
The Impact of Angiotensin-Converting Enzyme-2/Angiotensin 1-7 Axis in Establishing Severe COVID-19 Consequences
 , , ,
, , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
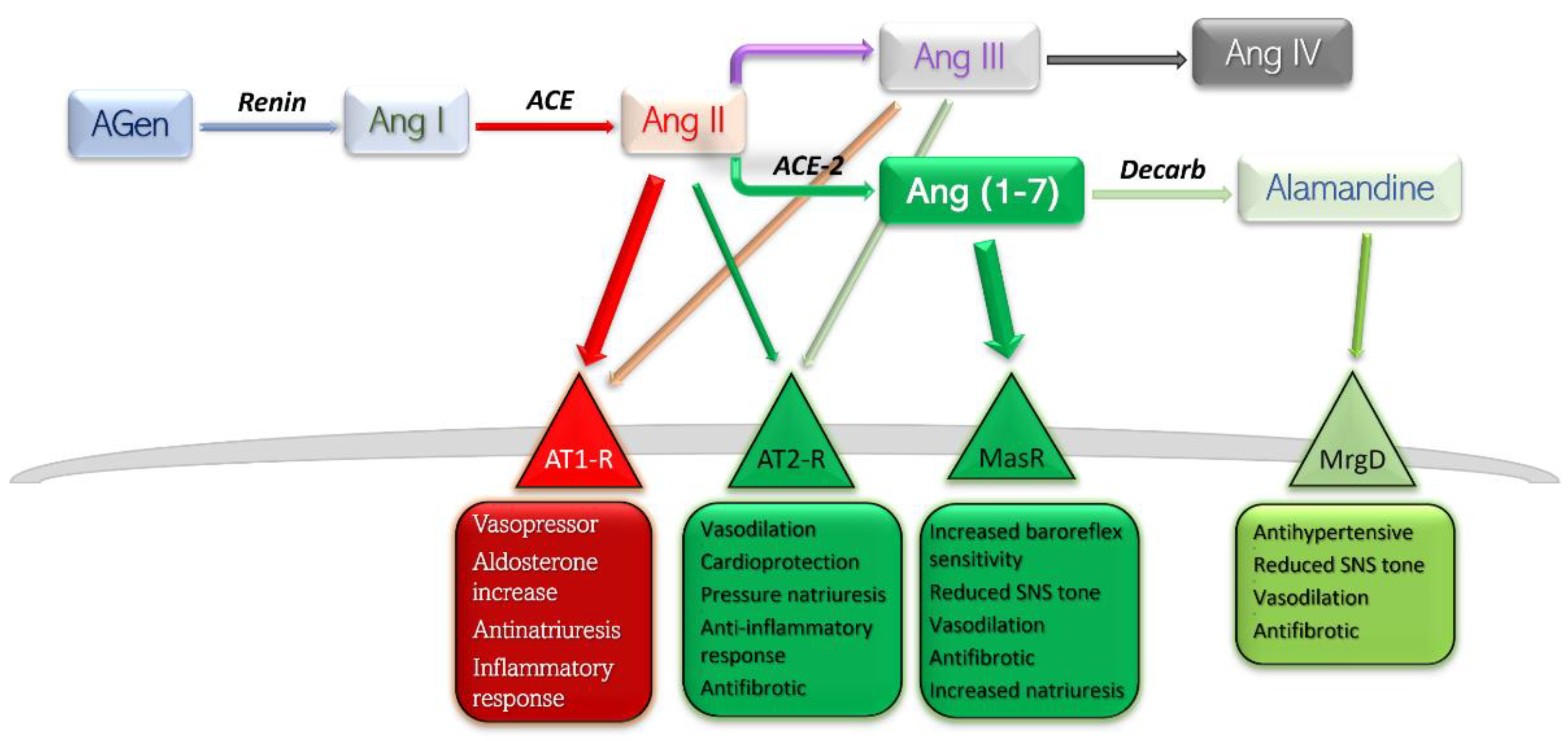
2. RAAS—The Classical View
3. RAAS—The Alternative Pathway
3.1. ACE2 Enzyme
3.2. SARS-CoV-2 and Its Receptor, ACE2
3.3. Angiotensin (1-9)
3.4. Angiotensin (1-7)
3.4.1. Intracellular Signalling Pathways Employed by Angiotensin (1-7)
Influence on Metabolism
Oxidative Stress
Arterial Blood Pressure
Apoptosis
3.4.2. Effects Mediated by Angiotensin (1-7) in Specific Organs
Brain
Cardiac Direct Involvement
Kidney
Vascular Actions
4. Medium and Long-Term Complications Determined by Infection with SARS-CoV-2
4.1. Brain
4.2. Heart
4.3. Renal
4.4. Vascular
5. COVID-19 Infection Severity: Proven Risk Factors and Their Link to ACE2 Expression
5.1. Age
5.2. Demographic Factors
5.3. Arterial Hypertension
5.4. Diabetes
5.5. Obesity
5.6. Pregnancy
6. Long-COVID Organ-Specific Management
6.1. CNS Complications Evaluation
6.2. Cardiovascular Complications Evaluation and Guidance
6.3. COVID-19-Related Chronic Kidney Disease Predictors
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hikmet, F.; Mear, L.; Edvinsson, A.; Micke, P.; Uhlen, M.; Lindskog, C. The protein expression profile of ACE2 in human tissues. Mol. Syst. Biol. 2020, 16, e9610. [Google Scholar] [CrossRef] [PubMed]
- Freitas, F.C.; Ferreira, P.H.B.; Favaro, D.C.; Oliveira, R.J. Shedding Light on the Inhibitory Mechanisms of SARS-CoV-1/CoV-2 Spike Proteins by ACE2-Designed Peptides. J. Chem. Inf. Model. 2021, 61, 1226–1243. [Google Scholar] [CrossRef] [PubMed]
- Mehrabadi, M.E.; Hemmati, R.; Tashakor, A.; Homaei, A.; Yousefzadeh, M.; Hemati, K.; Hosseinkhani, S. Induced dysregulation of ACE2 by SARS-CoV-2 plays a key role in COVID-19 severity. Biomed. Pharmacoter. 2021, 137. [Google Scholar] [CrossRef]
- Ahmadi Badi, S.; Tarashi, S.; Fateh, A.; Rohani, P.; Masotti, A.; Siadat, S.D. From the Role of Microbiota in Gut-Lung Axis to SARS-CoV-2 Pathogenesis. Mediators Inflamm. 2021, 2021, 6611222. [Google Scholar] [CrossRef]
- Gu, J.; Yin, J.; Zhang, M.; Li, J.; Wu, Y.; Chen, J.; Miao, H. Study on the Clinical Significance of ACE2 and Its Age-Related Expression. J. Inflamm. Res. 2021, 14, 2873–2882. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.D.; Karnik, S.S. Angiotensin Receptors: Structure, Function, Signaling and Clinical Applications. J. Cell. Sign. 2016, 1. [Google Scholar] [CrossRef]
- Carey, R.M. AT2 Receptors: Potential Therapeutic Targets for Hypertension. Am. J. Hypertens. 2017, 30, 339–347. [Google Scholar] [CrossRef]
- Li, X.C.; Widdop, R.E. AT2 receptor-mediated vasodilatation is unmasked by AT1 receptor blockade in conscious SHR. Br. J. Pharmacol. 2004, 142, 821–830. [Google Scholar] [CrossRef]
- Widdop, R.E.; Matrougui, K.; Levy, B.I.; Henrion, D. AT2 receptor-mediated relaxation is preserved after long-term AT1 receptor blockade. Hypertension 2002, 40, 516–520. [Google Scholar] [CrossRef]
- Siragy, H.M.; de Gasparo, M.; Carey, R.M. Angiotensin type 2 receptor mediates valsartan-induced hypotension in conscious rats. Hypertension 2002, 35, 1074–1077. [Google Scholar] [CrossRef] [Green Version]
- Gammelgaard, I.; Wamberg, S.; Bie, P. Systemic effects of angiotensin III in conscious dogs during acute double blockade of the renin-angiotensin-aldosterone-system. Acta Physiol. 2006, 188, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Tipnis, S.R.; Hooper, N.M.; Hyde, R.; Karran, E.; Christie, G.; Turner, A.J. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000, 275, 33238–33243. [Google Scholar] [CrossRef] [PubMed]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ. Res. 2000, 87, E1–E9. [Google Scholar] [CrossRef] [PubMed]
- Campbell, D.J.; Zeitz, C.J.; Esler, M.D.; Horowitz, J.D. Evidence against a major role for angiotensin converting enzyme-related carboxypeptidase (ACE2) in angiotensin peptide metabolism in the human coronary circulation. J. Hypertens. 2004, 22, 1971–1976. [Google Scholar] [CrossRef]
- Cornell, M.J.; Williams, T.A.; Lamango, N.S.; Coates, D.; Corvol, P.; Soubrier, F.; Hoheisel, J.; Lehrach, H.; Isaac, R.E. Cloning and Expression of an Evolutionary Conserved Single-domain Angiotensin Converting Enzyme from Drosophila melanogaster. J. Biol. Chem. 1995, 270, 13613–13619. [Google Scholar] [CrossRef]
- Lambert, D.W.; Yarski, M.; Warner, F.J.; Thornhill, P.; Parkin, E.T.; Smith, A.I.; Hooper, N.M.; Turner, A.J. Tumor necrosis factor-alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J. Biol. Chem. 2005, 280, 30113–30119. [Google Scholar] [CrossRef]
- Glass, W.G.; Subbarao, K.; Murphy, B.; Murphy, P.M. Mechanisms of host defense following severe acute respiratory syndrome-coronavirus (SARS-CoV) pulmonary infection of mice. J. Immunol. 2004, 173, 4030–4039. [Google Scholar] [CrossRef]
- Kadam, S.B.; Sukhramani, G.S.; Bishnoi, P.; Pable, A.A.; Barvkar, V.T. SARS-CoV-2, the pandemic coronavirus: Molecular and structural insights. J. Basic Microbiol. 2021, 61, 180–202. [Google Scholar] [CrossRef]
- Grasselli, G.; Pesenti, A.; Cecconi, M. Critical Care Utilization for the COVID-19 Outbreak in Lombardy, Italy: Early Experience and Forecast During an Emergency Response. JAMA 2020, 323, 1545–1546. [Google Scholar] [CrossRef]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94, e00127-20. [Google Scholar] [CrossRef] [PubMed]
- Ocaranza, M.P.; Moya, J.; Barrientos, V.; Alzamora, R.; Hevia, D.; Morales, C.; Pinto, M.; Escudero, N.; García, L.; Novoa, U.; et al. Angiotensin-(1-9) reverses experimental hypertension and cardiovascular damage by inhibition of the angiotensin converting enzyme/Ang II axis. J. Hypertens. 2014, 32, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Kramkowski, K.; Mogielnicki, A.; Leszczynska, A.; Buczko, W. Angiotensin-(1-9), the product of angiotensin I conversion in platelets, enhances arterial thrombosis in rats. J. Physiol. Pharmacol. 2010, 61, 317–324. [Google Scholar] [PubMed]
- Mogielnicki, A.; Kramkowski, K.; Hermanowicz, J.M.; Leszczynska, A.; Przyborowski, K.; Buczko, W. Angiotensin-(1-9) enhances stasis-induced venous thrombosis in the rat because of the impairment of fibrinolysis. J. Renin Angiotensin Aldosterone Syst. 2014, 15, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Calò, L.A.; Schiavo, S.; Davis, P.A.; Pagnin, E.; Mormino, P.; D’Angelo, A.; Pessina, A.C. Angiotensin II signaling via type 2 receptors in a human model of vascular hyporeactivity: Implications for hypertension. J. Hypertens. 2010, 28, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Vickers, C.; Hales, P.; Kaushik, V.; Dick, L.; Gavin, J.; Tang, J.; Godbout, K.; Parsons, T.; Baronas, E.; Hsieh, F. Hydrolysis of Biological Peptides by Human Angiotensin-converting Enzyme-related Carboxypeptidase. J. Biol. Chem. 2002, 277, 14838–14843. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.; Sampaio, W.O.; Alzamora, A.C.; Motta-Santos, D.; Alenina, N.; Bader, M.; Campagnole-Santos, M.J. The ACE2/Angiotensin-(1-7)/MAS Axis of the Renin-Angiotensin System: Focus on Angiotensin-(1-7). Physiol. Rev. 2018, 98, 505–553. [Google Scholar] [CrossRef]
- Chappell, M.C.; Allred, A.J.; Ferrario, C.M. Pathways of angiotensin-(1-7) metabolism in the kidney. Nephrol. Dial. Transplant. 2001, 16, 22–26. [Google Scholar] [CrossRef]
- Rice, G.I.; Thomas, D.A.; Grant, P.J.; Turner, A.J.; Hooper, N.M. Evaluation of angiotensin-converting enzyme (ACE), its homologue ACE2 and neprilysin in angiotensin peptide metabolism. Biochem. J. 2004, 383, 45–51. [Google Scholar] [CrossRef]
- Lautner, R.Q.; Villela, D.C.; Fraga-Silva, R.A.; Silva, N.; Verano-Braga, T.; Costa-Fraga, F.; Jankowski, J.; Jankowski, V.; Sousa, F.; Alzamora, A.; et al. Discovery and characterization of alamandine: A novel component of the renin-angiotensin system. Circ. Res. 2013, 112, 1104–1111. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, L.B.; Parreiras-E-Silva, L.T.; Bruder-Nascimento, T.; Duarte, D.A.; Simões, S.C.; Costa, R.M.; Rodríguez, D.Y.; Ferreira, P.; Silva, C.; Abrao, E.P.; et al. Ang-(1-7) is an endogenous β-arrestin-biased agonist of the AT1 receptor with protective action in cardiac hypertrophy. Sci. Rep. 2017, 7, 11903. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Hussain, T. Synergism between Angiotensin receptors ligands: Role of Angiotensin-(1-7) in modulating AT2 R agonist response on nitric oxide in kidney cells. Pharmacol. Res. Perspect. 2020, 8, e00667. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, M.C.; Giani, J.F.; Burghi, V.; Mayer, M.A.; Carranza, A.; Taira, C.A.; Dominici, F.P. The Mas receptor mediates modulation of insulin signaling by angiotensin-(1-7). Regul. Pept. 2012, 177, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.H.; Giani, J.F.; Burghi, V.; Miquet, J.G.; Qadri, F.; Braga, J.F.; Todiras, M.; Kotnik, K.; Alenina, N.; Dominici, F.P.; et al. Oral administration of angiotensin-(1-7) ameliorates type 2 diabetes in rats. J. Mol. Med. 2014, 92, 255–265. [Google Scholar] [CrossRef]
- Hashimoto, T.; Perlot, T.; Rehman, A.; Trichereau, J.; Ishiguro, H.; Paolino, M.; Sigl, V.; Hanada, T.; Hanada, R.; Lipinski, S. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature 2012, 487, 477–481. [Google Scholar] [CrossRef]
- Niu, M.J.; Yang, J.K.; Lin, S.S.; Ji, X.J.; Guo, L.M. Loss of angiotensin-converting enzyme 2 leads to impaired glucose homeostasis in mice. Endocrine 2008, 34, 56–61. [Google Scholar] [CrossRef]
- Villalobos, L.A.; San Hipólito-Luengo, Á.; Ramos-González, M.; Cercas, E.; Vallejo, S.; Romero, T.; Carraro, R.; Sanchez-Ferrer, C.F.; Peiro, C. The Angiotensin-(1-7)/Mas Axis Counteracts Angiotensin II-Dependent and -Independent Pro-inflammatory Signaling in Human Vascular Smooth Muscle Cells. Front. Pharmacol. 2016, 7. [Google Scholar] [CrossRef]
- Lovren, F.; Pan, Y.; Quan, A.; Teoh, H.; Wang, G.; Shukla, P.C.; Levitt, K.S.; Oudit, G.Y.; Al-Omran, M.; Stewart, D.J. Angiotensin converting enzyme-2 confers endothelial protection and attenuates atherosclerosis. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H1377–H1384. [Google Scholar] [CrossRef]
- Rabelo, L.A.; Todiras, M.; Nunes-Souza, V.; Qadri, F.; Szijártó, I.A.; Gollasch, M.; Penninger, J.M.; Bader, M.; Santos, R.A.; Alenina, N. Genetic Deletion of ACE2 Induces Vascular Dysfunction in C57BL/6 Mice: Role of Nitric Oxide Imbalance and Oxidative Stress. PLoS ONE 2016, 11, e0150255. [Google Scholar] [CrossRef]
- Wysocki, J.; Ortiz-Melo, D.I.; Mattocks, N.K.; Xu, K.; Prescott, J.; Evora, K.; Ye, M.; Sparks, M.A.; Haque, S.K.; Batlle, D. ACE2 deficiency increases NADPH-mediated oxidative stress in the kidney. Physiol. Rep. 2014, 2, e00264. [Google Scholar] [CrossRef]
- Shi, Y.; Lo, C.S.; Padda, R.; Abdo, S.; Chenier, I.; Filep, J.G.; Ingelfinger, J.R.; Zhang, S.L.; Chan, J.S.D. Angiotensin-(1–7) prevents systemic hypertension, attenuates oxidative stress and tubulointerstitial fibrosis, and normalizes renal angiotensin-converting enzyme 2 and Mas receptor expression in diabetic mice. Clin. Sci. 2015, 128, 649–663. [Google Scholar] [CrossRef] [PubMed]
- Zamilpa, R.; Chilton, R.J.; Lindsey, M.L. Tumor necrosis factor-alpha--converting enzyme roles in hypertension-induced hypertrophy: Look both ways when crossing the street. Hypertension 2009, 54, 471–472. [Google Scholar] [CrossRef] [PubMed]
- Knepper, M.A.; Brooks, H.L. Regulation of the sodium transporters NHE3, NKCC2 and NCC in the kidney. Curr. Opin. Nephrol. 2001, 10, 655–659. [Google Scholar] [CrossRef] [PubMed]
- Moritani, T.; Iwai, M.; Kanno, H.; Nakaoka, H.; Iwanami, J.; Higaki, T.; Ishii, E.; Horiuchi, M. ACE2 deficiency induced perivascular fibrosis and cardiac hypertrophy during postnatal development in mice. J. Am. Soc. Hypertens. 2013, 7, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Ren, X.; Zhao, M.; Zhou, B.; Han, Y. Angiotensin-(1-7) abrogates angiotensin II-induced proliferation, migration and inflammation in VSMCs through inactivation of ROS-mediated PI3K/Akt and MAPK/ERK signaling pathways. Sci. Rep. 2016, 6, 34621. [Google Scholar] [CrossRef]
- Gava, E.; Samad-Zadeh, A.; Zimpelmann, J.; Bahramifarid, N.; Kitten, G.T.; Santos, R.A.; Touyz, R.M.; Burns, K.D. Angiotensin-(1-7) activates a tyrosine phosphatase and inhibits glucose-induced signalling in proximal tubular cells. Nephrol. Dial. Transplant. 2009, 24, 1766–1773. [Google Scholar] [CrossRef]
- von Bohlen und Halbach, O.; Albrecht, D. The CNS renin-angiotensin system. Cell Tissue Res. 2006, 326, 599–616. [Google Scholar] [CrossRef]
- Doobay, M.F.; Talman, L.S.; Obr, T.D.; Tian, X.; Davisson, R.L.; Lazartigues, E. Differential expression of neuronal ACE2 in transgenic mice with overexpression of the brain renin-angiotensin system. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R373–R381. [Google Scholar] [CrossRef]
- Becker, L.K.; Etelvino, G.M.; Walther, T.; Santos, R.A.; Campagnole-Santos, M.J. Immunofluorescence localization of the receptor Mas in cardiovascular-related areas of the rat brain. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1416–H1424. [Google Scholar] [CrossRef]
- Xue, B.; Zhang, Z.; Johnson, R.F.; Guo, F.; Hay, M.; Johnson, A.K. Central endogenous angiotensin-(1-7) protects against aldosterone/NaCl-induced hypertension in female rats. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H699–H705. [Google Scholar] [CrossRef] [Green Version]
- Guimaraes, P.S.; Santiago, N.M.; Xavier, C.H.; Velloso, E.P.; Fontes, M.A.; Santos, R.A.; Campagnole-Santos, M.J. Chronic infusion of angiotensin-(1-7) into the lateral ventricle of the brain attenuates hypertension in DOCA-salt rats. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H393–H400. [Google Scholar] [CrossRef] [PubMed]
- de Moura, M.M.; dos Santos, R.A.; Campagnole-Santos, M.J.; Todiras, M.; Bader, M.; Alenina, N.; Haibara, A.S. Altered cardiovascular reflexes responses in conscious Angiotensin-(1-7) receptor Mas-knockout mice. Peptides 2010, 31, 1934–1939. [Google Scholar] [CrossRef] [PubMed]
- Heringer-Walther, S.; Batista, E.N.; Walther, T.; Khosla, M.C.; Santos, R.A.; Campagnole-Santos, M.J. Baroreflex improvement in shr after ace inhibition involves angiotensin-(1-7). Hypertension 2001, 37, 1309–1314. [Google Scholar] [CrossRef] [PubMed]
- Britto, R.R.; Santos, R.A.; Fagundes-Moura, C.R.; Khosla, M.C.; Campagnole-Santos, M.J. Role of angiotensin-(1-7) in the modulation of the baroreflex in renovascular hypertensive rats. Hypertension 1997, 30, 549–556. [Google Scholar] [CrossRef]
- Wu, Z.T.; Ren, C.Z.; Yang, Y.H.; Zhang, R.W.; Sun, J.C.; Wang, Y.K.; Su, D.F.; Wang, W.Z. The PI3K signaling-mediated nitric oxide contributes to cardiovascular effects of angiotensin-(1-7) in the nucleus tractus solitarii of rats. Nitric. Oxide 2016, 52, 56–65. [Google Scholar] [CrossRef]
- Guimaraes, P.S.; Oliveira, M.F.; Braga, J.F.; Nadu, A.P.; Schreihofer, A.; Santos, R.A.S.; Campagnole-Santos, M.J. Increasing Angiotensin-(1-7) Levels in the Brain Attenuates Metabolic Syndrome-Related Risks in Fructose-Fed Rats. Hypertension 2014, 63, 1078–1085. [Google Scholar] [CrossRef]
- Whitaker, A.M.; Molina, P.E. Angiotensin (1-7) contributes to nitric oxide tonic inhibition of vasopressin release during hemorrhagic shock in acute ethanol intoxicated rodents. Life Sci. 2013, 93, 623–629. [Google Scholar] [CrossRef]
- Walther, T.; Balschun, D.; Voigt, J.P.; Fink, H.; Zuschratter, W.; Birchmeier, C.; Ganten, D.; Bader, M. Sustained long term potentiation and anxiety in mice lacking the Mas protooncogene. J. Biol. Chem. 2013, 273, 11867–11873. [Google Scholar] [CrossRef]
- Almeida-Santos, A.F.; Kangussu, L.M.; Moreira, F.A.; Santos, R.A.; Aguiar, D.C.; Campagnole-Santos, M.J. Anxiolytic- and antidepressant-like effects of angiotensin-(1-7) in hypertensive transgenic (mRen2)27 rats. Clin. Sci. 2016, 130, 1247–1255. [Google Scholar] [CrossRef]
- Braszko, J.J.; Karwowska-Polecka, W.; Halicka, D.; Gard, P.R. Captopril and Enalapril Improve Cognition and Depressed Mood in Hypertensive Patients. J. Basic Clin. Physiol. Pharmacol. 2003, 14, 323–343. [Google Scholar] [CrossRef]
- Oscar, C.G.; de Figueiredo Muller-Ribeiro, F.C.; de Castro, L.G.; Lima, A.M.; Campagnolo-Santos, M.J.; Santos, R.A.S.; Xavier, C.H.; Fontes, M.A.P. Angiotensin-(1–7) in the basolateral amygdala attenuates the cardiovascular response evoked by acute emotional stress. Brain Res. 2015, 1594, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Souza, A.P.S.; Sobrinho, D.B.S.; Almeida, J.F.Q.; Alves, G.M.M.; Macedo, L.M.; Porto, J.E.; Vêncio, E.F.; Colugnati, D.B.; Santos, R.A.S.; Ferreira, A.J.; et al. Angiotensin II Type 1 receptor blockade restores angiotensin-(1-7)-induced coronary vasodilation in hypertrophic rat hearts. Clin. Sci. 2013, 125, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.A.; Lopez Verrilli, M.A.; Gomez, K.A.; Nakagawa, P.; Peña, C.; Arranz, C.; Gironacci, M.M. Angiotensin-(1-7) upregulates cardiac nitric oxide synthase in spontaneously hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H1205–H1211. [Google Scholar] [CrossRef] [PubMed]
- Gomes, E.R.M.; Santos, R.A.S.; Guatimosim, S. Angiotensin-(1-7)-Mediated Signaling in Cardiomyocytes. Int. J. Hypertens. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.J.; Moraes, P.L.; Foureaux, G.; Andrade, A.B.; Santos, R.A.; Almeida, A.P. The angiotensin-(1-7)/Mas receptor axis is expressed in sinoatrial node cells of rats. J. Histochem. Cytochem. 2011, 59, 761–768. [Google Scholar] [CrossRef]
- De Mello, W.C.; Ferrario, C.M.; Jessup, J.A. Beneficial versus harmful effects of Angiotensin (1-7) on impulse propagation and cardiac arrhythmias in the failing heart. J. Renin Angiotensin Aldosterone Syst. 2007, 8, 74–80. [Google Scholar] [CrossRef]
- Lin, L.; Liu, X.; Xu, J.; Weng, L.; Ren, J.F.; Ge, J.; Zou, Y. Mas receptor mediates cardioprotection of angiotensin-(1-7) against Angiotensin II-induced cardiomyocyte autophagy and cardiac remodelling through inhibition of oxidative stress. J. Cell. Mol. Med. 2016, 20, 48–57. [Google Scholar] [CrossRef]
- McCollum, L.T.; Gallagher, P.E.; Tallant, E.A. Angiotensin-(1-7) abrogates mitogen-stimulated proliferation of cardiac fibroblasts. Peptides 2012, 34, 380–388. [Google Scholar] [CrossRef]
- Chang, R.L.; Lin, J.W.; Kuo, W.W.; Hsieh, D.J.; Yeh, Y.L.; Shen, C.Y.; Day, C.H.; Ho, T.J.; Viswanadha, V.P.; Huang, C.Y. Angiotensin-(1-7) attenuated long-term hypoxia-stimulated cardiomyocyte apoptosis by inhibiting HIF-1α nuclear translocation via Mas receptor regulation. Growth Factors 2016, 34, 11–18. [Google Scholar] [CrossRef]
- Donoghue, M.; Wakimoto, H.; Maguire, C.T.; Acton, S.; Hales, P.; Stagliano, N.; Fairchild-Huntress, V.; Xu, J.; Lorenz, J.N.; Kadambi, V. Heart block, ventricular tachycardia, and sudden death in ACE2 transgenic mice with downregulated connexins. J. Mol. Cell. Cardiol. 2003, 35, 1043–1053. [Google Scholar] [CrossRef]
- Wysocka, M.B.; Pietraszek-Gremplewicz, K.; Nowak, D. The Role of Apelin in Cardiovascular Diseases, Obesity and Cancer. Front. Physiol. 2018, 9, 557. [Google Scholar] [CrossRef] [PubMed]
- Schiavone, M.T.; Santos, R.A.; Brosnihan, K.B.; Khosla, M.C.; Ferrario, C.M. Release of vasopressin from the rat hypothalamo-neurohypophysial system by angiotensin-(1-7) heptapeptide. Proc. Natl. Acad. Sci. USA 1988, 85, 4095–4098. [Google Scholar] [CrossRef] [PubMed]
- Baracho, N.C.; Simões-e-Silva, A.C.; Khosla, M.C.; Santos, R.A. Effect of selective angiotensin antagonists on the antidiuresis produced by angiotensin-(1-7) in water-loaded rats. Braz. J. Med. Biol. Res. 1998, 31, 1221–1227. [Google Scholar] [CrossRef] [PubMed]
- Magaldi, A.J.; Cesar, K.R.; de Araújo, M.; Simões e Silva, A.C.; Santos, R.A. Angiotensin-(1-7) stimulates water transport in rat inner medullary collecting duct: Evidence for involvement of vasopressin V2 receptors. Pflugers Arch. 2003, 447, 223–230. [Google Scholar] [CrossRef]
- Ren, Y.; Garvin, J.L.; Carretero, O.A. Vasodilator action of angiotensin-(1-7) on isolated rabbit afferent arterioles. Hypertension 2002, 39, 799–802. [Google Scholar] [CrossRef]
- van Twist, D.J.; Houben, A.J.; de Haan, M.W.; Mostard, G.J.; Kroon, A.A.; de Leeuw, P.W. Angiotensin-(1-7)-induced renal vasodilation in hypertensive humans is attenuated by low sodium intake and angiotensin II co-infusion. Hypertension 2013, 62, 789–793. [Google Scholar] [CrossRef]
- Li, P.; Chappell, M.C.; Ferrario, C.M.; Brosnihan, K.B. Angiotensin-(1-7) augments bradykinin-induced vasodilation by competing with ACE and releasing nitric oxide. Hypertension 1997, 29, 394–400. [Google Scholar] [CrossRef]
- Tallant, E.A.; Clark, M.A. Molecular mechanisms of inhibition of vascular growth by angiotensin-(1-7). Hypertension 2003, 42, 574–579. [Google Scholar] [CrossRef]
- Fraga-Silva, R.A.; Costa-Fraga, F.P.; De Sousa, F.B.; Alenina, N.; Bader, M.; Sinisterra, R.D.; Santos, R.A. An orally active formulation of angiotensin-(1-7) produces an antithrombotic effect. Clinics 2011, 66, 837–841. [Google Scholar] [CrossRef]
- Langeveld, B.; van Gilst, W.H.; Tio, R.A.; Zijlstra, F.; Roks, A.J. Angiotensin-(1-7) attenuates neointimal formation after stent implantation in the rat. Hypertension 2005, 45, 138–141. [Google Scholar] [CrossRef] [Green Version]
- Sui, Y.B.; Chang, J.R.; Chen, W.J.; Zhao, L.; Zhang, B.H.; Yu, Y.R.; Tang, C.S.; Yin, X.H.; Qi, Y.F. Angiotensin-(1-7) inhibits vascular calcification in rats. Peptides 2013, 42, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Stavrou, E.; Schmaier, A.A.; Grobe, N.; Morris, M.; Chen, A.; Nieman, M.T.; Adams, G.N.; LaRusch, G.; Zhou, Y.; et al. Angiotensin 1-7 and Mas decrease thrombosis in Bdkrb2-/- mice by increasing NO and prostacyclin to reduce platelet spreading and glycoprotein VI activation. Blood 2013, 121, 3023–3032. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, W.O.; Nascimento, A.A.S.; Santos, R.A.S. Regulation of Cardiovascular Signaling by Kinins and Products of Similar Converting Enzyme Systems. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H1985–H1994. [Google Scholar] [CrossRef]
- Sampaio, W.O.; Henrique de Castro, C.; Santos, R.A.S.; Schiffrin, E.L.; Touyz, R.M. Angiotensin-(1-7) counterregulates angiotensin II signaling in human endothelial cells. Hypertension 2007, 50, 1093–1098. [Google Scholar] [CrossRef] [PubMed]
- Verano-Braga, T.; Schwämmle, V.; Sylvester, M.; Passos-Silva, D.G.; Peluso, A.A.B.; Etelvino, G.M.; Santos, R.A.S.; Roepstorff, P. Time-resolved quantitative phosphoproteomics: New insights into Angiotensin-(1-7) signaling networks in human endothelial cells. J. Proteome Res. 2012, 11, 3370–3381. [Google Scholar] [CrossRef]
- Pernomian, L.; Gomes, M.S.; Restini, C.B.; de Oliveira, A.M. MAS-mediated antioxidant effects restore the functionality of angiotensin converting enzyme 2-angiotensin-(1-7)-MAS axis in diabetic rat carotid. BioMed Res. Int. 2014, 2014, 640329. [Google Scholar] [CrossRef]
- Zhang, F.; Ren, J.; Chan, K.; Chen, H. Angiotensin-(1-7) regulates Angiotensin II-induced VCAM-1 expression on vascular endothelial cells. Biochem. Biophys. Res. Commun. 2013, 430, 642–646. [Google Scholar] [CrossRef]
- Rentzsch, B.; Todiras, M.; Iliescu, R.; Popova, E.; Campos, L.A.; Oliveira, M.L.; Baltatu, O.C.; Santos, R.A.; Bader, M. Transgenic angiotensin-converting enzyme 2 overexpression in vessels of SHRSP rats reduces blood pressure and improves endothelial function. Hypertension 2008, 52, 967–973. [Google Scholar] [CrossRef]
- Alenina, N.; Xu, P.; Rentzsch, B.; Patkin, E.L.; Bader, M. Genetically altered animal models for Mas and angiotensin-(1-7). Exp. Physiol. 2008, 93, 528–537. [Google Scholar] [CrossRef]
- Regenhardt, R.W.; Mecca, A.P.; Desland, F.; Ritucci-Chinni, P.F.; Ludin, J.A.; Greenstein, D.; Banuelos, C.; Bizon, J.L.; Reinhard, M.K.; Sumners, C. Centrally administered angiotensin-(1-7) increases the survival of stroke-prone spontaneously hypertensive rats. Exp. Physiol. 2014, 99, 442–453. [Google Scholar] [CrossRef]
- Helms, J.; Kremer, S.; Merdji, H.; Clere-Jehl, R.; Schenck, M.; Kummerlen, C.; Collange, O.; Boulay, C.; Fafi-Kremer, S.; Ohana, M.; et al. Neurologic Features in Severe SARS-CoV-2 Infection. N. Engl. J. Med. 2020, 382, 2268–2270. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.C.; Kern, F.; Losada, P.M.; Agam, M.R.; Maat, C.A.; Schmartz, G.P.; Fehlmann, T.; Stein, J.A.; Schaum, N.; Lee, D.P.; et al. Dysregulation of brain and choroid plexus cell types in severe COVID-19. Nature 2021, 595, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Chertow, D.; Stein, S.; Ramelli, S.; Grazioli, A.; Chung, J.Y.; Singh, M.; Yinda, C.K.; Winkler, C.; Dickey, J.; Ylaya, K.; et al. SARS-CoV-2 infection and persistence throughout the human body and brain. Res. Sq. 2021; preprint under review. [Google Scholar] [CrossRef]
- Paterson, R.W.; Brown, R.L.; Benjamin, L.; Nortley, R.; Wiethoff, S.; Bharucha, T.; Jayaseelan, D.L.; Kumar, G.; Raftopoulos, R.E.; Zambreanu, L.; et al. The emerging spectrum of COVID-19 neurology: Clinical, radiological and laboratory findings. Brain 2020, 143, 3104–3120. [Google Scholar] [CrossRef] [PubMed]
- Lechien, J.R.; Chiesa-Estomba, C.M.; De Siati, D.R.; Horoi, M.; Le Bon, S.D.; Rodriguez, A.; Dequanter, D.; Blecic, S.; El Afia, F.; Distinguin, L.; et al. Olfactory and gustatory dysfunctions as a clinical presentation of mild-to-moderate forms of the coronavirus disease (COVID-19): A multicenter European study. Eur. Arch. Otorhinolaryngol. 2020, 277, 2251–2261. [Google Scholar] [CrossRef] [PubMed]
- Postma, E.M.; Smeets, P.A.M.; Boek, W.M.; Boesveldt, S. Investigating morphological changes in the brain in relation to etiology and duration of olfactory dysfunction with voxel-based morphometry. Sci. Rep. 2021, 11, 12704. [Google Scholar] [CrossRef]
- Carmichael, S.T.; Clugnet, M.C.; Price, J.L. Central olfactory connections in the macaque monkey. J. Comp. Neurol. 1994, 346, 403–434. [Google Scholar] [CrossRef]
- Douaud, G.; Lee, S.; Alfaro-Almagro, F.; Arthofer, C.; Wang, C.; McCarthy, P.; Lange, F.; Andersson, J.; Griffanti, L.; Duff, E.; et al. SARS-CoV-2 is associated with changes in brain structure in UK Biobank. Nature 2022, 604, 697–707. [Google Scholar] [CrossRef]
- Metzger, R.; Bader, M.; Ludwig, T.; Berberich, C.; Bunnemann, B.; Ganten, D. Expression of the mouse and rat mas proto-oncogene in the brain and peripheral tissues. FEBS lett. 1995, 357, 27–32. [Google Scholar] [CrossRef]
- Hellner, K.; Walther, T.; Schubert, M.; Albrecht, D. Angiotensin-(1-7) enhances LTP in the hippocampus through the G-protein-coupled receptor Mas. Mol. Cell Neurosci. 2005, 2, 427–435. [Google Scholar] [CrossRef]
- Valenzano, A.; Scarinci, A.; Monda, V.; Sessa, F.; Messina, A.; Monda, M.; Precenzano, F.; Mollica, M.P.; Carotenuto, M.; Messina, G.; et al. The Social Brain and Emotional Contagion: COVID-19 Effects. Medicina 2020, 56, 640. [Google Scholar] [CrossRef] [PubMed]
- Hellmuth, J.; Barnett, T.A.; Asken, B.M.; Kelly, J.D.; Torres, L.; Stephens, M.L.; Greenhouse, B.; Martin, J.N.; Chow, F.C.; Deeks, E.; et al. Persistent COVID-19-associated neurocognitive symptoms in non-hospitalized patients. J. Neurovirol. 2021, 27, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Chaar, L.J.; Alves, T.P.; Batista Junior, A.M.; Michelini, L.C. Early Training-Induced Reduction of Angiotensinogen in Autonomic Areas-The Main Effect of Exercise on Brain Renin-Angiotensin System in Hypertensive Rats. PLoS ONE 2015, 10, e0137395. [Google Scholar] [CrossRef] [PubMed]
- Kar, S.; Gao, L.; Zucker, I.H. Exercise training normalizes ACE and ACE2 in the brain of rabbits with pacing-induced heart failure. J. Appl. Physiol. 2010, 108, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Renaud-Charest, O.; Lui, L.M.W.; Eskander, S.; Ceban, F.; Ho, R.; Di Vincenzo, J.D.; Rosenblat, J.D.; Lee, Y.; Subramaniapillai, M.; McIntyre, R.S. Onset and frequency of depression in post-COVID-19 syndrome: A systematic review. Psychiatr. Res. 2021, 144, 129–137. [Google Scholar] [CrossRef] [PubMed]
- da Silva Lopes, L.; Silva, R.O.; de Sousa Lima, G.; de Araújo Costa, A.C.; Barros, D.F.; Silva-Néto, R.P. Is there a common pathophysiological mechanism between COVID-19 and depression? Acta Neurol. Belg. 2021, 121, 1117–1122. [Google Scholar] [CrossRef]
- Mazza, M.G.; De Lorenzo, R.; Conte, C.; Poletti, S.; Vai, B.; Bollettini, I.; Melloni, E.; Furlan, R.; Ciceri, F.; Rovere-Querini, P. Anxiety and depression in COVID-19 survivors: Role of inflammatory and clinical predictors. Brain Behav. Immun. 2020, 89, 594–600. [Google Scholar] [CrossRef]
- Li, B.; Yang, J.; Zhao, F.; Zhi, L.; Wang, X.; Liu, L.; Bi, Z.; Zhao, Y. Prevalence and impact of cardiovascular metabolic diseases on COVID-19 in China. Clin. Res. Cardiol. 2020, 109, 531–538. [Google Scholar] [CrossRef]
- Guo, T.; Fan, Y.; Chen, M.; Wu, X.; Zhang, L.; He, T.; Wang, H.; Wan, J.; Wang, X.; Lu, Z. Cardiovascular Implications of Fatal Outcomes of Patients with Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020, 5, 811–818. [Google Scholar] [CrossRef]
- Chen, C.; Zhou, Y.; Wang, D.W. SARS-CoV-2: A potential novel etiology of fulminant myocarditis. Herz 2020, 45, 230–232. [Google Scholar] [CrossRef] [Green Version]
- Immazio, M.; Klingel, K.; Kindermann, I.; Brucato, A.; De Rosa, F.G.; Adler, Y.; De Ferrari, G.M. COVID-19 pandemic and troponin: Indirect myocardial injury, myocardial inflammation or myocarditis? Heart 2020, 106, 1127–1131. [Google Scholar] [CrossRef]
- Lippi, G.; Lavie, C.J.; Sanchis-Gomar, F. Cardiac troponin I in patients with coronavirus disease 2019 (COVID-19): Evidence from a meta-analysis. Prog. Cardiovasc. Dis. 2020, 63, 390–391. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, B.A.; Torres, P.; Ramos-Tuarez, F.; Dewaswala, N.; Abdallah, A.; Chen, K.; Qader, M.A.; Job, R.; Aboulenain, S.; Dziadkwiec, K.; et al. Cardiac Troponin-I and COVID-19: A Prognostic Tool for In-Hospital Mortality. Cardiol. Res. 2020, 11, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus-Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Kochi, A.N.; Tagliari, A.P.; Forleo, G.B.; Fassini, G.M.; Tondo, C. Cardiac and arrhythmic complications in patients with COVID-19. J. Cardiovasc. Electrophysiol. 2020, 31, 1003–1008. [Google Scholar] [CrossRef]
- Fu, E.L.; Janse, R.J.; de Jong, Y.; van der Endt, V.H.W.; Milders, J.; van der Willik, E.M.; de Rooij, E.N.M.; Dekkers, O.M.; Rotmans, J.I.; van Diepen, M. Acute kidney injury and kidney replacement therapy in COVID-19: A systematic review and meta-analysis. Clin. Kidney J. 2020, 13, 550–563. [Google Scholar] [CrossRef]
- Golmai, P.; Larsen, C.P.; DeVita, M.V.; Wahl, S.J.; Weins, A.; Rennke, H.G.; Bijol, V.; Rosenstock, J.L. Histopathologic and Ultrastructural Findings in Postmortem Kidney Biopsy Material in 12 Patients with AKI and COVID-19. J. Am. Soc. Nephrol. 2020, 31, 1944–1947. [Google Scholar] [CrossRef]
- Akilesh, S.; Nast, C.C.; Yamashita, M.; Henriksen, K.; Charu, V.; Troxell, M.L.; Kambham, N.; Bracamonte, E.; Houghton, D.; Ahmed, N.I.; et al. Multicenter Clinicopathologic Correlation of Kidney Biopsies Performed in COVID-19 Patients Presenting with Acute Kidney Injury or Proteinuria. Am. J. Kidney Dis. 2021, 77, 82–93. [Google Scholar] [CrossRef]
- Sharma, P.; Uppal, N.N.; Wanchoo, R.; Shah, H.H.; Yang, Y.; Parikh, R.; Khanin, Y.; Madireddy, V.; Larsen, C.P.; Jhaveri, K.D.; et al. COVID-19-Associated Kidney Injury: A Case Series of Kidney Biopsy Findings. J. Am. Soc. Nephrol. 2020, 31, 1948–1958. [Google Scholar] [CrossRef]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Rapkiewicz, A.V.; Mai, X.; Carsons, S.E.; Pittaluga, S.; Kleiner, D.E.; Berger, J.S. Megakaryocytes and platelet-fibrin thrombi characterize multi-organ thrombosis at autopsy in COVID-19: A case series. EClinicalMedicine 2020, 24. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, Y.; Wang, X.; Yang, L.; Li, H.; Wang, Y.; Liu, M.; Zhao, X.; Xie, Y.; Yang, Y.; et al. SARS-CoV-2 binds platelet ACE2 to enhance thrombosis in COVID-19. J. Hematol. Oncol. 2020, 13, 120. [Google Scholar] [CrossRef] [PubMed]
- Cantaluppi, V.; Quercia, A.D.; Dellepiane, S.; Ferrario, S.; Camussi, G.; Biancone, L. Interaction between systemic inflammation and renal tubular epithelial cells. Nephrol. Dial. Transplant. 2014, 29, 2004–2011. [Google Scholar] [CrossRef] [PubMed]
- Gurkan, S.; Cabinian, A.; Lopez, V.; Bhaumik, M.; Chang, J.M.; Rabson, A.B.; Mundel, P. Inhibition of type I interferon signalling prevents TLR ligand-mediated proteinuria. J. Pathol. 2013, 231, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Cugno, M.; Meroni, P.L.; Gualtierotti, R.; Griffini, S.; Grovetti, E.; Torri, A.; Panigada, M.; Aliberti, S.; Blasi, F.; Tedesco, F. Complement activation in patients with COVID-19: A novel therapeutic. J. Allergy Clin. Immunol. 2020, 146, 215–217. [Google Scholar] [CrossRef]
- Giamarellos-Bourboulis, E.J.; Netea, M.G.; Rovina, N.; Akinosoglou, K.; Antoniadou, A.; Antonakos, N.; Damoraki, G.; Gkavogianni, T.; Adami, M.E.; Katsaounou, P.; et al. Complex Immune Dysregulation in COVID-19 Patients with Severe Respiratory Failure. Cell Host Microbe 2020, 27, 992–1000. [Google Scholar] [CrossRef]
- Richardson, K.L.; Jain, A.; Evans, J.; Uzun, O. Giant coronary artery aneurysm as a feature of coronavirus-related inflammatory syndrome. BMJ Case Rep. 2021, 14, e238740. [Google Scholar] [CrossRef]
- Navaeifar, M.R.; Shahbaznejad, L.; Sadeghi Lotfabadi, A.; Rezai, M.S. COVID-19-Associated Multisystem Inflammatory Syndrome Complicated with Giant Coronary Artery Aneurysm. Case Rep. Pediatr. 2021, 2021, 8836403. [Google Scholar] [CrossRef]
- Pick, J.M.; Wang, S.; Wagner-Lees, S.; Badran, S.; Szmuszkovicz, R.J.; Wong, P.; Votava-Smith, J. Abstract 17092: Coronary Artery Aneurysms Are More Common in Post-COVID-19 Multisystem Inflammatory Syndrome in Children (MIS-C) Than Pre-Pandemic Kawasaki Disease. Circulation 2020, 142, 17092. [Google Scholar] [CrossRef]
- Zhang, C.; Shen, L.; Le, K.J.; Pan, M.M.; Kong, L.C.; Gu, Z.C.; Xu, H.; Zhang, Z.; Ge, W.H.; Lin, H.W. Incidence of Venous Thromboembolism in Hospitalized Coronavirus Disease 2019 Patients: A Systematic Review and Meta-Analysis. Front. Cardiovasc. Med. 2020, 7, 151. [Google Scholar] [CrossRef]
- Birkeland, K.; Zimmer, R.; Kimchi, A.; Kedan, I. Venous Thromboembolism in Hospitalized COVID-19 Patients: Systematic Review. Interact. J. Med. Res. 2020, 9, e22768. [Google Scholar] [CrossRef]
- Bangalore, S.; Sharma, A.; Slotwiner, A.; Yatskar, L.; Harari, R.; Shah, B.; Ibrahim, H.; Friedman, G.H.; Thompson, C.; Alviar, C.L. ST-Segment Elevation in Patients with COVID-19—A Case Series. N. Engl. J. Med. 2020, 382, 2478–2480. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, S.; Mendes, M.C.; Camargo Martins, A.P.; Borges, N.H.; Godoy, T.M.; Miggiolaro, A.; da Silva Dezidério, F.; Machado-Souza, C.; de Noronha, L. Endothelial Dysfunction and Thrombosis in Patients With COVID-19-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2404–2407. [Google Scholar] [CrossRef] [PubMed]
- Götzinger, F.; Santiago-García, B.; Noguera-Julián, A.; Lanaspa, M.; Lancella, L.; Calò Carducci, F.I.; Gabrovska, N.; Velizarova, S.; Prunk, P.; Osterman, V.; et al. COVID-19 in children and adolescents in Europe: A multinational, multicentre cohort study. Lancet Child Adolesc. Health 2020, 4, 653–661. [Google Scholar] [CrossRef]
- King, J.A.; Whitten, T.A.; Bakal, J.A.; McAlister, F.A. Symptoms associated with a positive result for a swab for SARS-CoV-2 infection among children in Alberta. CMAJ 2021, 193, E1–E9. [Google Scholar] [CrossRef]
- Lu, X.; Zhang, L.; Du, H.; Zhang, J.; Li, Y.Y.; Qu, J.; Zhang, W.; Wang, Y.; Bao, S.; Li, Y.; et al. Chinese Pediatric Novel Coronavirus Study Team (2020). SARS-CoV-2 Infection in Children. N. Engl. J. Med. 2020, 382, 1663–1665. [Google Scholar] [CrossRef] [PubMed]
- Midulla, F.; Cristiani, L.; Mancino, E. Will children reveal their secret? The coronavirus dilemma. Eur. Respir. J. 2020, 55. [Google Scholar] [CrossRef]
- Nikolopoulou, G.B.; Maltezou, H.C. COVID-19 in Children: Where do we Stand? Arch. Med. Res. 2022, 53, 1–8. [Google Scholar] [CrossRef]
- Castagnoli, R.; Votto, M.; Licari, A.; Brambilla, I.; Bruno, R.; Perlini, S.; Rovida, F.; Baldanti, F.; Marseglia, G.L. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Infection in Children and Adolescents: A Systematic Review. JAMA Pediatr. 2020, 174, 882–889. [Google Scholar] [CrossRef]
- Siebach, M.K.; Piedimonte, G.; Ley, S.H. COVID-19 in childhood: Transmission, clinical presentation, complications and risk factors. Pediatr. Pulmonol. 2021, 56, 1342–1356. [Google Scholar] [CrossRef]
- Bastolla, U.; Chambers, P.; Abia, D.; Garcia-Bermejo, M.L.; Fresno, M. Is COVID-19 Severity Associated with ACE2 Degradation? Front. Drug. Discov. 2022, 1, 789710. [Google Scholar] [CrossRef]
- Dong, Y.; Mo, X.; Hu, Y.; Qi, X.; Jiang, F.; Jiang, Z.; Tong, S. Epidemiology of COVID-19 Among Children in China. Pediatrics 2020, 145, e20200702. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Hu, N.; Lou, J.; Chen, K.; Kang, X.; Xiang, Z.; Chen, H.; Wang, D.; Liu, N.; Liu, D.; et al. Characteristics of COVID-19 infection in Beijing. J. Infect. 2020, 80, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Kolberg, E.S.; Wickstrøm, K.; Tonby, K.; Dyrhol-Riise, A.M.; Holten, A.R.; Amundsen, E.K. Serum ACE as a prognostic biomarker in COVID-19: A case series. APMIS 2021, 129, 237–238. [Google Scholar] [CrossRef]
- Rieder, M.; Wirth, L.; Pollmeier, L.; Jeserich, M.; Goller, I.; Baldus, N.; Schmid, B.; Busch, H.J.; Hofmann, M.; Kern, W.; et al. Serum ACE2, Angiotensin II, and Aldosterone Levels Are Unchanged in Patients With COVID-19. Am. J. Hypertens 2021, 34, 278–281. [Google Scholar] [CrossRef]
- Gao, Y.D.; Ding, M.; Dong, X.; Zhang, J.J.; Kursat Azkur, A.; Azkur, D.; Gan, H.; Sun, Y.L.; Fu, W.; Li, W.; et al. Risk factors for severe and critically ill COVID-19 patients: A review. Allergy 2021, 76, 428–455. [Google Scholar] [CrossRef]
- Kuo, C.L.; Pilling, L.C.; Atkins, J.C.; Masoli, J.; Delgado, J.; Tignanelli, C.; Kuchel, G.; Melzer, D.; Beckman, K.B.; Levine, M. COVID-19 severity is predicted by earlier evidence of accelerated aging. medRxiv 2020. [Google Scholar] [CrossRef]
- Ran, J.; Song, Y.; Zhuang, Z.; Han, L.; Zhao, S.; Cao, P.; Geng, Y.; Xu, L.; Qin, J.; He, D.; et al. Blood pressure control and adverse outcomes of COVID-19 infection in patients with concomitant hypertension in Wuhan, China. Hypertens Res. 2020, 43, 1267–1276. [Google Scholar] [CrossRef]
- AlGhatrif, M.; Tanaka, T.; Moore, A.Z.; Bandinelli, S.; Lakatta, E.G.; Ferrucci, L. Age-associated difference in circulating ACE2, the gateway for SARS-COV-2, in humans: Results from the InCHIANTI study. GeroScience 2021, 43, 619–627. [Google Scholar] [CrossRef]
- Ferrucci, L.; Corsi, A.; Lauretani, F.; Bandinelli, S.; Bartali, B.; Taub, D.D.; Guralnik, J.M.; Longo, D.L. The origins of age-related proinflammatory state. Blood 2015, 105, 2294–2299. [Google Scholar] [CrossRef]
- Akhtar, S.; Benter, I.F.; Danjuma, M.I.; Doi, S.; Hasan, S.S.; Habib, A.M. Pharmacotherapy in COVID-19 patients: A review of ACE2-raising drugs and their clinical safety. J. Drug. Target. 2020, 28, 683–699. [Google Scholar] [CrossRef] [PubMed]
- Emilsson, V.; Gudmundsson, E.F.; Aspelund, T.; Jonsson, B.G.; Gudjonsson, A.; Launer, L.J.; Jennings, L.L.; Gudmundsdottir, V.; Gudnason, V. Antihypertensive medication uses and serum ACE2 levels: ACEIs/ARBs treatment does not raise serum levels of ACE2. medRxiv, 2020; not certified by peer review. [Google Scholar] [CrossRef]
- Gao, C.; Cai, Y.; Zhang, K.; Zhou, L.; Zhang, Y.; Zhang, X.; Li, Q.; Li, W.; Yang, S.; Zhao, X.; et al. Association of hypertension and antihypertensive treatment with COVID-19 mortality: A retrospective observational study. Eur. Heart J. 2020, 41, 2058–2066. [Google Scholar] [CrossRef] [PubMed]
- Fosbøl, E.L.; Butt, J.H.; Østergaard, L.; Andersson, C.; Selmer, C.; Kragholm, K.; Schou, M.; Phelps, M.; Gislason, G.H.; Gerds, T.A.; et al. Association of Angiotensin-Converting Enzyme Inhibitor or Angiotensin Receptor Blocker Use With COVID-19 Diagnosis and Mortality. JAMA 2020, 324, 168–177. [Google Scholar] [CrossRef]
- Summers, K.L.; Marleau, A.M.; Mahon, J.L.; McManus, R.; Hramiak, I.; Singh, B. Reduced IFN-alpha secretion by blood dendritic cells in human diabetes. Clin. Immunol. 2020, 121, 81–89. [Google Scholar] [CrossRef]
- Hu, R.; Xia, C.Q.; Butfiloski, E.; Clare-Salzle, M. Effect of high glucose on cytokine production by human peripheral blood immune cells and type I interferon signaling in monocytes: Implications for the role of hyperglycemia in the diabetes inflammatory process and host defense against infection. Clin. Immunol. 2018, 195, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Roca-Ho, H.; Riera, M.; Palau, V.; Pascual, J.; Soler, M.J. Characterization of ACE and ACE2 Expression within Different Organs of the NOD Mouse. Int. J. Mol. Sci. 2017, 18, 563. [Google Scholar] [CrossRef]
- Qeadan, F.; Mensah, N.A.; Tingey, B.; Stanford, J.B. The risk of clinical complications and death among pregnant women with COVID-19 in the Cerner COVID-19 cohort: A retrospective analysis. BMC Pregnancy Childbirth 2021, 21, 305. [Google Scholar] [CrossRef]
- Harb, J.; Debs, N.; Rima, M.; Wu, Y.; Cao, Z.; Kovacic, H.; Fajloun, Z.; Sabatier, J.M. SARS-CoV-2, COVID-19, and Reproduction: Effects on Fertility, Pregnancy, and Neonatal Life. Biomedicines 2022, 10, 1775. [Google Scholar] [CrossRef]
- Gupta, P.; Kumar, S.; Sharma, S.S. SARS-CoV-2 prevalence and maternal-perinatal outcomes among pregnant women admitted for delivery: Experience from COVID-19-dedicated maternity hospital in Jammu, Jammu and Kashmir (India). J. Med. Virol. 2021, 93, 5505–5514. [Google Scholar] [CrossRef]
- Mirbeyk, M.; Saghazadeh, A.; Rezaei, N. A systematic review of pregnant women with COVID-19 and their neonates. Arch. Gynecol. Obstet. 2021, 304, 5–38. [Google Scholar] [CrossRef]
- Thornton, P.; Douglas, J. Coagulation in pregnancy. Best Pract. Res. Clin. Obstet. Gynaecol. 2010, 24, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Mehandru, S.; Merad, M. Pathological sequelae of long-haul COVID. Nat. Immunol. 2022, 23, 194–202. [Google Scholar] [CrossRef]
- Khazaal, S.; Harb, J.; Rima, M.; Annweiler, C.; Wu, Y.; Cao, Z.; Khattar, Z.A.; Legros, C.; Kovacic, H.; Fajloun, Z.; et al. The Pathophysiology of Long COVID throughout the Renin-Angiotensin System. Molecules 2022, 27, 2903. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Huang, L.; Wang, Y.; Li, X.; Ren, L.; Gu, X.; Kang, L.; Guo, L.; Liu, M.; Zhou, X.; et al. 6-month consequences of COVID-19 in patients discharged from hospital: A cohort study. Lancet 2021, 397, 220–232. [Google Scholar] [CrossRef]
- Ledford, H. Can drugs reduce the risk of long COVID? What scientists know so far. Nature 2022, 604, 21–22. [Google Scholar] [CrossRef] [PubMed]
- Kaseda, E.T.; Levine, A.J. Post-traumatic stress disorder: A differential diagnostic consideration for COVID-19 survivors. Clin. Neuropsychol. 2020, 34, 1498–1514. [Google Scholar] [CrossRef] [PubMed]
- Asadi-Pooya, A.A.; Akbari, A.; Emami, A.; Lotfi, M.; Rostamihhoseinkhani, M.; Nemati, H.; Barzegar, Z.; Kabiri, M.; Zeraatpisheh, Z.; Farjoud-Kouhanjani, M.; et al. Long COVID syndrome-associated brain fog. J. Med. Virol. 2022, 94, 979–984. [Google Scholar] [CrossRef]
- Mikkelsen, M.E.; Christie, J.D.; Lanken, P.N.; Biester, R.C.; Thompson, B.T.; Bellamy, S.L.; Localio, A.R.; Demissie, E.; Hopkins, R.O.; Angus, D.C. The Adult Respiratory Distress Syndrome Cognitive Outcomes Study Long-Term Neuropsychological Function in Survivors of Acute Lung Injury. Am. J. Respir. Crit. Care Med. 2012, 185, 1307–1315. [Google Scholar] [CrossRef] [Green Version]
- Desai, A.D.; Lavelle, M.; Buorsiquot, B.C.; Wan, E.Y. Long-term complications of COVID-19. Am. J. Physiol. Cell. Physiol. 2022, 322, 1–11. [Google Scholar] [CrossRef]
- Hellgren, L.; Thornberg, U.B.; Samuelsson, K.; Levi, R.; Divanoglou, A.; Blystad, I. Brain MRI and neuropsychological findings at long-term follow-up after COVID-19 hospitalisation: An observational cohort study. BMJ Open 2021, 11, e055164. [Google Scholar] [CrossRef]
- Samuelson, K.W.; Bartel, A.; Valadez, R.; Jordan, J.T. PTSD symptoms and perception of cognitive problems: The roles of posttraumatic cognitions and trauma coping self-efficacy. Psychol. Trauma 2017, 9, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Richter, D.; Guasti, L.; Koehler, F.; Squizzato, A.; Nistri, S.; Christodorescu, R.; Dievart, F.; Gaudio, G.; Asteggiano, R.; Ferrini, M. Late phase of COVID-19 pandemic in General Cardiology. A position paper of the ESC Council for Cardiology Practice. ESC Heart Fail. 2021, 8, 3483–3494. [Google Scholar] [CrossRef] [PubMed]
- Goitein, O.; Matetzky, S.; Beinart, R.; Di Segni, E.; Hod, H.; Bentancur, A.; Konen, E. Acute myocarditis: Noninvasive evaluation with cardiac MRI and transthoracic echocardiography. AJR Am. J. Roentgenol. 2009, 192, 254–258. [Google Scholar] [CrossRef]
- Han, Y.; Chen, T.; Bryant, J.; Bucciarelli-Ducci, C.; Dyke, C.; Eliott, M.D.; Ferrari, V.A.; Friedrich, M.G.; Lawton, C.; Manning, W.J.; et al. Society for Cardiovascular Magnetic Resonance (SCMR) guidance for the practice of cardiovascular magnetic resonance during the COVID-19 pandemic. J. Cardiovasc. Magn. Reson. 2020, 22, 26. [Google Scholar] [CrossRef] [PubMed]
- Mele, D.; Flamigni, F.; Rapezzi, C.; Ferrari, R. Myocarditis in COVID-19 patients: Current problems. Intern. Emerg. Med. 2021, 16, 1123–1139. [Google Scholar] [CrossRef] [PubMed]
- Siripanthong, B.; Nazarian, S.; Muser, D.; Deo, R.; Santangeli, R.; Khanji, M.Y.; Cooper, L.T.; Chahal, C.A.A. Recognizing COVID-19-related myocarditis: The possible pathophysiology and proposed guideline for diagnosis and management. Heart Rhythm. 2020, 17, 1463–1471. [Google Scholar] [CrossRef]
- Santoro, F.; Monitillo, F.; Raimondo, P.; Lopizzo, A.; Brindicci, G.; Gilio, M.; Musaico, F.; Mazzola, M.; Vestito, D.; Di Benedetto, R.; et al. QTc Interval Prolongation and Life-Threatening Arrhythmias During Hospitalization in Patients with Coronavirus Disease 2019 (COVID-19): Results from a Multicenter Prospective Registry. Clin. Infect. Dis. 2021, 73, 4031–4038. [Google Scholar] [CrossRef]
- Giustino, G.; Pinney, S.P.; Lala, A.; Reddy, V.Y.; Johnston-Cox, H.A.; Mechanick, J.I.; Halperin, J.L.; Fuster, V. Coronavirus and Cardiovascular Disease, Myocardial Injury, and Arrhythmia: JACC Focus Seminar. J. Am. Coll. Cardiol. 2020, 76, 2011–2023. [Google Scholar] [CrossRef]
- Dherange, P.; Lang, J.; Qian, P.; Oberfeld, B.; Sauer, W.H.; Koplan, B.; Tedrow, U. Arrhythmias and COVID-19: A Review. JACC Clin. Electrophysiol. 2020, 6, 1193–1204. [Google Scholar] [CrossRef]
- Rudski, L.; Januzzi, J.L.; Rigolin, V.H.; Bohula, E.A.; Blankstein, R.; Patel, A.R.; Bucciarelli-Ducci, C.; Vorovich, E.; Mukheriee, M.; Rao, S.V.; et al. Multimodality Imaging in Evaluation of Cardiovascular Complications in Patients With COVID-19. J. Am. Coll. Cardiol. 2020, 76, 1345–1357. [Google Scholar] [CrossRef]
- Zoghbi, W.A.; DiCarli, M.F.; Blankstein, R.; Choi, A.D.; Dilsizian, V.; Flaschkampf, F.A.; Geske, J.B.; Grayburn, P.A.; Jaffer, F.A.; Kwong, R.Y.; et al. Multimodality Cardiovascular Imaging in the Midst of the COVID-19 Pandemic. JACC Cardiovasc. Imaging 2020, 13, 1615–1626. [Google Scholar] [CrossRef] [PubMed]
- Bozkurt, B.; Kovacs, R.; Harrington, B. Joint HFSA/ACC/AHA Statement Addresses Concerns Re: Using RAAS Antagonists in COVID-19. J. Card. Fail. 2020, 26, 370. [Google Scholar] [CrossRef] [PubMed]
- D‘Alto, M.; Romeo, E.; Argiento, P.; Pavelescu, A.; Melot, C.; D‘Andrea, A.; Corerra, A.; Bossone, E.; Calabro, R.; Russo, M.G.; et al. Echocardiographic prediction of pre- versus postcapillary pulmonary hypertension. J. Am. Soc. Echocardiogr. 2015, 28, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Sadeghipour, P.; Talasz, A.H.; Rashidi, F.; Sharif-Kashani, B.; Beigmohammadi, M.T.; Farrokhpour, M.; Sezavar, S.H.; Payandemehr, P.; Dabbagh, A.; Moghadam, K.G.; et al. Effect of Intermediate-Dose vs Standard-Dose Prophylactic Anticoagulation on Thrombotic Events, Extracorporeal Membrane Oxygenation Treatment, or Mortality Among Patients With COVID-19 Admitted to the Intensive Care Unit: The INSPIRATION Randomized Clinical Trial. JAMA 2021, 325, 1620–1630. [Google Scholar] [CrossRef] [PubMed]
- Leentjens, J.; van Haaps, T.F.; Wessels, P.F.; Schutgens, R.E.G.; Middeldrop, S. COVID-19-associated coagulopathy and antithrombotic agents-lessons after 1 year. Lancet Haematol. 2021, 8, 524–533. [Google Scholar] [CrossRef]
- Chawla, L.S.; Eggers, P.W.; Star, R.A.; Kimmel, P.L. Acute Kidney Injury and Chronic Kidney Disease as Interconnected Syndromes. N. Engl. J. Med. 2014, 371, 58–66. [Google Scholar] [CrossRef]
- Yende, S.; Parikh, C.R. Long COVID and kidney disease. Nat. Rev. Nephrol. 2021, 17, 792–793. [Google Scholar] [CrossRef]
- He, L.; Wei, Q.; Liu, J.; Yi, M.; Liu, Y.; Liu, H.; Sun, L.; Peng, Y.; Liu, F.; Venkatachalam, M.A.; et al. AKI on CKD: Heightened injury, suppressed repair, and the underlying mechanisms. Kidney Int. 2017, 92, 1071–1083. [Google Scholar] [CrossRef]
- Venkatachalam, M.A.; Griffin, K.A.; Lan, R.; Geng, H.; Saikumar, P.; Bidani, A.K. Acute kidney injury: A springboard for progression in chronic kidney disease. Am. J. Physiol. Renal. Physiol. 2010, 298, 1078–1094. [Google Scholar] [CrossRef]
- Al-Aly, Z.; Xie, Y.; Bowe, B. High-dimensional characterization of post-acute sequelae of COVID-19. Nature 2021, 594, 259–264. [Google Scholar] [CrossRef]
- Maldonado, D.; Ray, J.; Lin, X.B.; Salem, F.; Brown, M.; Bansal, I. COVAN Leading to ESKD Despite Minimal COVID Symptoms. J. Investig. Med. High Impact. Case Rep. 2022, 10, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Delanaye, P.; Huart, J.; Boquegneau, A.; Jouret, F. Long-term effects of COVID-19 on kidney function. Lancet 2021, 397, 1807. [Google Scholar] [CrossRef]
- Delanaye, P.; Jager, K.J.; Bokenkamp, A.; Christensson, A.; Douburg, L.; Eriksen, B.O.; Gaillard, F.; Gambaro, G.; van der Giet, M.; Glassock, R.J.; et al. CKD: A Call for an Age-Adapted Definition. J. Am. Soc. Nephrol. 2019, 30, 1785–1805. [Google Scholar] [CrossRef] [PubMed]
- Bruchfeld, A. The COVID-19 pandemic: Consequences for nephrology. Nat. Rev. Nephrol. 2021, 17, 81–82. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maranduca, M.A.; Tanase, D.M.; Cozma, C.T.; Dima, N.; Clim, A.; Pinzariu, A.C.; Serban, D.N.; Serban, I.L. The Impact of Angiotensin-Converting Enzyme-2/Angiotensin 1-7 Axis in Establishing Severe COVID-19 Consequences. Pharmaceutics 2022, 14, 1906. https://doi.org/10.3390/pharmaceutics14091906
Maranduca MA, Tanase DM, Cozma CT, Dima N, Clim A, Pinzariu AC, Serban DN, Serban IL. The Impact of Angiotensin-Converting Enzyme-2/Angiotensin 1-7 Axis in Establishing Severe COVID-19 Consequences. Pharmaceutics. 2022; 14(9):1906. https://doi.org/10.3390/pharmaceutics14091906
Chicago/Turabian StyleMaranduca, Minela Aida, Daniela Maria Tanase, Cristian Tudor Cozma, Nicoleta Dima, Andreea Clim, Alin Constantin Pinzariu, Dragomir Nicolae Serban, and Ionela Lacramioara Serban. 2022. "The Impact of Angiotensin-Converting Enzyme-2/Angiotensin 1-7 Axis in Establishing Severe COVID-19 Consequences" Pharmaceutics 14, no. 9: 1906. https://doi.org/10.3390/pharmaceutics14091906