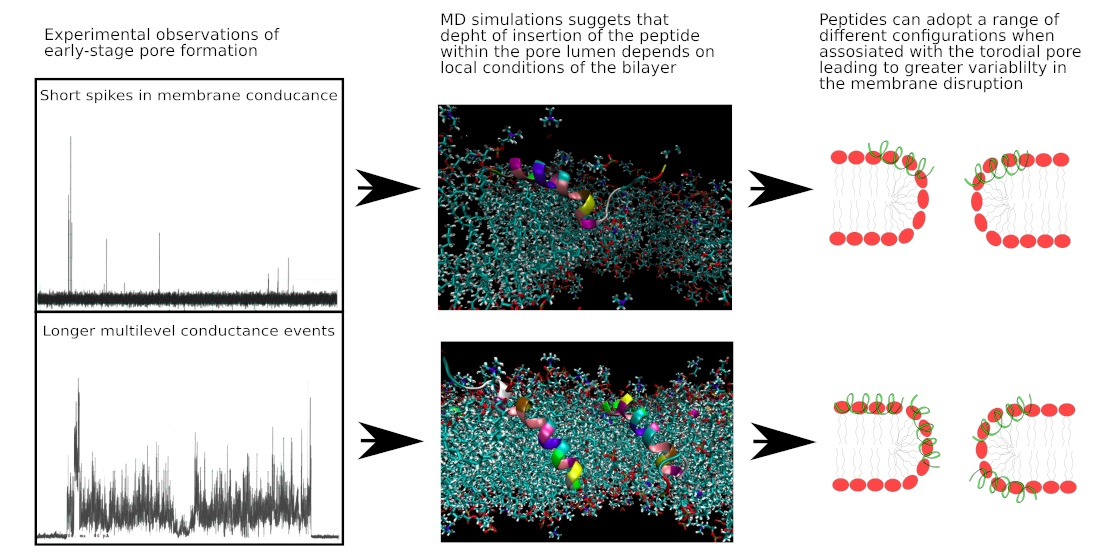
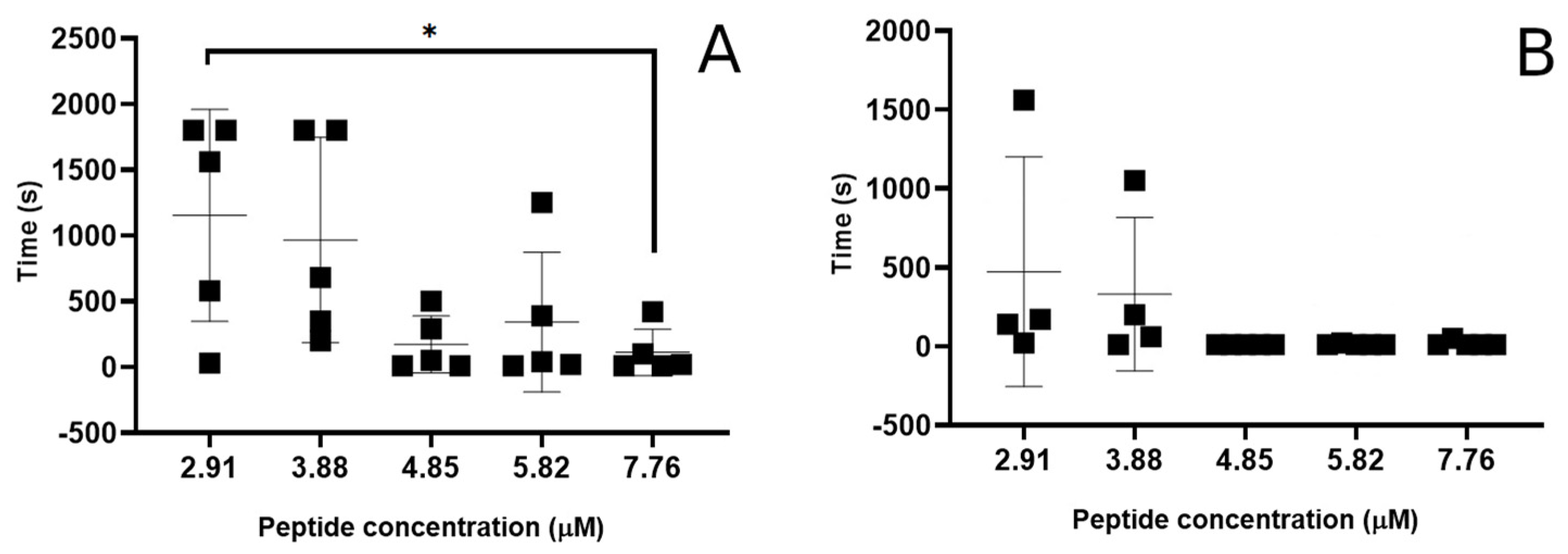
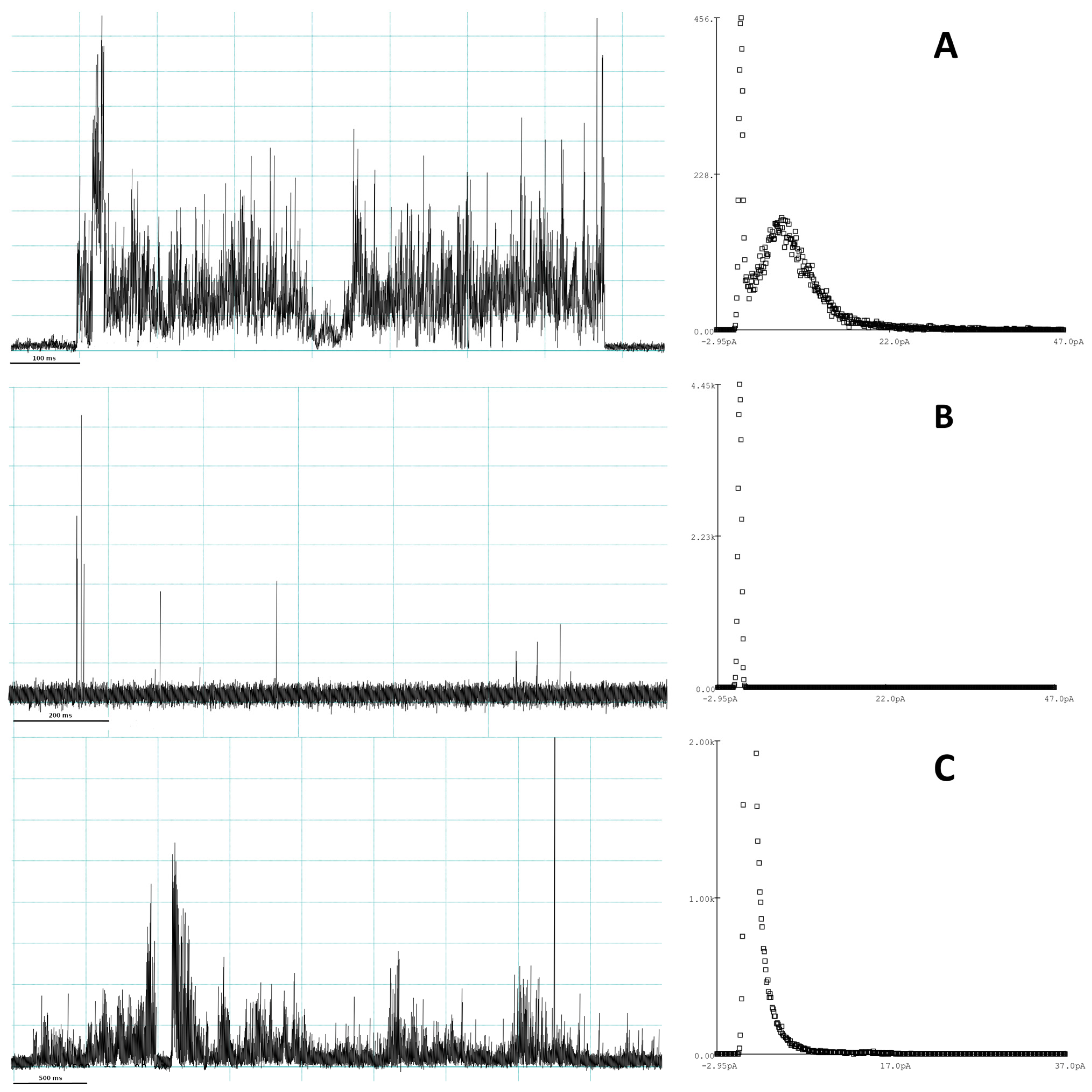
3.1. Evaluation of Peptide-Induced Membrane Disruption Using Patch-Clamp Electrophysiology
3.2. Investigation of Interactions between Smp24 and Lipid Bilayers Using MD Simulation
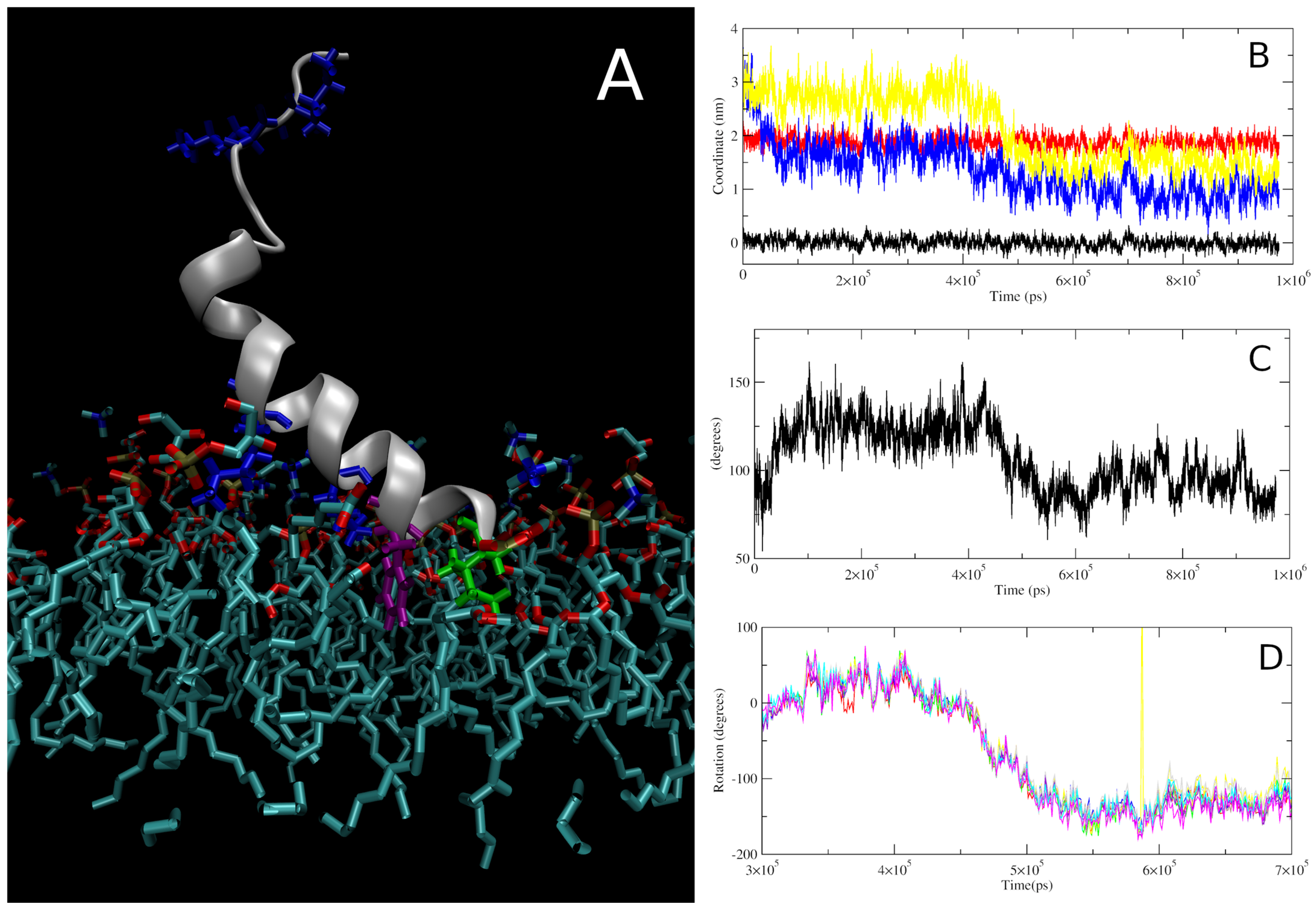
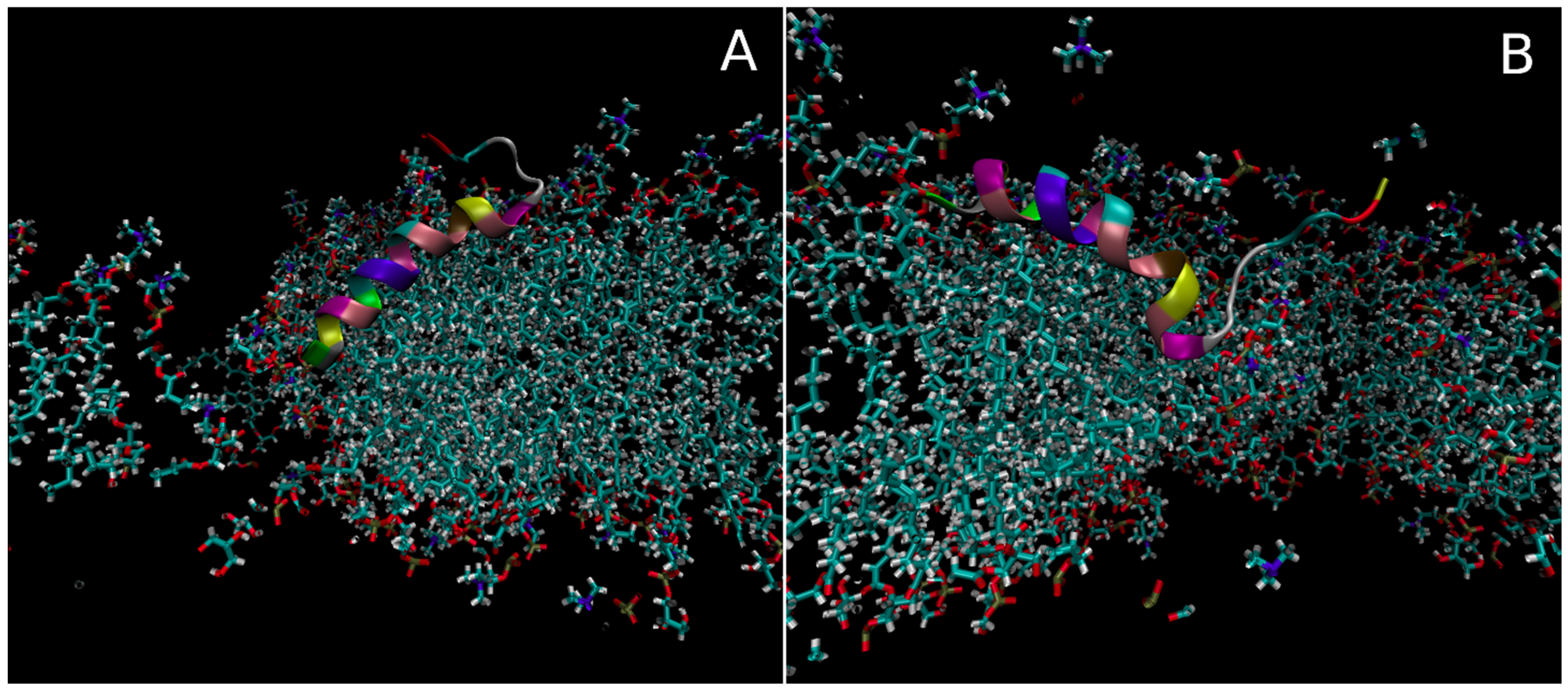
3.2.1. Insertion of Smp24 into Bilayers
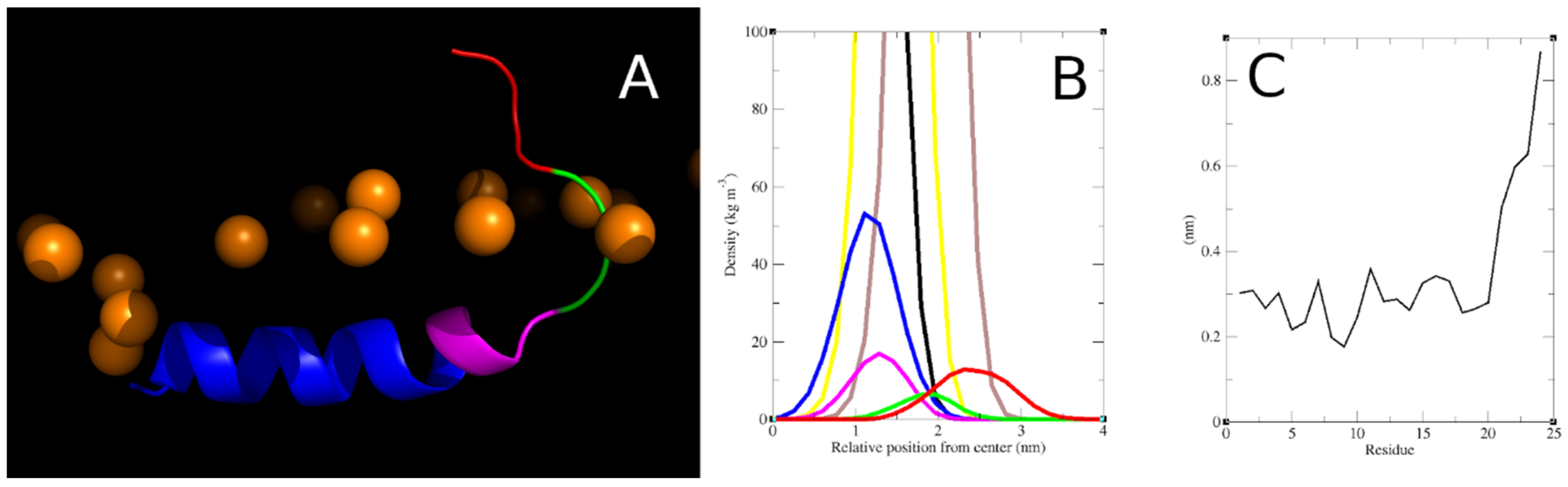
3.2.2. Structure, Orientation and Position of the Inserted Smp24
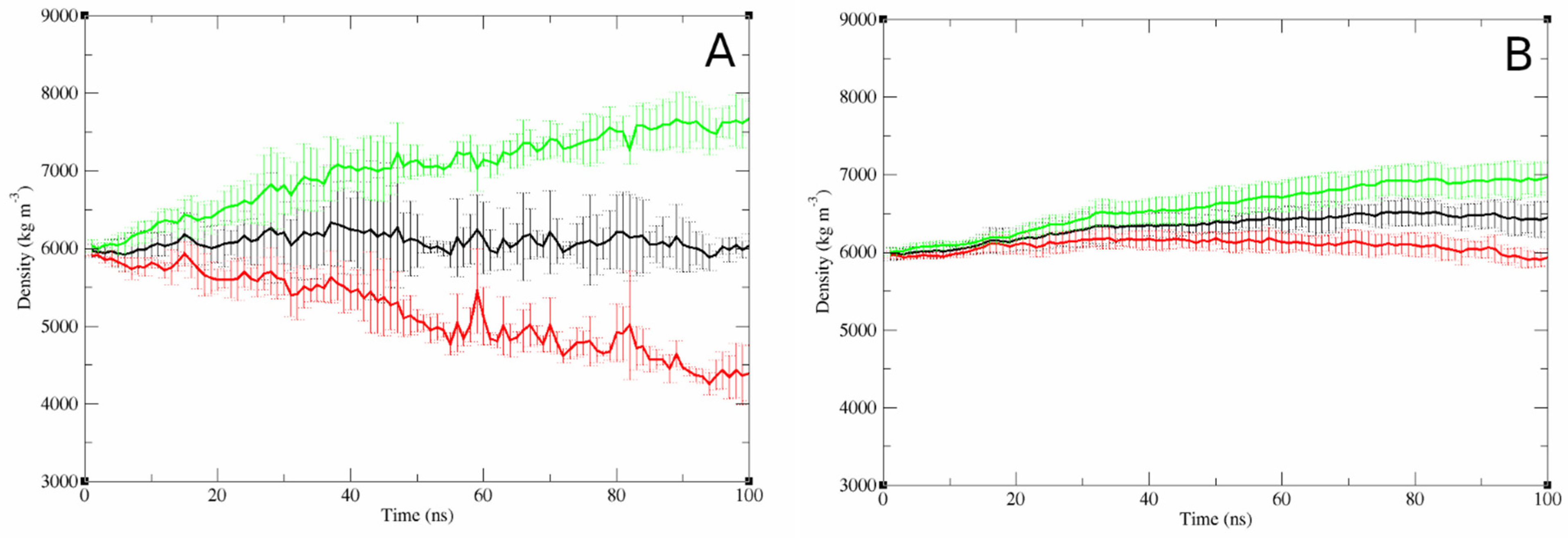
3.2.3. Concentration-Dependent Effects of Bilayer Properties
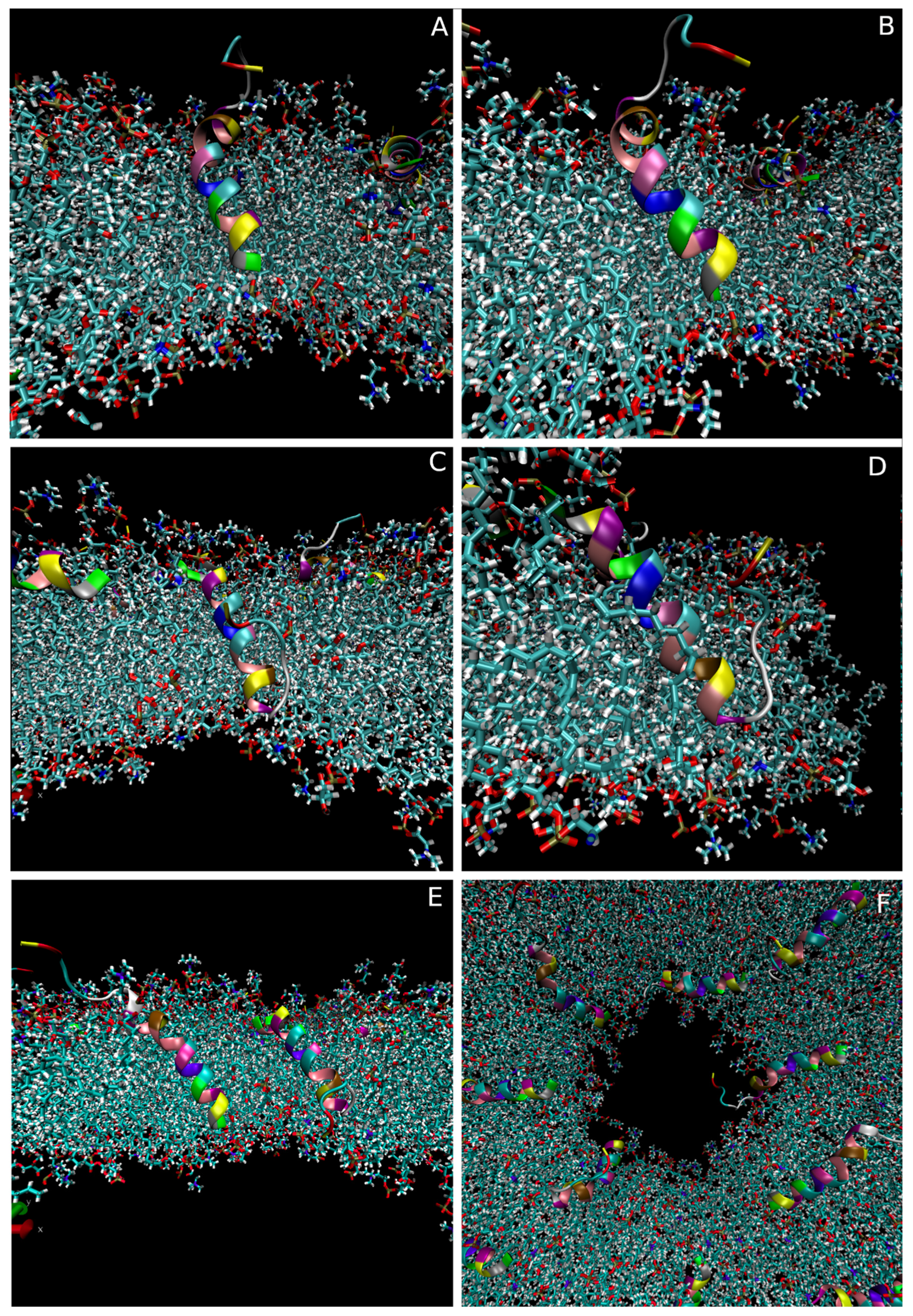
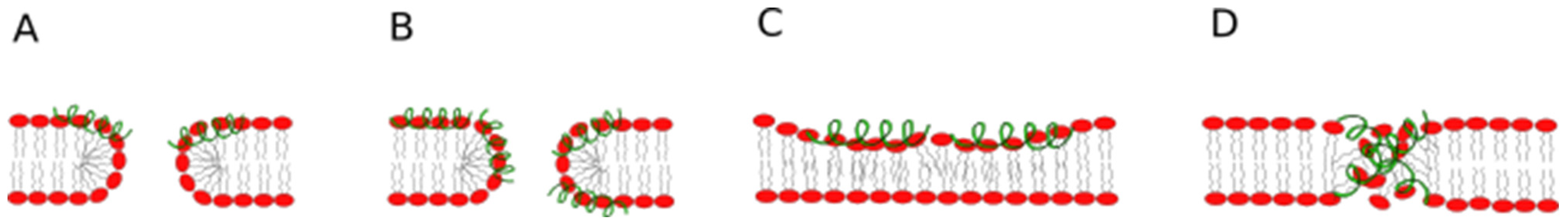
3.2.4. Modelling of the Pore-Associated Peptide Configurations
4. Discussion
4.1. Experimental Investigation of the Mechanisms of Pore Formation of Smp24
4.2. MD Simulation-Derived Structure of Smp24
4.3. Molecular Mechanisms and Structure behind Early-Stage Smp24-Induced Pores
4.3.1. Concentration-Dependent Membrane Disruption
4.3.2. Molecular-Level Structures Corresponding to Disruption Events
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations; Government of the United Kingdom: London, UK, 2016.
- Lopes, B.S.; Hanafiah, A.; Nachimuthu, R.; Muthupandian, S.; Md Nesran, Z.N.; Patil, S. The Role of Antimicrobial Peptides as Antimicrobial and Antibiofilm Agents in Tackling the Silent Pandemic of Antimicrobial Resistance. Molecules 2022, 27, 2995. [Google Scholar] [CrossRef]
- Assoni, L.; Milani, B.; Carvalho, M.R.; Nepomuceno, L.N.; Waz, N.T.; Guerra, M.E.S.; Converso, T.R.; Darrieux, M. Resistance Mechanisms to Antimicrobial Peptides in Gram-Positive Bacteria. Front. Microbiol. 2020, 11, 2362. [Google Scholar] [CrossRef]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef]
- Bechinger, B. The SMART model: Soft Membranes Adapt and Respond, also Transiently, in the presence of antimicrobial peptides. J. Pept. Sci. 2015, 21, 346–355. [Google Scholar] [CrossRef]
- Henderson, J.M.; Waring, A.J.; Separovic, F.; Lee, K.Y.C. Antimicrobial Peptides Share a Common Interaction Driven by Membrane Line Tension Reduction. Biophys. J. 2016, 111, 2176–2189. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.L.; Abdel-Rahman, M.A.; Strong, P.N.; Tawfik, M.M.; Miller, K. Characterisation of three alpha-helical antimicrobial peptides from the venom of Scorpio maurus palmatus. Toxicon 2016, 117, 30–36. [Google Scholar] [CrossRef]
- Rawson, K.M.; Lacey, M.M.; Strong, P.N.; Miller, K. Improving the Therapeutic Index of Smp24, a Venom-Derived Antimicrobial Peptide: Increased Activity against Gram-Negative Bacteria. Int. J. Mol. Sci. 2022, 23, 7979. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, L.S.; Pande, J.; Shekhtman, A. Helical Structure of Recombinant Melittin. J. Phys. Chem. B 2019, 123, 356–368. [Google Scholar] [CrossRef]
- Gesell, J.; Zasloff, M.; Opella, S.J. Two-dimensional 1 H NMR experiments show that the 23-residue magainin antibiotic peptide is an α α-helix in dodecylphosphocholine micelles, sodium dodecylsulfate micelles, and trifluoroethanol/water solution. J. Biomol. NMR 1997, 9, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Timmons, P.B.; O’Flynn, D.; Conlon, J.M.; Hewage, C.M. Structural and positional studies of the antimicrobial peptide brevinin-1BYa in membrane-mimetic environments. J. Pept. Sci. 2019, 25, e3208. [Google Scholar] [CrossRef]
- Scott Perrin, B., Jr.; Tian, Y.; Fu, R.; Grant, C.V.; Chekmenev, E.Y.; Wieczorek, W.E.; Dao, A.E.; Hayden, R.M.; Burzynski, C.M.; Venable, R.M.; et al. High-Resolution Structures and Orientations of Antimicrobial Peptides Piscidin 1 and Piscidin 3 in Fluid Bilayers Reveal Tilting, Kinking, and Bilayer Immersion. J. Am. Chem. Soc. 2014, 136, 3491–3504. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.; Lee, Y.; Park, C.; Lee, K.; Kim, S.; Choi, B. Structural and functional implications of a proline residue in the antimicrobial peptide gaegurin. Eur. J. Biochem. 1999, 266, 665–674. [Google Scholar] [CrossRef]
- Harrison, P.L.; Heath, G.R.; Johnson, B.R.G.; Abdel-Rahman, M.A.; Strong, P.N.; Evans, S.D.; Miller, K. Phospholipid dependent mechanism of smp24, an α-helical antimicrobial peptide from scorpion venom. Biochim. Et Biophys. Acta-Biomembr. 2016, 1858, 2737–2744. [Google Scholar] [CrossRef] [PubMed]
- Brüggemann, A.; Farre, C.; Haarmann, C.; Haythornthwaite, A.; Kreir, M.; Stoelzle, S.; George, M.; Fertig, N. Planar patch clamp: Advances in electrophysiology. Methods Mol. Biol. 2008, 491, 165–176. [Google Scholar]
- Sondermann, M.; George, M.; Fertig, N.; Behrends, J.C. High-resolution electrophysiology on a chip: Transient dynamics of alamethicin channel formation. Biochim. Et Biophys. Acta (BBA)-Biomembr. 2006, 1758, 545–551. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, T.; Wei, D.; Strandberg, E.; Ulrich, A.S.; Ulmschneider, J.P. How reliable are molecular dynamics simulations of membrane active antimicrobial peptides? Biochim. Et Biophys. Acta (BBA)-Biomembr. 2014, 1838, 2280–2288. [Google Scholar] [CrossRef] [PubMed]
- Jafari, M.; Mehrnejad, F.; Aghdami, R.; Chaparzadeh, N.; Razaghi Moghadam Kashani, Z.; Doustdar, F. Identification of the Crucial Residues in the Early Insertion of Pardaxin into Different Phospholipid Bilayers. J. Chem. Inf. Model. 2017, 57, 929–941. [Google Scholar] [CrossRef]
- Jahangiri, S.; Jafari, M.; Arjomand, M.; Mehrnejad, F. Molecular insights into the interactions of GF-17 with the gram-negative and gram-positive bacterial lipid bilayers. J. Cell. Biochem. 2018, 119, 9205–9216. [Google Scholar] [CrossRef]
- Ramos-Martín, F.; Herrera-León, C.; Antonietti, V.; Sonnet, P.; Sarazin, C.; D’Amelio, N. Antimicrobial Peptide K11 Selectively Recognizes Bacterial Biomimetic Membranes and Acts by Twisting Their Bilayers. Pharmaceuticals 2020, 14, 1. [Google Scholar] [CrossRef]
- Annaval, T.; Ramos-Martín, F.; Herrera-León, C.; Adélaïde, M.; Antonietti, V.; Buchoux, S.; Sonnet, P.; Sarazin, C.; D’Amelio, N. Antimicrobial Bombinin-like Peptide 3 Selectively Recognizes and Inserts into Bacterial Biomimetic Bilayers in Multiple Steps. J. Med. Chem. 2021, 64, 5185–5197. [Google Scholar] [CrossRef]
- Leontiadou, H.; Mark, A.E.; Marrink, S.J. Antimicrobial Peptides in Action. J. Am. Chem. Soc. 2006, 128, 12156–12161. [Google Scholar] [CrossRef]
- Sengupta, D.; Leontiadou, H.; Mark, A.E.; Marrink, S.J. Toroidal pores formed by antimicrobial peptides show significant disorder. Biochim. Et Biophys. Acta (BBA)-Biomembr. 2008, 1778, 2308–2317. [Google Scholar] [CrossRef]
- Sun, D.; Forsman, J.; Woodward, C.E. Multistep Molecular Dynamics Simulations Identify the Highly Cooperative Activity of Melittin in Recognizing and Stabilizing Membrane Pores. Langmuir 2015, 31, 9388–9401. [Google Scholar] [CrossRef]
- Lamiable, A.; Thevenet, P.; Rey, J.; Vavrusa, M.; Derreumaux, P.; Tuffery, P. PEP-FOLD3: Faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 2016, 44, W449. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Buchoux, S. FATSLiM: A fast and robust software to analyze MD simulations of membranes. Bioinformatics 2017, 33, 133–134. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Starr, C.G.; Guha, S.; Wimley, W.C.; Ulmschneider, M.B.; Ulmschneider, J.P. Tuning of a Membrane-Perforating Antimicrobial Peptide to Selectively Target Membranes of Different Lipid Composition. J. Membr. Biol. 2021, 254, 75–96. [Google Scholar] [CrossRef]
- Sohlenkamp, C.; Geiger, O. Bacterial membrane lipids: Diversity in structures and pathways. FEMS Microbiol. Rev. 2016, 40, 133–159. [Google Scholar] [CrossRef]
- Chui, J.K.W.; Fyles, T.M. Ionic conductance of synthetic channels: Analysis, lessons, and recommendations. Chem. Soc. Rev. 2011, 41, 148–175. [Google Scholar] [CrossRef]
- Saint, N.; Cadiou, H.; Bessin, Y.; Molle, G. Antibacterial peptide pleurocidin forms ion channels in planar lipid bilayers. Biochim. Et Biophys. Acta (BBA)-Biomembr. 2002, 1564, 359–364. [Google Scholar] [CrossRef]
- Duclohier, H.; Molle, G.; Spach, G. Antimicrobial peptide magainin I from Xenopus skin forms anion-permeable channels in planar lipid bilayers. Biophys. J. 1989, 56, 1017–1021. [Google Scholar] [CrossRef]
- Manzo, G.; Hind, C.K.; Ferguson, P.M.; Amison, R.T.; Hodgson-Casson, A.C.; Ciazynska, K.A.; Weller, B.J.; Clarke, M.; Lam, C.; Man, R.C.H.; et al. A pleurocidin analogue with greater conformational flexibility, enhanced antimicrobial potency and in vivo therapeutic efficacy. Commun. Biol. 2020, 3, 697. [Google Scholar] [CrossRef] [PubMed]
- Manzo, G.; Ferguson, P.M.; Gustilo, V.B.; Hind, C.K.; Clifford, M.; Bui, T.T.; Drake, A.F.; Atkinson, R.A.; Sutton, J.M.; Batoni, G.; et al. Minor sequence modifications in temporin B cause drastic changes in antibacterial potency and selectivity by fundamentally altering membrane activity. Sci. Rep. 2019, 9, 1385. [Google Scholar] [CrossRef]
- Manzo, G.; Ferguson, P.M.; Hind, C.K.; Clifford, M.; Gustilo, V.B.; Ali, H.; Bansal, S.S.; Bui, T.T.; Drake, A.F.; Atkinson, R.A.; et al. Temporin L and aurein 2.5 have identical conformations but subtly distinct membrane and antibacterial activities. Sci. Rep. 2019, 9, 10934. [Google Scholar] [CrossRef] [PubMed]
- Fennouri, A.; Mayer, S.F.; Schroeder, T.B.H.; Mayer, M. Single channel planar lipid bilayer recordings of the melittin variant MelP5. Biochim. Et Biophys. Acta (BBA)-Biomembr. 2017, 1859, 2051–2057. [Google Scholar] [CrossRef]
- Watanabe, H.; Kawano, R. Channel Current Analysis for Pore-forming Properties of an Antimicrobial Peptide, Magainin 1, Using the Droplet Contact Method. Anal. Sci. 2016, 32, 57–60. [Google Scholar] [CrossRef]
- Ashrafuzzaman, M.; Andersen, O.S.; McElhaney, R.N. The Antimicrobial Peptide Gramicidin S Permeabilizes Phospholipid Bilayer Membranes Without Forming Discrete Ion Channels. Biochim. Et Biophys. Acta 2008, 1778, 2814. [Google Scholar] [CrossRef]
- Bechinger, B. The structure, dynamics and orientation of antimicrobial peptides in membranes by multidimensional solid-state NMR spectroscopy. Biochim. Et Biophys. Acta (BBA)-Biomembr. 1999, 1462, 157–183. [Google Scholar] [CrossRef]
- Yamaguchi, S.; Huster, D.; Waring, A.; Lehrer, R.I.; Kearney, W.; Tack, B.F.; Hong, M. Orientation and dynamics of an antimicrobial peptide in the lipid bilayer by solid-state NMR spectroscopy. Biophys. J. 2001, 81, 2203. [Google Scholar] [CrossRef]
- Jeong, J.H.; Kim, J.S.; Choi, S.S.; Kim, Y. NMR Structural Studies of Antimicrobial Peptides: LPcin Analogs. Biophys. J. 2016, 110, 423–430. [Google Scholar] [CrossRef]
- Corzo, G.; Escoubas, P.; Villegas, E.; Barnham, K.J.; He, W.; Norton, R.S.; Nakajima, T. Characterization of unique amphipathic antimicrobial peptides from venom of the scorpion Pandinus imperator. Biochem. J. 2001, 359, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Guilhelmelli, F.; Vilela, N.; Smidt, K.S.; Oliveira, M.A.d.; Álvares, A.d.C.M.; Rigonatto, M.C.L.; Costa, P.H.d.S.; Tavares, A.H.; Freitas, S.M.d.; Nicola, A.M.; et al. Activity of Scorpion Venom-Derived Antifungal Peptides against Planktonic Cells of Candida spp. and Cryptococcus neoformans and Candida albicans Biofilms. Front. Microbiol. 2016, 7, 1844. [Google Scholar] [CrossRef] [PubMed]
- Neves, R.C.d.; Mortari, M.R.; Schwartz, E.F.; Kipnis, A.; Junqueira-Kipnis, A.P. Antimicrobial and Antibiofilm Effects of Peptides from Venom of Social Wasp and Scorpion on Multidrug-Resistant Acinetobacter baumannii. Toxins 2019, 11, 216. [Google Scholar] [CrossRef] [PubMed]
- Grage, S.L.; Afonin, S.; Kara, S.; Buth, G.; Ulrich, A.S. Membrane thinning and thickening induced by membrane-active amphipathic peptides. Front. Cell Dev. Biol. 2016, 4, 65. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.Y.; Lee, M.T.; Huang, H.W. Evidence for Membrane Thinning Effect as the Mechanism for Peptide-Induced Pore Formation. Biophys. J. 2003, 84, 3751. [Google Scholar] [CrossRef]
- Bechinger, B.; Lohner, K. Detergent-like actions of linear amphipathic cationic antimicrobial peptides. Biochim. Et Biophys. Acta (BBA)-Biomembr. 2006, 1758, 1529–1539. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Peptides | Peptide-to-Lipid Ratio | Bilayer Size (nm2) | Area per Lipid (nm2) | Bilayer Thickness (nm) | Average Shift in the Lipid Order Parameters of C13–18 Relative to the Bilayer-Only Simulation (Scd) | Lateral Peptide Diffusion Constant (10−7 cm2/s) |
|---|---|---|---|---|---|---|
| 0 | na | 13.64 | 0.696 ± 0.006 | 3.797 ± 0.029 | na | na |
| 4 | 1:144 | 14.21 | 0.707 ± 0.006 | 3.789 ± 0.030 | 2.9 × 10−4 ± 6.85 × 10−4 | 1.52 ± 0.52 |
| 9 | 1:48 | 12.62 | 0.736 ± 0.007 | 3.752 ± 0.030 | −5.36 × 10−3 ± 1.51 × 10−3 | 0.99 ± 0.37 |
| 16 | 1:32 | 14.20 | 0.758 ± 0.006 | 3.711 ± 0.029 | −1.02 × 10−2 ± 1.53 × 10−3 | 0.32 ± 0.26 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bertelsen, M.; Lacey, M.M.; Nichol, T.; Miller, K. Mechanistic Insight into the Early Stages of Toroidal Pore Formation by the Antimicrobial Peptide Smp24. Pharmaceutics 2023, 15, 2399. https://doi.org/10.3390/pharmaceutics15102399
Bertelsen M, Lacey MM, Nichol T, Miller K. Mechanistic Insight into the Early Stages of Toroidal Pore Formation by the Antimicrobial Peptide Smp24. Pharmaceutics. 2023; 15(10):2399. https://doi.org/10.3390/pharmaceutics15102399
Chicago/Turabian StyleBertelsen, Magnus, Melissa M. Lacey, Tim Nichol, and Keith Miller. 2023. "Mechanistic Insight into the Early Stages of Toroidal Pore Formation by the Antimicrobial Peptide Smp24" Pharmaceutics 15, no. 10: 2399. https://doi.org/10.3390/pharmaceutics15102399