1. Introduction
Mitochondrial membrane protein-associated neurodegeneration (MPAN) is a rare form of neurodegeneration with brain iron accumulation (NBIA) [
1]. Patients with MPAN present with progressive spastic paraparesis, psychiatric disturbances, and slow cognitive decline, variably accompanied by motor neuropathy, extrapyramidal, and visual symptoms during the disease course [
2]. MPAN is associated with autosomal recessive (AR) and dominant (AD) pathogenic variants in the gene
C19orf12 (*614297) [
2,
3,
4,
5,
6]. The encoded protein is evolutionarily conserved [
2]. Downregulation of the two
C19orf12 orthologs in fruit flies leads to neurodegeneration comparable to humans [
7]. Additionally, in zebrafish embryos, the downregulation of
C19orf12 negatively affects neuronal and muscular development [
8].
In humans, the gene encodes a membrane-associated protein that localizes to mitochondria, endoplasmic reticulum (ER), and endoplasmic reticulum–mitochondria contact sites [
2,
9,
10]. According to the public database GTEx (Analysis Release V8) [
11],
C19orf12 is ubiquitously expressed, with highest expression levels in adipose tissue, brain, and muscle.
So far, the function of C19orf12 is unknown, though the protein has been associated with a potential role in lipid metabolism. In 2011, Hartig et al. showed that
C19orf12 expression in whole blood is co-regulated with genes involved in fatty acid biogenesis and valine, leucine, and isoleucine degradation. The authors also showed that
C19orf12 expression increases continuously during the differentiation of healthy pre-adipocytes into fully mature adipocytes [
2]. A recent study in fruit flies investigated the functional interplay between C19orf12 and PLA2G6. Over-expression of
C19orf12 in
PLA2G6-deficient flies rescued acyl-chain shortening in phospholipids caused by loss of
PLA2G6 [
12]. Furthermore, Drecourt et al. revealed that mutations in
C19orf12 and other NBIA genes are associated with reduced palmitoylation of the transferrin receptor, leading to its impaired recycling and elevated cytosolic iron accumulation in patients’ fibroblasts [
13].
A role for C19orf12 in autophagy has been proposed by Venco et al. [
10]. The study reported that over-expression of
C19orf12 stimulates lipidation of the autophagy marker LC3-I to its active form LC3-II. The authors also showed that fibroblasts derived from one MPAN patient carrying the homozygous G58S variant were more susceptible to oxidative stress-induced cell death and exhibited increased mitochondrial calcium compared with control cells, suggesting that the mutation impairs mitochondrial function and calcium homeostasis [
10].
More recently, both recessive and dominant pathogenic variants in
C19orf12 have been associated with impaired mitochondrial respiration, reduced mitochondrial length and cellular adenosine 5′-triphosphate (ATP), as well as increased iron, reactive oxygen species (ROS), and lipid peroxidation, conferring MPAN cells an increased susceptibility to ferroptosis, which was rescued upon treatment with the iron chelator DFO [
14].
Considering the variety of phenotypes associated with C19orf12 deficiency and the need to identify a robust functional target for the assessment of therapeutic options for MPAN patients and provide them with rational treatment, we re-evaluated most of the reported phenotypes in a larger sample cohort.
We provide evidence that impaired autophagy is the only significant readout in MPAN fibroblasts and can be used to screen small molecules with potential therapeutic effects in MPAN.
2. Materials and Methods
2.1. Fibroblasts Cell Lines and Culturing Condition
A list of primary skin fibroblasts used in this study and their variants in
C19orf12 can be found in
Table S1. Control cells over-expressing C19orf12 (33281-T-C19orf12) were generated as described in [
15] by cloning the C19orf12 isoform NM_001031726.3 in the pLenti6.3/V5™-TOPO plasmid (Invitrogen, Waltham, MA, USA, K531520).
Commercial cells included NHDF (Lonza, Basel, Switzerland, CC-2509), HEK293FT (Invitrogen, R70007), HeLa, Hep G2, and SH-SY5Y (ATCC, Manassas, VA, USA).
Cells were cultured in high glucose Dulbecco’s Modified Eagle Medium (Life Technologies, Carlsbad, CA, USA, 41966029) supplemented with 10% fetal bovine serum (Life Technologies, 10270106), 50 U/mL penicillin/streptomycin (Life Technologies, 15070063), and 200 μM uridine (Sigma, St. Louis, MI, USA, U3750), at 37 °C in a 5% CO2 humidified atmosphere. All cell lines tested negative for mycoplasma contamination using the MycoAlert Mycoplasma detection kit (Lonza, LT07-118), according to the manufacturer’s instructions.
2.2. Protein Overexpression and Purification
2.2.1. Preparation of Expression Constructs
Human C19orf12 long isoform (Q9NSK7-1; residues 1-152; 152 amino acids long), medium isoform (Q9NSK7-4; residues 12-152; 141 amino acids long), and short isoform (Q9NSK7-2; residues 79-152; 77 amino acids long) were amplified by PCR using Pfu polymerase and cloned into pETM-11, an expression vector containing an N-terminal His
6-tag followed by a TEV protease cleavage site [
16]. All expression constructs were verified by sequencing.
2.2.2. Protein Expression and Purification
The constructs for the long and medium isoforms were transformed into
E. coli strain Rosetta2 (DE3) and cultured at 20 °C in 2-L flasks containing 500 mL ZYM 5052 auto-induction medium [
17], 100 µg/mL kanamycin, and 33 µg/mL chloramphenicol. The construct for the short isoform was transformed into
E. coli strain Rosetta2 (DE3) pLysS and cultured in 2-L flasks containing 500 mL of ZYM 5052 and 100 µg/mL kanamycin, and 33 µg/mL chloramphenicol. Cells were harvested by centrifugation after reaching saturation.
For the purification of long and medium isoforms, cells from 1 L of culture were resuspended in 60 mL lysis buffer (50 mM Tris-HCl, 300 mM NaCl, 20 mM imidazole, 10 mM MgCl2, 10 µg/mL DNaseI, 1 mM AEBSF.HCl, 1 mg/mL lysozyme, 0.01% (v/v) 1-thioglycerol, pH 8.0) containing 1% (w/v) DDM, incubated for 10 min on the bench and lysed by sonication. The lysate was clarified by centrifugation (40,000× g) and filtration (0.45 µM). The supernatant was applied to a 3-mL Ni-NTA column (Qiagen, Hilden, Germany), equilibrated in binding buffer (50 mM Tris-HCl, 300 mM NaCl, 20 mM imidazole, 0.01% (v/v) 1-thioglycerol, 0.01% (w/v) DDM, pH 8.0). The column was washed 2 times with 25 mL binding buffer. Bound proteins were eluted with elution buffer (50 mM Tris-HCl, 300 mM NaCl, 300 mM imidazole, 0.01% (w/v) DDM, 0.01% (v/v) 1-thioglycerol, pH 8.0). Fractions containing the target proteins were pooled and dialyzed overnight at 4 °C against 1 L SEC buffer (50 mM Tris-HCl, 300 mM NaCl, 0.01% (w/v) DDM, 0.01% (v/v) 1-thioglycerol, pH 8.0) in the presence of His-tagged TEV protease in a 1:25 molar ratio (TEV:protein). As described above, the cleaved-off proteins were further purified by affinity chromatography, and the flow-through and protein-containing wash fractions were pooled and concentrated to less than 5 mL. This was subsequently subjected to size exclusion chromatography using a HiLoad Superdex 200 16/600 GL column (Cytiva, Emeryville, CA, USA), equilibrated in SEC buffer. The elution fractions containing the C19orf12 protein were collected and concentrated to approx. 10 mg/mL and stored at −80 °C.
For the purification of the short isoform, cells from 1 L of the culture were resuspended in 60 mL lysis buffer (50 mM Tris-HCl, 300 mM NaCl, 20 mM imidazole, 10 mM MgCl2, 10 µg/mL DNaseI, 1 mM AEBSF.HCl, 0.2% (v/v) NP-40, 1 mg/mL lysozyme, 0.01% (v/v) 1-thioglycerol, pH 8.0), and lysed by sonication. The lysate was clarified by centrifugation (40,000× g) and filtration (0.2 µM). The purification was performed as described above using buffers without DDM.
Protein concentrations were determined by measuring the absorbance at 280 nm using the extinction coefficient of 13980 (long and medium isoforms) and 8696.07 (short isoform) M−1·cm−1, respectively.
2.3. Multi-Well Plate-Based Analyses
2.3.1. Quantification of Intracellular Calcium in Fibroblasts
Cells were seeded in black 96-well plates with black bottoms at a density of 20,000 cells/well and left to adhere overnight. The following day, intracellular calcium was measured using the Cal-520 AM kit (Abcam, Cambridge, UK, ab171868). Cells were washed with HHBS (AAT Bioquest, Pleasanton, CA, USA, 20011) and incubated with 10 µM Cal-520 in HHBS supplemented with 10% FBS for 90 min at 37 °C, followed by 30 min at room temperature. Afterward, half of the wells were treated for 3 min with 100 µM ATP (Sigma, A6419) in HHBS, while the other half were incubated with HHBS only. Fluorescence was measured using the microplate reader with the following parameters: excitation/emission = 490/525 nm.
Afterward, cells were washed with PBS (Life Technologies, 14190094) and mechanically lysed using 1× mammalian lysis buffer (Abcam, ab179835) freshly supplemented with protease inhibitors (Merck, Rahway, NJ, USA, 535140) by pipetting up and down and scraping the bottom of the wells with the pipette tips. After 15 min of incubation, cells were centrifuged at 435× g for 5 min, and the supernatant was transferred to a clean plate for protein quantification.
2.3.2. Quantification of Intracellular ROS in Fibroblasts
Cells were seeded in black 96-well plates with black bottoms at 20,000 cells/well density and left to adhere overnight. The following day, ROS were measured using the DCFDA/H2DCFDA—Cellular ROS Assay Kit (Abcam, ab113851). Cells were washed once with 1× buffer, then incubated with 25 µM DCFDA in 1× buffer for 45 min at 37 °C. Afterward, cells were washed once with 1x buffer, and half of the wells were treated with 50 µM tBHP in 1× buffer supplemented with 10% FBS. Next, 1× buffer + 10% FBS was added to the other half of the wells. After 3 h of incubation at 37 °C, cells were washed with PBS, and fluorescence was measured in the microplate reader at excitation/emission = 485/535 nm. Cells were lysed for protein quantification.
2.3.3. Quantification of Reduced Glutathione in Fibroblasts
Cells were seeded in transparent 96-well plates at a density of 20,000 cells/well and left to adhere overnight. The following day, cells were lysed, and protein concentration was measured with Bradford. Reduced glutathione in cell lysates was measured using GSH/GSSG Ratio Detection Assay Kit (Abcam, ab138881). A GSH standard curve was prepared according to the manufacturer’s instructions in a black 96-well plate, to which the cell lysates were added. The GSH Assay Mixture was added to each GSH standard and cell lysate sample. After 10 min of incubation, fluorescence was measured with the microplate reader at excitation/emission = 490/520 nm.
2.3.4. Quantification of ATP in Fibroblasts
Cells were seeded in transparent 96-well plates at a density of 20,000 cells/well and left to adhere overnight. The following day, cells were lysed using boiling 100 mM Tris, 4 mM EDTA pH 7.5. After 2 min of incubation, cells were centrifuged at 1000× g for 1 min, and the supernatant was transferred to black 96-well plates. According to the manufacturer’s instructions, total ATP was quantified using ATP Bioluminescence Assay Kit CLS II (Roche, Basel, Switzerland, 11699695001). Luminescence was measured using the microplate reader at 10 s integration time.
2.3.5. Oxygen Consumption Assay
Cells were seeded in Seahorse XF96 Cell culture microplates (Agilent Technologies, Santa Clara, CA, USA, 102416-100) at 20,000 cells/well density and left to adhere overnight. On the same day, Seahorse XF96 Sensor Cartridges (Agilent Technologies, 102416-100) were hydrated by adding sterile water to each well of the utility plate. The following day, water was removed, and Seahorse XF Calibrant solution (Agilent Technologies, 102416-100) was added to the Sensor Cartridge and incubated for 45–60 min in a 37 °C incubator without CO2. Cells were washed with medium (4.5 g/L glucose, 1 mM pyruvate, pH 7.4) and incubated with the same solution for 30 min in a 37 °C incubator without CO2. Oxygen consumption rate (OCR) was measured during 3 cycles of 2 min mixing, 2 min waiting, and 3 min measuring at the basal state and in the presence of 1 µM oligomycin, 0.4 µM FCCP, and 2 µM/2.5 µM of rotenone/antimycin A. At the end of the measurements, cells were washed with PBS and the plate was stored at −80 °C. The following day, the number of cells was counted using the CyQUANT™ Cell Proliferation Assay according to the manufacturer’s instructions.
Fluorescence values (i.e., calcium, ROS, and GSH) and luminescence values (i.e., ATP) were normalized to proteins with a Bradford assay (Bio-Rad, Hercules, CA, USA, 5000006) using a plate reader (Cytation 3, BioTek, Winooski, VT, USA). Oxygen consumption rates (OCR) were normalized to cell numbers with CyQUANT™ Cell Proliferation Assay (Invitrogen, C7026).
To control for inter-plate variability, control NHDF cells were seeded in each plate of each independent experiment. Within each plate, fluorescence, luminescence, and OCR values of control and MPAN samples were normalized to proteins or cell number normalized NHDF average values (i.e., fold change vs. NHDF).
In each assay, fold change values are shown in box plots. Within each box, horizontal black lines denote median values. Boxes extend from the 25th to the 75th percentile of each group’s distribution and the whiskers denote adjacent values. Dots outside of the whiskers denote outliers. p values were calculated with an independent sample t-test. All p values were two-sided with a significance level of 0.05. * p < 0.05, ** p < 0.01, *** p < 0.001.
2.4. Electron Microscopy and Evaluation of Mitochondria Morphology
Cells were seeded in 6-well plates at a density of 90,000 cells/well and left to adhere overnight. The following day, cells were fixed in 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer pH 7.4 (Electron Microscopy Sciences, Hatfield, PA, USA, 16537-15) for 1 h at room temperature, washed three times with 0.2 M sodium cacodylate buffer (Electron Microscopy Sciences, 11650), and fixed in 1% osmium tetroxide, 1.5% potassium ferrocyanide in 0.1 M cacodylate for 1 h on ice. After several washes in distilled water, samples were “en bloc” stained with 0.5% uranyl acetate in water overnight at 4 °C. Finally, samples were dehydrated in a graded ethanol series (30%, 50%,70%, 80%, 90%, 96%, 5 min each, and 3 washes with absolute ethanol, 10 min each). Samples were then infiltrated in a 1:1 ethanol/ Epon 812 solution for 2 h, in 100% Epon twice for 1 h each; then the monolayer cells were covered with a layer of Epon and polymerized in an oven at 60 °C for 48 h. At the end of the polymerization, the plastic was separated from the Epon block by mechanically breaking the plastic, leaving the cell monolayer facing up on the resin block. A portion of the specimen was glued on top of an Epon block and mounted on a Leica Ultracut UCT ultramicrotome. Ultrathin (70–90 nm) sections were then collected on copper grids and stained with uranyl acetate and lead citrate. Grids were examined with a transmission electron microscope (TALOS L120C ThermoScientific, Waltham, MA, USA) at 120 kV. Mitochondria were segmented manually from the EM images. For each mitochondrion mask of every image, size and shape properties were measured with the eccentricity and maximum Feret diameter functions on Matlab.
2.5. Autophagy Treatments in Fibroblasts and Cell Lysis
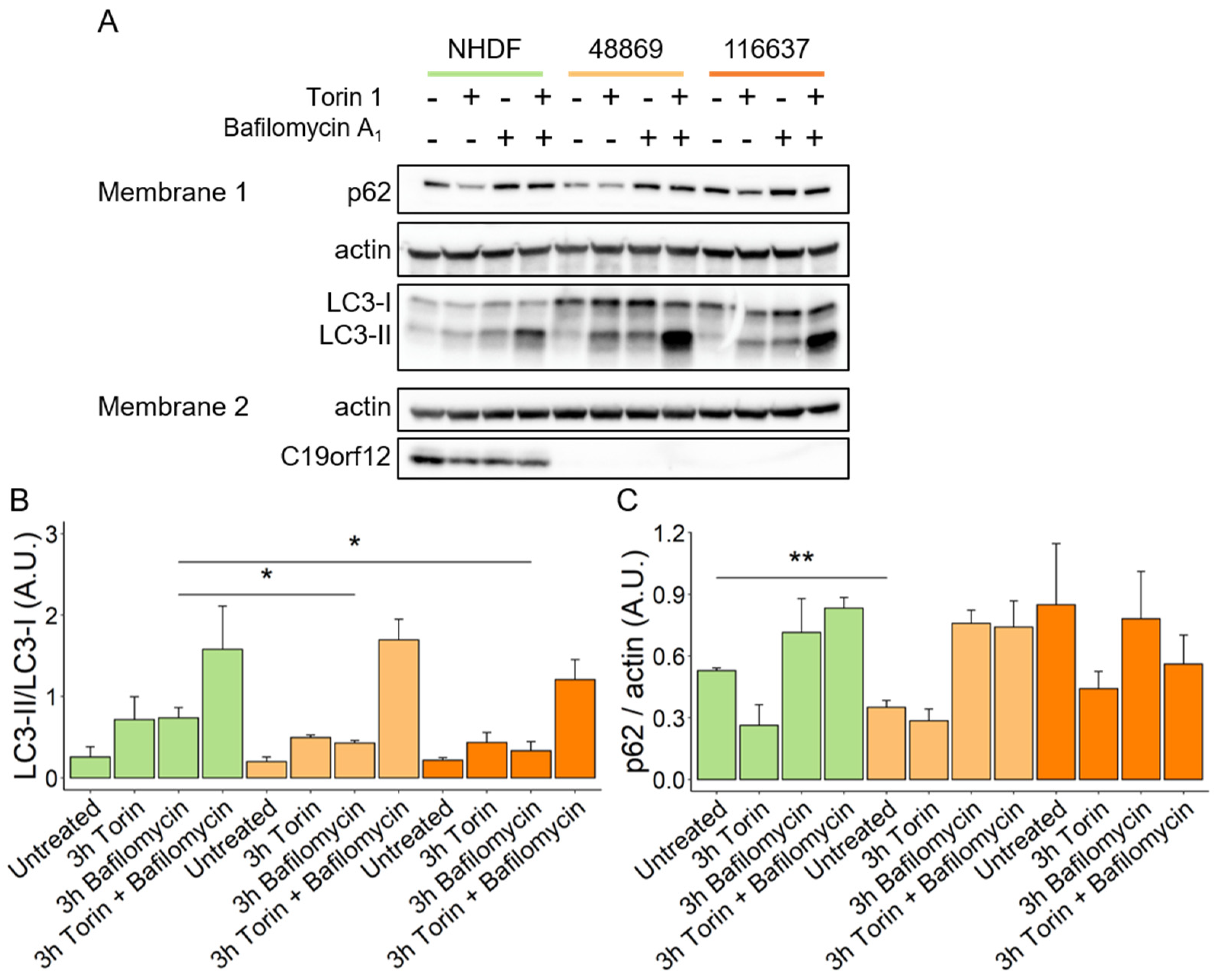
Confluent fibroblasts were treated in the following conditions: 100 nM Torin (Cell Signaling Technology, Danvers, MA, USA, 14379S) for 3 h at 37 °C, 100 nM Bafilomycin A1 (Santa Cruz Biotechnology, Dallas, TX, USA, sc-201550) for 3 h at 37 °C, 100 nM Torin and 100 nM Bafilomycin for 3 h at 37 °C, 20 µM carbonyl cyanide m-chlorophenyl hydrazine (CCCP) (Sigma, C2759) for 24 h at 37 °C, and 20 µM CCCP and 100 nM Bafilomycin for 24 and 3 h, respectively, at 37 °C. Before each treatment fibroblasts were washed with PBS.
Afterward, fibroblasts were washed with PBS, detached using trypsin (Life Technologies, 25300054), and centrifuged at 500× g for 3 min and 30 s. Cell pellets were resuspended in PBS and centrifuged at 1000× g for 5 min at 4 °C. Washed pellets were lysed in RIPA buffer (50 mM Tris-HCl, 150 mM NaCl, 1% (v/v) NP-40, 0.1% (w/v) SDS, 0.5% (w/v) deoxycholate, pH 8.0) with freshly added protease inhibitors (Merck, 535140), and incubated for 1 h at 4 °C while constantly spinning. Cell lysates were centrifuged at 15,000× g for 15 min at 4 °C, and supernatants were transferred to clean tubes. Protein concentration was determined with a Bradford assay.
2.6. Protein Extraction from Tissue Samples
Autoptic or bioptic frozen human tissues were homogenized in 15 volumes of RIPA buffer (Tris-HCl 50 mM pH 7.5, NaCl 150 mM, EDTA 5 mM, NP40 1%, SDS 0.1%, and sodium deoxycholate 0.5%) in the presence of complete protease inhibitors (Roche, 11697498001) using a Dounce homogenizer. Homogenates were incubated on ice for 30 min, cleared by centrifugation at 10,000× g at 4 °C for 10 min, and then the protein concentration was determined by Bio-Rad protein assay dye reagent (Bio-Rad, 5000006).
2.7. Sample Preparation, SDS-PAGE, and Western Blot
Protein samples were reduced using 4× Laemmli buffer (8% SDS, 40% Glycerol, 0.02% Bromophenol blue, 250 mM Tris-HCl pH 6.8, 0.1 M DTT) and heated at 50 °C for 10 min. Separation of proteins occurred via SDS-PAGE. Samples were loaded on polyacrylamide gels (Bio-Rad, 4561046; or produced
in-house) and transferred on a PVDF membrane (Bio-Rad, 1704156). After blotting, membranes were washed 3 times with TBS-T (20 mM Tris-HCl, 150 mM NaCl, 1% Tween20) and blocked for 5 min using EveryBlot buffer (Bio-Rad, 12010020). Membranes were incubated with primary antibodies overnight at 4 °C and with secondary antibodies or primary antibodies against housekeeping genes for 1 h at room temperature. Membranes were washed three times with TBS-T between each incubation and before detection. Each antibody was diluted in EveryBlot buffer at the following dilutions: anti-LC3 (Sigma, L7543; 1:20,000), anti-p62 (Progen, Heidelberg, Germany, GP62-C; 1:1000), anti-C19orf12 (produced
in-house; 1:2000), anti-SDHA (Abcam, ab14715, 1:1000), anti-actin (Santa Cruz, ac-15; 1:50,000), anti-tubulin (Merck, T5168, 1:20,000), anti-rabbit (BioLegend, San Diego, CA, USA, 406401; 1:1000), anti-mouse (Jackson Immuno Research, 115-036-062; 1:10,000), and anti-guinea pig (Abcam, ab130746; 1:1000). Immunological detection was performed using an ECL solution (GE Healthcare, Chicago, IL, USA, RPN2236) and images acquired at Fusion FX (Vilber) instrument. Quantification was performed using Fiji software v. 2.3.0/1.53f [
18].
2.8. Evaluation of Autophagy in Fibroblasts by Immunocytochemistry
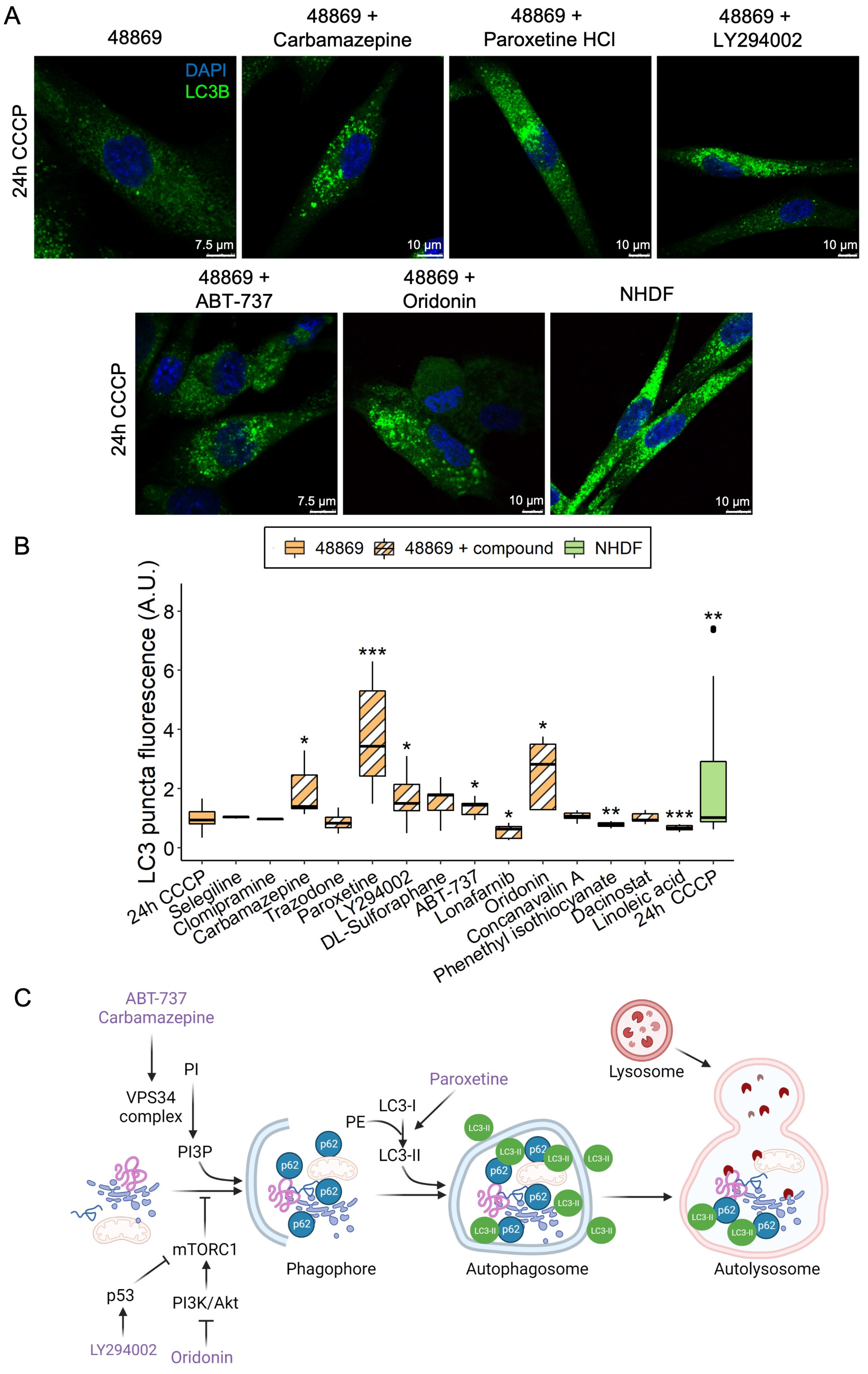
Fibroblasts were seeded on a Permanox slide (Thermo Fisher Scientific, Waltham, MA, USA, 177402) at 7000 cells/well density and left to adhere. Subsequently, cells were treated with 20 μM CCCP for 3, 6, or 24 h and fixed with 4% PFA. Cells were permeabilized using a 0.1% NP-40 solution in PBS, and primary and secondary antibodies were diluted in a 2% BSA 0.1% NP-40 solution in PBS at a 1:200 and 1:500 dilution, respectively. LC3 was immuno-stained with the corresponding antibody (LC3, Sigma, L7543) and detected with a fluorescent-labeled anti-rabbit secondary antibody (Thermo Fisher Scientific, A11034). Nuclei were stained with DAPI (Thermo Fisher Scientific, P36931), and images were acquired by confocal microscopy (Leica TCS SP5). The quantification of LC3 puncta total fluorescence was performed using the Fiji software v. 2.3.0/1.53f using the formula integrated density/area of cell × average mean of background fluorescence to calculate the corrected total cell fluorescence (CTCF).
2.9. Screening of Compounds in Fibroblasts by Immunocytochemistry
Fibroblasts were seeded as described in the section above. Cells were simultaneously treated for 24 h with 20 µM CCCP and an autophagy-modulating compound (see
Table S2 for the list of compounds and working concentration). Fixing, staining, and image processing were performed as previously described.
4. Discussion
In this study, we employed a large number of MPAN fibroblasts from patients with AR and AD forms and controls to identify phenotypes reliably shared in MPAN for testing pharmacological treatments. We focused our investigations on mitochondria-related phenotypes given the subcellular localization of C19orf12. Fibroblasts from carriers of AR MPAN were used as controls to match the genetic background of the patients to the best extent possible. Because of the large size of our sample cohort, we used high-throughput assays to investigate parameters whenever multi-well-based approaches were available. Investigations requiring confocal microscopy, electron microscopy, and Western blotting were instead performed on a smaller number of samples due to the limited throughput intrinsic to the methods.
Under the tested experimental conditions, we found no evidence of mitochondrial dysfunction in MPAN fibroblasts, as the investigated parameters (calcium handling, mitochondrial bioenergetics and morphology, ROS generation, and antioxidant defense) were not consistently impaired in all patients. We did not explore iron and ferritin levels, however, the absence of increased ROS, and normal GSH levels, spoke against eliciting ferroptosis in MPAN fibroblasts.
The absence of a consistent mitochondrial impairment in MPAN fibroblasts in our study is probably due to the fact that we used cells from multiple individuals, thus increasing interindividual variability and producing data with larger variation, when compared with studies that used repeated measures in smaller data sets, as in [
14]. While our approach likely neglects subtle defects, it has the advantage of identifying robust phenotypes shared by all MPAN fibroblasts.
A significant increase in intracellular calcium was identified only in the cell line with C19orf12
G58S/G58S mutations after ATP stimulation. A similar result was reported in a previous study [
10] using fibroblasts from the same donor, assessed with a different approach (fluorogenic dye Cal520 for total cellular calcium in the current study vs. aequorin for mitochondria-associated calcium in Venco et al. [
10]). We exclude the association of elevated calcium level with the localization of the G58S mutation in a specific C19orf12 domain, in this case the predicted transmembrane domain, as an additional mutation also clustering in the transmembrane domain (G65V), was not accompanied by high intracellular calcium levels. We therefore speculate that the elevated calcium level in this isolated cell line stems from either a specific effect of the G58S mutation or additional variants in genes relevant to calcium homeostasis. Of note, C19orf12
G58S/G58S fibroblasts in the current and two previous studies [
10,
14] were retrieved from the Telethon Network of Genetic Biobanks, indicating that fibroblasts from the same donor were employed in all studies. Future studies in fibroblasts from a different donor carrying the same variant and trio sequencing in patient 117912 and carriers will help clarify this aspect.
In the current study, we also point out that the effects of AD MPAN mutations on mitochondrial cellular phenotypes are indistinguishable from those of AR mutations, providing evidence that the overlap of clinical and pathology aspects in AD and AR MPAN patients [
3] is also substantiated by overlap at a functional level.
By investigating autophagy in naïve MPAN fibroblasts, we corroborated the finding that C19orf12 intervenes in the initiation of the autophagy process by supporting the conversion of LC3-I to LC3-II without binding directly to LC3 [
10] and, possibly, by promoting the maintenance of a pool of functioning mitochondria. Although the accumulation of dysfunctional mitochondria upon CCCP treatment did not reach the threshold for statistical significance in MPAN fibroblasts, results in neurons could be different. Fibroblasts can dilute damaged mitochondria through cell division, in addition to removing them via autophagy, while neuronal cells, being non-dividing cells, rely mainly on autophagy to clear dysfunctional organelles [
19]. Investigating clearance of mitochondria in patient-derived neuronal cells could provide compelling evidence of an additional role in mitophagy for C19orf12, besides a role in initiating autophagy.
As the conversion of LC3-I to LC3-II relies on the conjugation of LC3-I to a phosphatidylethanolamine (PE) moiety, defective lipidation of LC3 in MPAN may point to a primary role for C19orf12 in the conjugation of PE to LC3-I, or in the synthesis of PE between ER and mitochondria. Considering C19orf12 localization at the mitochondria, ER, and ER-mitochondria contact sites, C19orf12 could tether mitochondria and ER to allow efficient transfer of the PE precursor, phosphatidylserine, from ER to mitochondria, its conversion to PE in mitochondria and transfer back to the ER for phosphatidylcholine production. By integrating studies in cellular models with dynamic lipid metabolism and structural biology, we hope to unambiguously clarify the function of C19orf12 soon.
Despite the fact that the primary function of C19orf12 is still to be defined, mutations in
C19orf12 result in impaired activation of LC3. Therefore, any pharmacological treatment restoring LC3 activity could be used as a therapeutic compound for MPAN. To this end, we screened 14 autophagy-modulating compounds in MPAN fibroblasts and identified five that were able to increase the LC3 levels in patient fibroblasts: carbamazepine, LY294002, ABT-737, oridonin, and paroxetine. Of note, LY294002 and paroxetine may also have an inhibitory effect on autophagy [
20,
21], suggesting a dual effect for these compounds, similarly to DL-Sulforaphane, also tested in our screening [
22,
23]. Still, the dual effect seems to depend on the cell type tested [
24], and the concentration of the compound, as reported for LY294002 [
20,
25].
Although the five hits take part in various biological processes (anticonvulsant, antidepressant, anti-tumoral, and inhibitors of phosphoinositide 3-kinases and Bcl-2), based on a literature search, they have a shared effect in stimulating the initial steps of the autophagy process. Oridonin and LY294002 inhibit the mechanistic target of rapamycin complex 1 (mTORC1) by inhibiting the PI3K/Akt pathway or activating p53, respectively [
25,
26]. Carbamazepine and ABT-737 promote the activity of VPS34 complex [
27,
28]. Paroxetine stimulates LC3-I conversion to LC3-II [
29,
30,
31]. Notably, carbamazepine, oridonin, and paroxetine are molecules approved by the Food and Drug Administration, implying a realistic clinical translatability in MPAN, as opposed to LY294002 and ABT-737, which still require further validation.
Although fibroblasts cannot recapitulate tissue-specific metabolic aspects, conserved processes such as autophagy can be reliably investigated due to the conservation of the autophagy machinery and the prominent expression of the 141 aa isoform in fibroblasts, as well as in all other investigated cell types and tissues. In conclusion, while further research is needed to identify the primary function of C19orf12, our study moves one step closer to understanding the consequences of C19orf12 dysfunction at the cellular level and to establishing a rational therapy for MPAN.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





