
Quality by Design-Driven Zeta Potential Optimisation Study of Liposomes with Charge Imparting Membrane Additives

 ,
,  , , , ,
, , , ,  and
and
Abstract
:1. Introduction
1.1. Liposomal Formulations
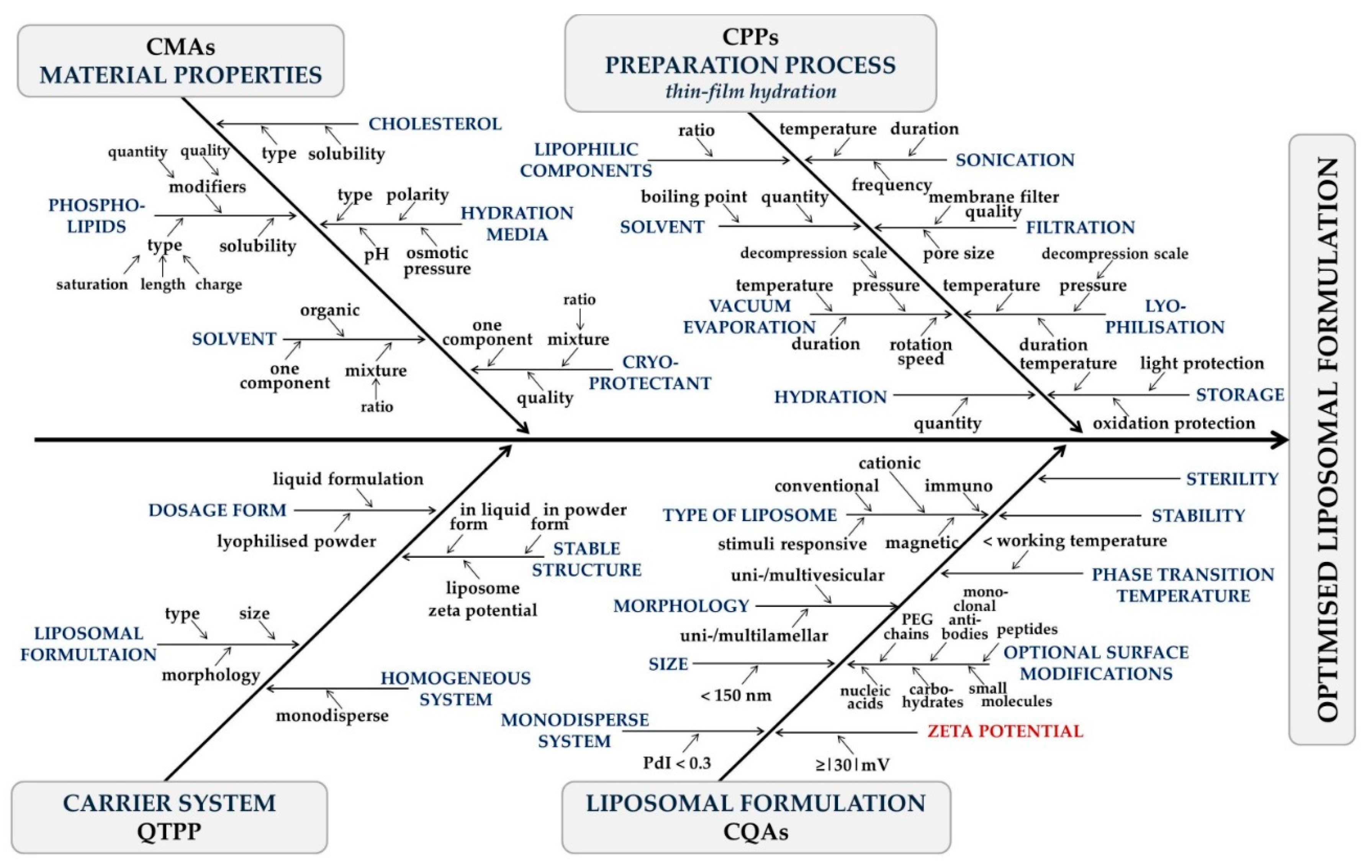
1.2. Quality by Design-Based Design and Development
1.3. Importance of Zeta Potential
1.4. Modification of Zeta Potential
2. Materials and Methods
2.1. Materials
2.2. Methods
2.2.1. Factorial Design-Based Experiment Design for Zeta Potential Optimisation
2.2.2. Preparation of Liposomes
2.2.3. Characterisation of Liposomes
Vesicle Size and Zeta Potential Analysis
Atomic Force Microscopy (AFM)
Transmission Electron Microscopy (TEM)
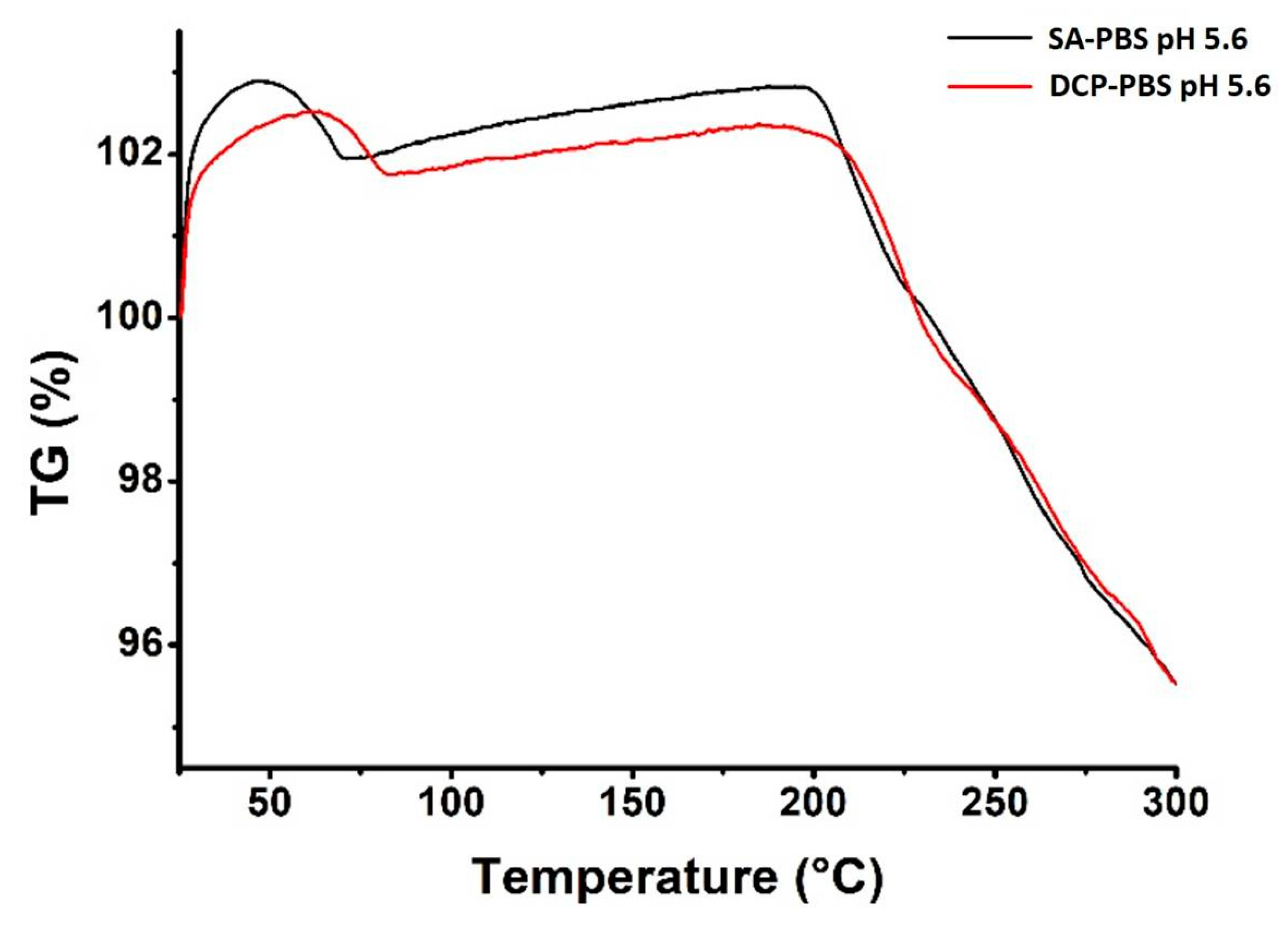
Thermal Analysis
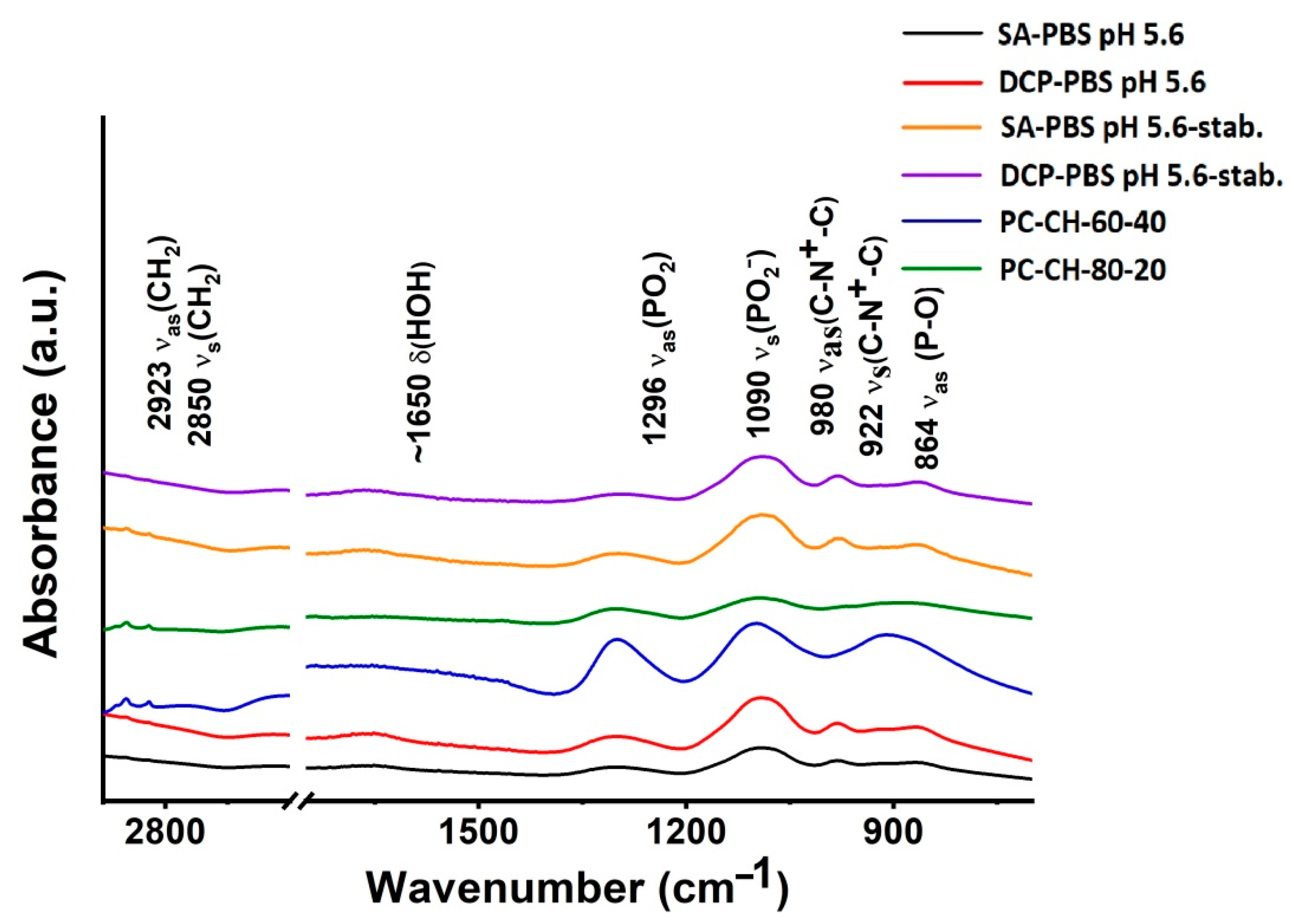
Fourier Transform Infrared Spectroscopy (FT-IR)
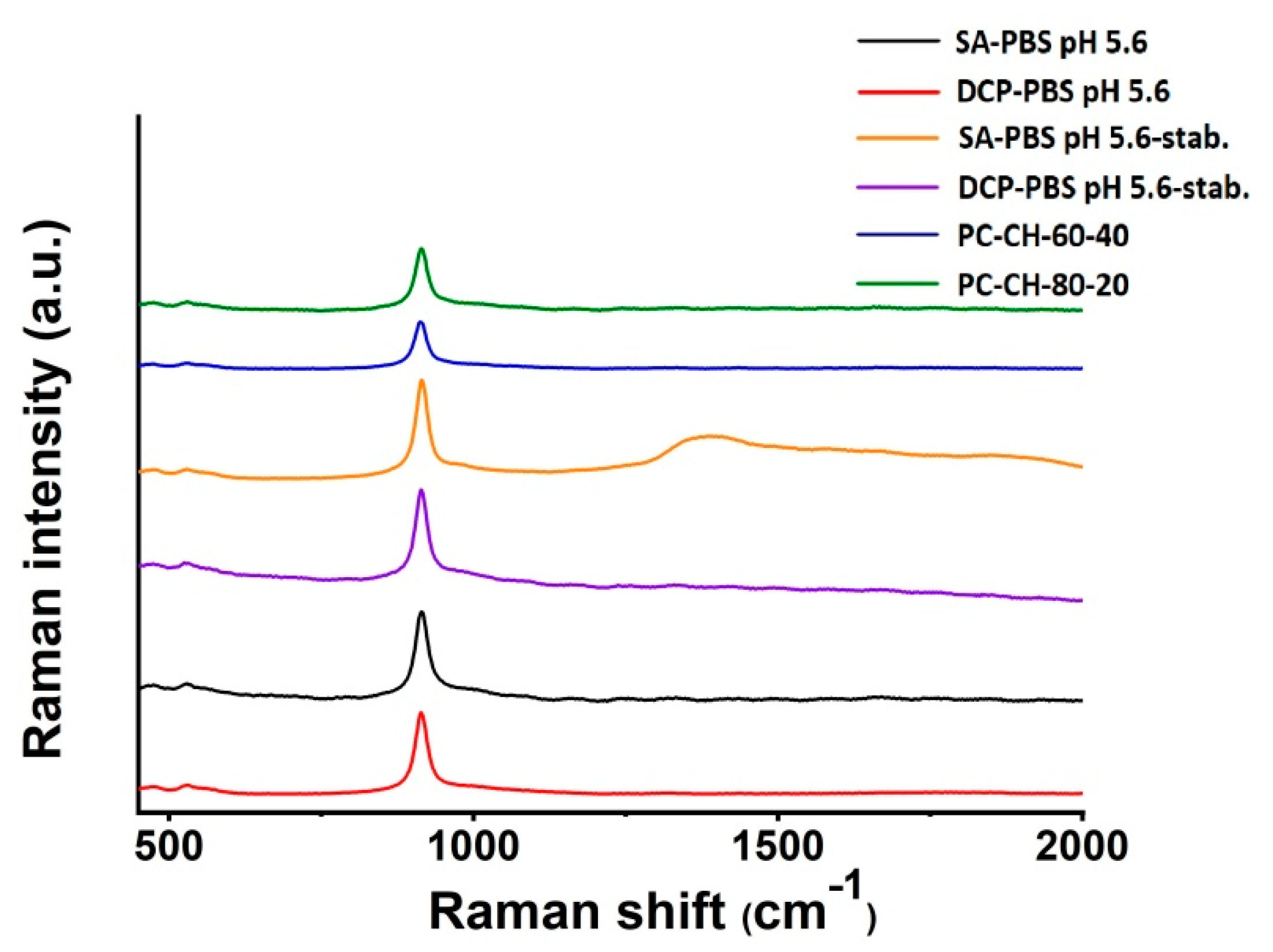
Raman Spectroscopy
Residual Ethanol Measurement via Gas Chromatography-Mass Spectrometry (GC-MS)
2.2.4. Physical Stability Studies
2.2.5. Statistical Analysis
3. Results
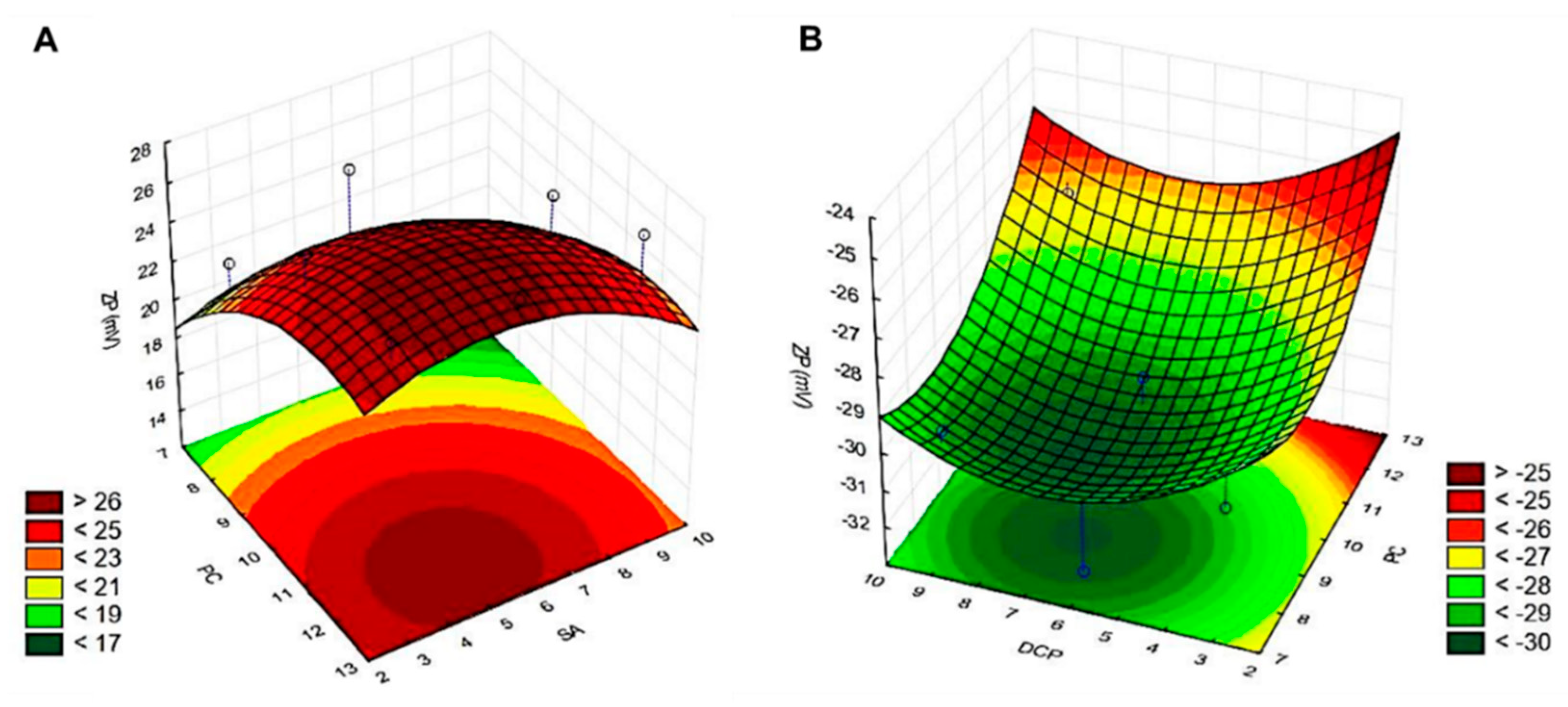
3.1. Factorial Design-Based Experimental Design for Zeta Potential Optimisation
3.2. Characterisation of Liposomes
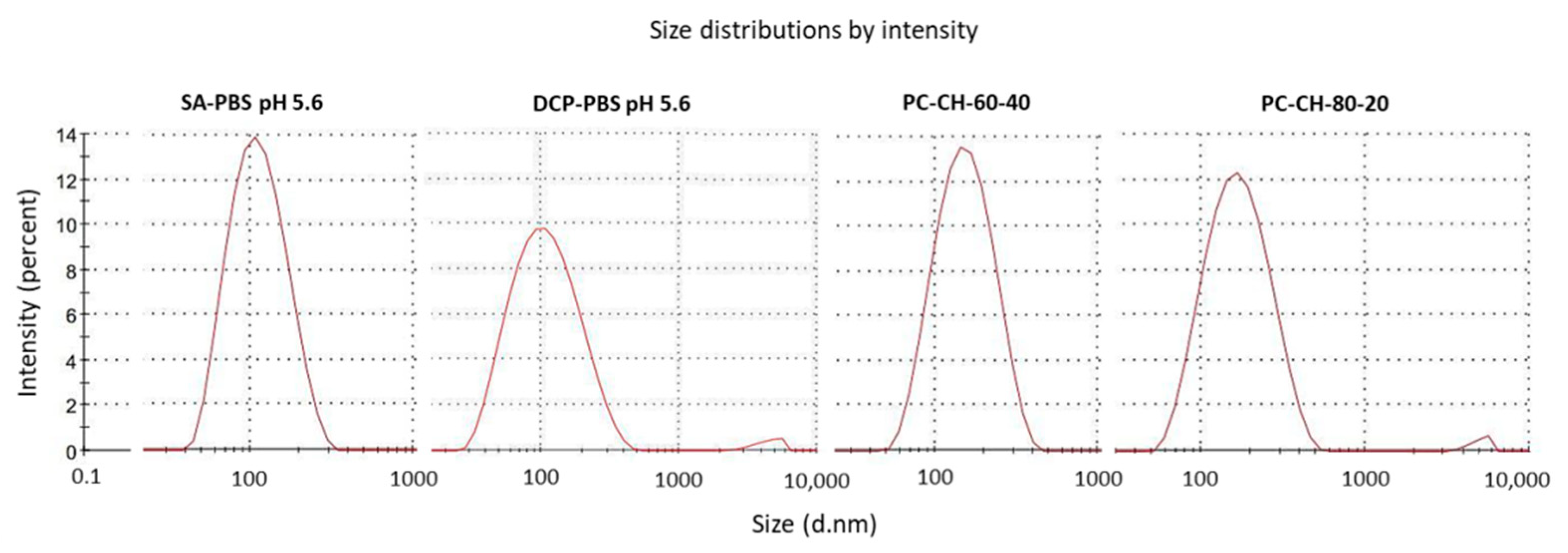
3.2.1. Vesicle Size and Zeta Potential Analysis
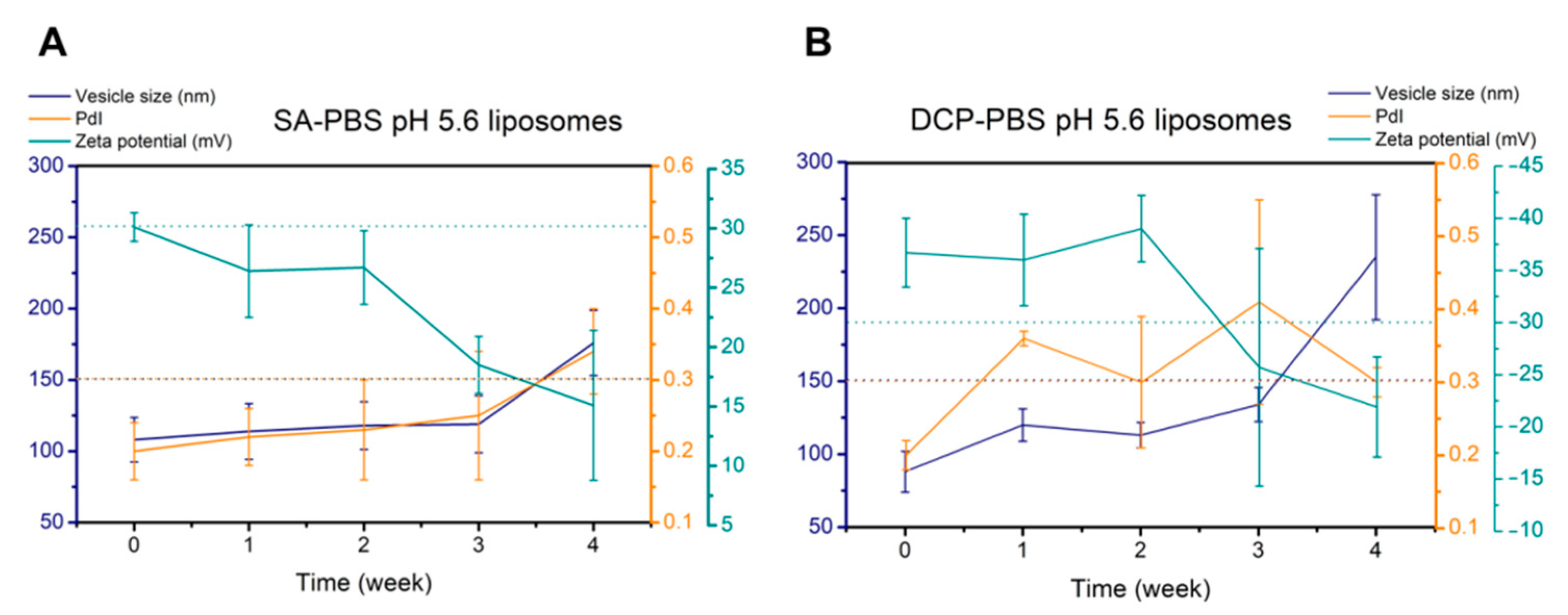
3.2.2. Physical Stability Studies
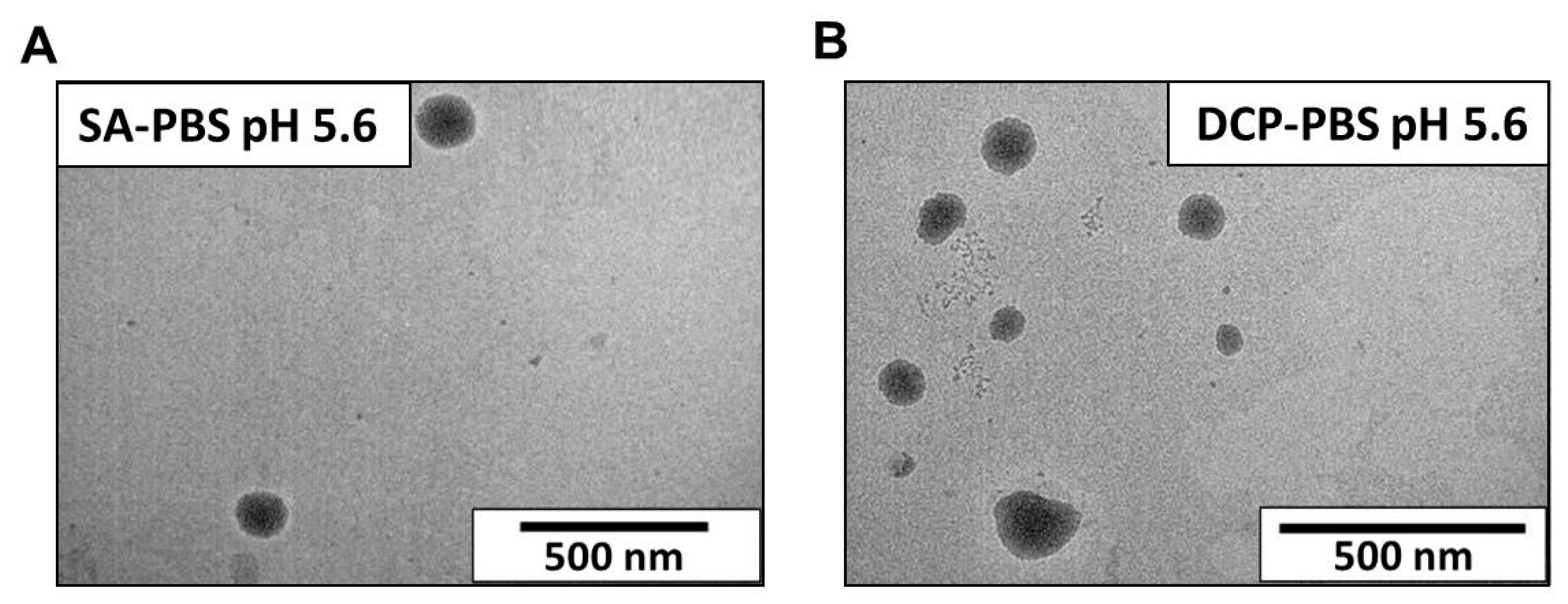
3.2.3. Transmission Electron Microscopy (TEM)
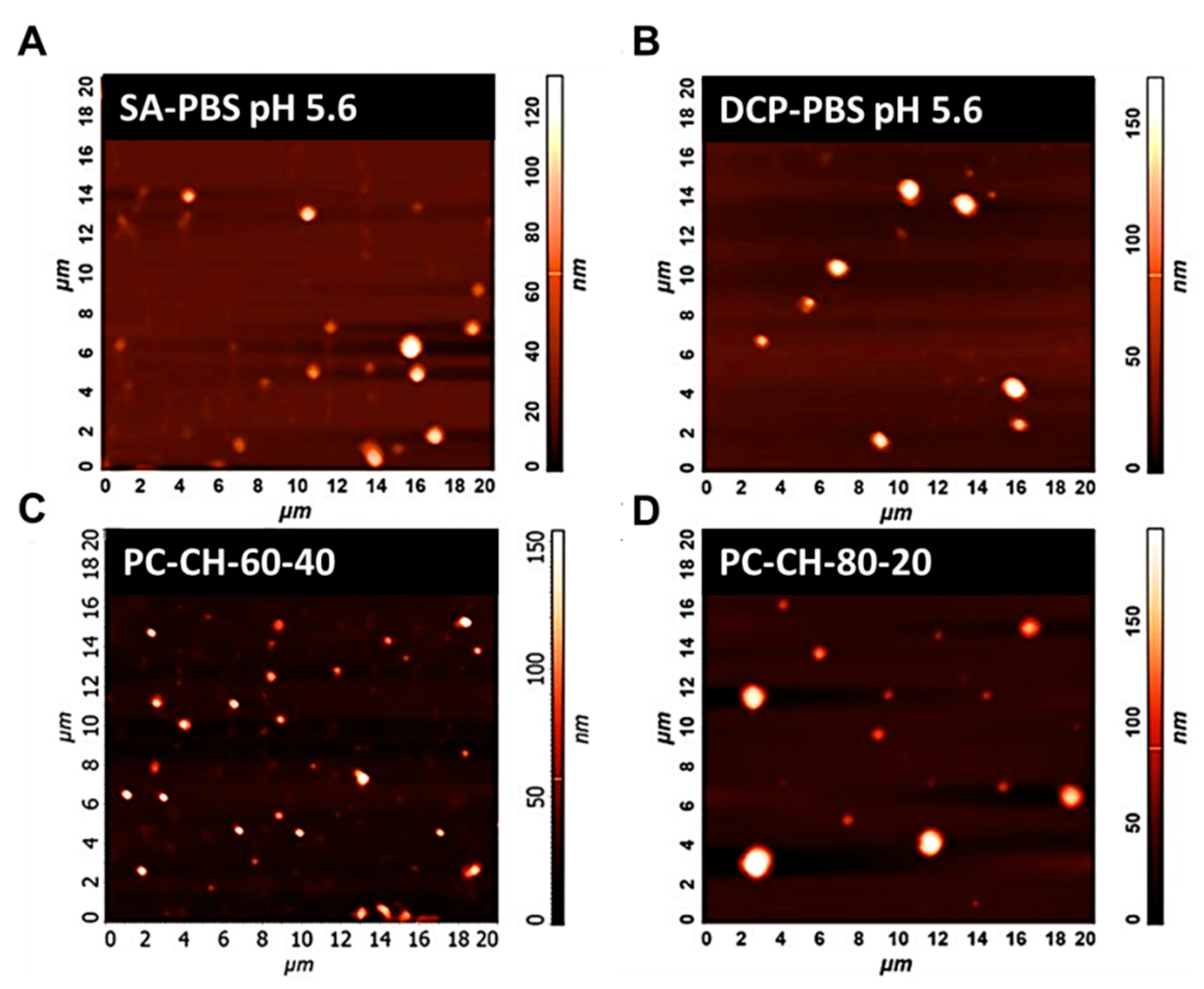
3.2.4. Atomic Force Microscopy (AFM)
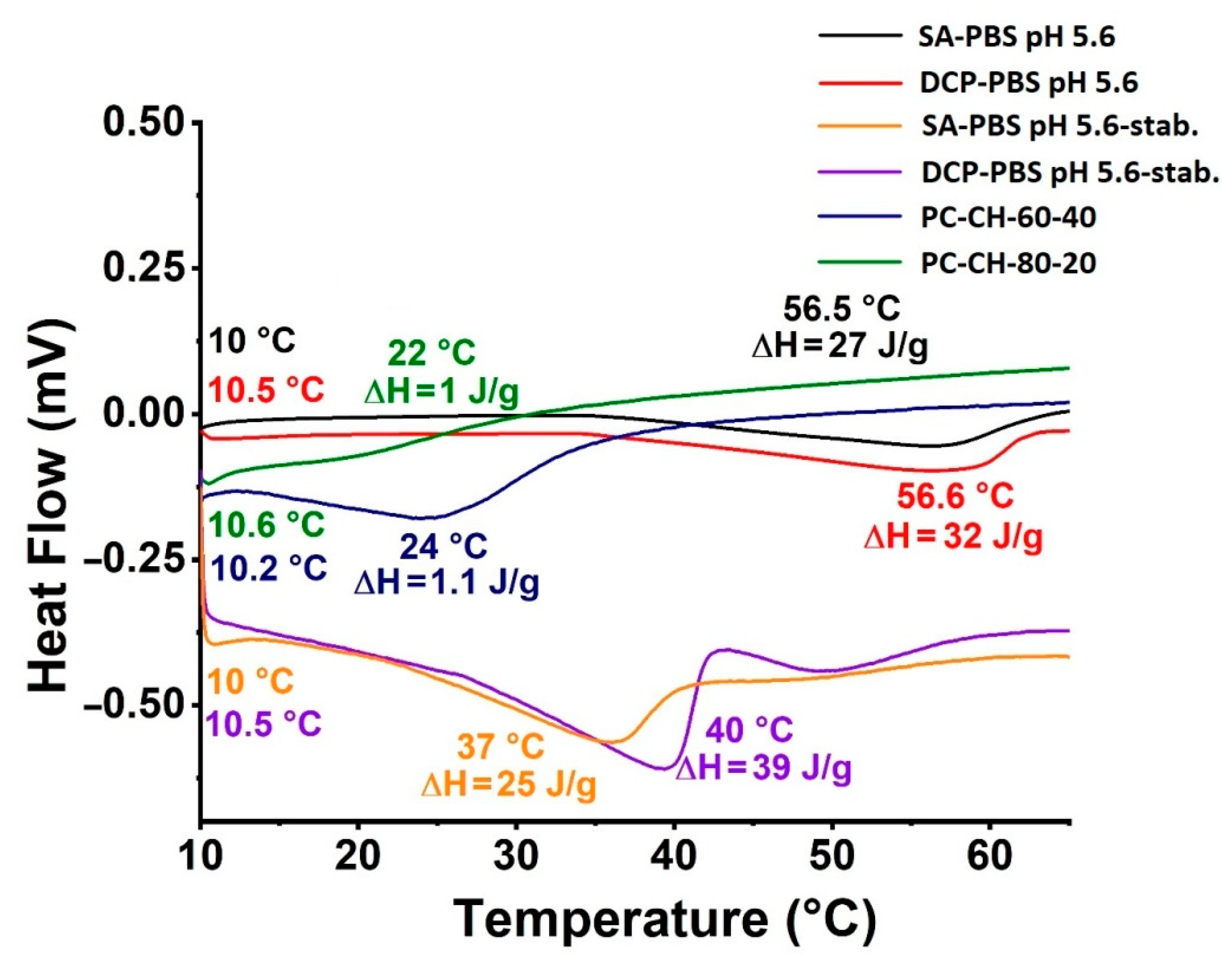
3.2.5. Thermal Analysis
3.2.6. Fourier Transformed Infrared Spectroscopy (FT-IR)
3.2.7. Raman Spectroscopy
3.2.8. Residual Ethanol Measurement via Gas Chromatography-Mass Spectrometry (GC-MS)
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ΔHm | enthalpy changes |
| AFM | atomic force microscopy |
| ANOVA | analysis of variance |
| API | active pharmaceutical ingredients |
| BBB | blood-brain barrier |
| CH | cholesterol |
| CMAs | Critical Material Attributes |
| CPPs | Critical Process Parameters |
| CQAs | Critical Quality Attributes |
| DCP | dicetyl phosphate |
| DLS | dynamic light scattering |
| DoE | Design of Experiments |
| DOPC | 1,2-dioleoyl-sn-glycero-3-phosphocholine |
| DOPE | 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine |
| DPPE | 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine |
| DS | Design Space |
| DSC | differential scanning calorimetry |
| DSPC | 1,2-distearoyl-sn-glycero-3-phosphocholine |
| DSPE-PEG2000 | 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-n-[methoxy(polyethyleneglycol)-2000] |
| EMA | European Medicines Agency |
| EPC | egg phosphatidylcholine |
| FITC | fluorescein isothiocyanate |
| FT-IR | Fourier transform infrared spectroscopy |
| GC-MS | gas chromatography-mass spectrometry |
| GDNF | glial cell-line derived neurotrophic factor |
| ICH | International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use |
| KCl | potassium chloride |
| K2HPO4 | dipotassium phosphate |
| KH2PO4 | potassium dihydrogen phosphate |
| MIR | mid-infrared spectroscopy |
| Na2HPO4 × 2 H2O | disodium hydrogen phosphate dihydrate |
| NaCl | sodium chloride |
| NBCDs | non-biologically complex drugs |
| PBS pH 5.6 | phosphate-buffered saline pH 5.6 |
| PBS pH 7.4 | phosphate-buffered saline pH 7.4 |
| PC | L-α-phosphatidylcholine |
| PdI | polydispersity index |
| QbD | Quality by Design |
| QTPP | Quality Target Product Profile |
| RA | Risk Assessment |
| RES | reticuloendothelial system |
| SA | stearylamine |
| Saline solution | sodium chloride physiological solution |
| Span 60 | sorbitan monostearate |
| SPC | soy phosphatidylcholine |
| Tg | glass transition temperature |
| TG | thermogravimetric measurements |
| TGA | thermogravimetric analysis |
| Tm | phase transition temperature |
| Tween 20 | sorbitan monolaurate/Polysorbate 20 |
| Tween 80 | sorbitan monooleate/Polysorbate 80 |
References
- Daraee, H.; Etemadi, A.; Kouhi, M.; Alimirzalu, S.; Akbarzadeh, A. Application of liposomes in medicine and drug delivery. Artif. Cells Nanomed. Biotechnol. 2016, 44, 381–391. [Google Scholar] [CrossRef]
- Li, M.; Du, C.; Guo, N.; Teng, Y.; Meng, X.; Sun, H.; Li, S.; Yu, P.; Galons, H. Composition design and medical application of liposomes. Eur. J. Med. Chem. 2019, 164, 640–653. [Google Scholar] [CrossRef] [PubMed]
- Alavi, M.; Karimi, N.; Safaei, M. Application of various types of liposomes in drug delivery systems. Adv. Pharm. Bull. 2017, 7, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Nakhaei, P.; Margiana, R.; Bokov, D.O.; Abdelbasset, W.K.; Jadidi Kouhbanani, M.A.; Varma, R.S.; Marofi, F.; Jarahian, M.; Beheshtkhoo, N. Liposomes: Structure, Biomedical Applications, and Stability Parameters With Emphasis on Cholesterol. Front. Bioeng. Biotechnol. 2021, 9, 705886. [Google Scholar] [CrossRef]
- Milton Harris, J.; Chess, R.B. Effect of pegylation on pharmaceuticals. Nat. Rev. Drug Discov. 2003, 2, 214–221. [Google Scholar] [CrossRef]
- Bozzuto, G.; Molinari, A. Liposomes as nanomedical devices. Int. J. Nanomed. 2015, 10, 975–999. [Google Scholar] [CrossRef]
- Nisini, R.; Poerio, N.; Mariotti, S.; De Santis, F.; Fraziano, M. The multirole of liposomes in therapy and prevention of infectious diseases. Front. Immunol. 2018, 9, 155. [Google Scholar] [CrossRef] [PubMed]
- Zylberberg, C.; Matosevic, S. Pharmaceutical liposomal drug delivery: A review of new delivery systems and a look at the regulatory landscape. Drug Deliv. 2016, 23, 3319–3329. [Google Scholar] [CrossRef]
- Schellekens, H.; Stegemann, S.; Weinstein, V.; De Vlieger, J.S.B.; Flühmann, B.; Mühlebach, S.; Gaspar, R.; Shah, V.P.; Crommelin, D.J.A. How to regulate nonbiological complex drugs (NBCD) and their follow-on versions: Points to consider. AAPS J. 2014, 16, 15–21. [Google Scholar] [CrossRef]
- European Medicine Agency. Reflection Paper on the Data Requirements for Intravenous Liposomal Products Developed with Reference to an Innovator Liposomal Product; EMA/CHMP/806058/2009/Rev. 02; European Medicines Agency: Amsterdam, The Netherlands, 2013; pp. 1–13.
- Bangham, A.D.; Standish, M.M.; Watkins, J.C. Diffusion of univalent ions across the lamellae of swollen phospholipids. J. Mol. Biol. 1965, 13, 238–252. [Google Scholar] [CrossRef]
- Nayak, D.; Tippavajhala, V.K. A comprehensive review on preparation, evaluation and applications of deformable liposomes. Iran. J. Pharm. Res. 2021, 20, 186–205. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Hou, S.; Tong, P.; Li, J. Liposomes: Preparation, Characteristics, and Application Strategies in Analytical Chemistry. Crit. Rev. Anal. Chem. 2022, 52, 392–412. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Seth, A.; Wibowo, N.; Zhao, C.X.; Mitter, N.; Yu, C.; Middelberg, A.P.J. Nanoparticle vaccines. Vaccine 2014, 32, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.X. Pharmaceutical quality by design: Product and process development, understanding, and control. Pharm. Res. 2008, 25, 781–791. [Google Scholar] [CrossRef]
- Csóka, I.; Pallagi, E.; Paál, T.L. Extension of quality-by-design concept to the early development phase of pharmaceutical R&D processes. Drug Discov. Today 2018, 23, 1340–1343. [Google Scholar] [CrossRef] [PubMed]
- Sipos, B.; Katona, G.; Csóka, I. A Systematic, Knowledge Space-Based Proposal on Quality by Design-Driven Polymeric Micelle Development. Pharmaceutics 2021, 13, 702. [Google Scholar] [CrossRef]
- ICH Harmonised Tripartite Guideline. Pharmaceutical Development Q8. 2009, pp. 1–28. Available online: https://database.ich.org/sites/default/files/Q8_R2_Guideline.pdf (accessed on 15 March 2022).
- ICH Harmonised Tripartite Guideline. ICH Quality Risk Management Q9. 2005, pp. 1–20. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/international-conference-harmonisation-technical-requirements-registration-pharmaceuticals-human-use_en-3.pdf (accessed on 15 March 2022).
- EMA ICH Guideline Q10 on Pharmaceutical Quality System. EMEA/CHMP/ICH/214732/2007. European Medicines Agency. 2015, pp. 1–20. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/international-conference-harmonisation-technical-requirements-registration-pharmaceuticals-human_en.pdf (accessed on 15 March 2022).
- Németh, Z.; Pallagi, E.; Dobó, D.G.; Csóka, I. A proposed methodology for a risk assessment-based liposome development process. Pharmaceutics 2020, 12, 1164. [Google Scholar] [CrossRef] [PubMed]
- Németh, Z.; Pallagi, E.; Dobó, D.G.; Kozma, G.; Kónya, Z.; Csóka, I. An updated risk assessment as part of the QbD-based liposome design and development. Pharmaceutics 2021, 13, 1071. [Google Scholar] [CrossRef]
- Smith, M.C.; Crist, R.M.; Clogston, J.D.; McNeil, S.E. Zeta potential: A case study of cationic, anionic, and neutral liposomes. Anal. Bioanal. Chem. 2017, 409, 5779–5787. [Google Scholar] [CrossRef]
- Clogston, J.D.; Patri, A.K. Zeta potential measurement. Methods Mol. Biol. 2011, 697, 63–70. [Google Scholar] [CrossRef]
- Xu, R. Progress in nanoparticles characterization: Sizing and zeta potential measurement. Particuology 2008, 6, 112–115. [Google Scholar] [CrossRef]
- Gumustas, M.; Sengel-Turk, C.T.; Gumustas, A.; Ozkan, S.A.; Uslu, B. Effect of Polymer-Based Nanoparticles on the Assay of Antimicrobial Drug Delivery Systems; Elsevier Inc.: Amsterdam, The Netherlands, 2017; ISBN 9780323527255. [Google Scholar]
- Rabinovich-Guilatt, L.; Couvreur, P.; Lambert, G.; Goldstein, D.; Benita, S.; Dubernet, C. Extensive surface studies help to analyse zeta potential data: The case of cationic emulsions. Chem. Phys. Lipids 2004, 131, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Honary, S.; Zahir, F. Effect of zeta potential on the properties of nano-drug delivery systems—A review (Part 1). Trop. J. Pharm. Res. 2013, 12, 255–264. [Google Scholar] [CrossRef]
- Samimi, S.; Maghsoudnia, N.; Eftekhari, R.B.; Dorkoosh, F. Lipid-Based Nanoparticles for Drug Delivery Systems; Elsevier Inc.: Amsterdam, The Netherlands, 2018; ISBN 9780128140321. [Google Scholar]
- Honary, S.; Zahir, F. Effect of zeta potential on the properties of nano-drug delivery systems—A review (Part 2). Trop. J. Pharm. Res. 2013, 12, 265–273. [Google Scholar] [CrossRef]
- Das, S.; Chaudhury, A. Recent advances in lipid nanoparticle formulations with solid matrix for oral drug delivery. AAPS PharmSciTech 2011, 12, 62–76. [Google Scholar] [CrossRef] [PubMed]
- Sikora, A.; Bartczak, D.; Geißler, D.; Kestens, V.; Roebben, G.; Ramaye, Y.; Varga, Z.; Palmai, M.; Shard, A.G.; Goenaga-Infante, H.; et al. A systematic comparison of different techniques to determine the zeta potential of silica nanoparticles in biological medium. Anal. Methods 2015, 7, 9835–9843. [Google Scholar] [CrossRef]
- Sahay, G.; Alakhova, D.Y.; Kabanov, A.V. Endocytosis of nanomedicines. J. Control. Release 2010, 145, 182–195. [Google Scholar] [CrossRef]
- Hu, Y.-J.; Ju, R.-J.; Zeng, F.; Qi, X.-R.; Lu, W.-L. Liposomes in Drug Delivery: Status and Advances. In Liposome-Based Drug Delivery Systems; Springer: Berlin/Heidelberg, Germany, 2021; pp. 3–24. [Google Scholar] [CrossRef]
- Villasmil-Sánchez, S.; Drhimeur, W.; Ospino, S.C.S.; Rabasco Alvarez, A.M.; González-Rodríguez, M.L. Positively and negatively charged liposomes as carriers for transdermal delivery of sumatriptan: In vitro characterization. Drug Dev. Ind. Pharm. 2010, 36, 666–675. [Google Scholar] [CrossRef]
- Jeon, H.S.; Seo, J.E.; Kim, M.S.; Kang, M.H.; Oh, D.H.; Jeon, S.O.; Jeong, S.H.; Choi, Y.W.; Lee, S. A retinyl palmitate-loaded solid lipid nanoparticle system: Effect of surface modification with dicetyl phosphate on skin permeation in vitro and anti-wrinkle effect in vivo. Int. J. Pharm. 2013, 452, 311–320. [Google Scholar] [CrossRef]
- Ingle, S.G.; Pai, R.V.; Monpara, J.D.; Vavia, P.R. Liposils: An effective strategy for stabilizing Paclitaxel loaded liposomes by surface coating with silica. Eur. J. Pharm. Sci. 2018, 122, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Henna Lu, F.S.; Nielsen, N.S.; Timm-Heinrich, M.; Jacobsen, C. Oxidative stability of marine phospholipids in the liposomal form and their applications. Lipids 2011, 46, 3–23. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; Bano, M.; Bisen, A.C.; Verma, S.; Sanap, S.N.; Kishor, R.; Shukla, P.K.; Bhatta, R.S. Topical corneal targeted sustained release amphotericin B liposomal formulation for the treatment of fungal keratitis and its PK-PD evaluation. J. Drug Deliv. Sci. Technol. 2020, 60, 101944. [Google Scholar] [CrossRef]
- Kotyńska, J.; Figaszewski, Z.A. Adsorption equilibria at interface separating electrolyte solution and phosphatidylcholine-stearylamine liposome membrane. Biophys. Chem. 2007, 127, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Jagwani, S.; Jalalpure, S.; Dhamecha, D.; Jadhav, K.; Bohara, R. Pharmacokinetic and Pharmacodynamic Evaluation of Resveratrol Loaded Cationic Liposomes for Targeting Hepatocellular Carcinoma. ACS Biomater. Sci. Eng. 2020, 6, 4969–4984. [Google Scholar] [CrossRef]
- Jeengar, M.K.; Kurakula, M.; Patil, P.; More, A.; Sistla, R.; Parashar, D. Effect of Cationic Lipid Nanoparticle Loaded siRNA with Stearylamine against Chikungunya Virus. Molecules 2022, 27, 1170. [Google Scholar] [CrossRef]
- Lai, W.F.; Wong, W.T.; Rogach, A.L. Molecular design of layer-by-layer functionalized liposomes for oral drug delivery. ACS Appl. Mater. Interfaces 2020, 12, 43341–43351. [Google Scholar] [CrossRef]
- Vhora, I.; Lalani, R.; Bhatt, P.; Patil, S.; Patel, H.; Patel, V.; Misra, A. Colloidally Stable Small Unilamellar Stearyl Amine Lipoplexes for Effective BMP-9 Gene Delivery to Stem Cells for Osteogenic Differentiation. AAPS PharmSciTech 2018, 19, 3550–3560. [Google Scholar] [CrossRef]
- He, K.; Liu, J.; Gao, Y.; Hao, Y.; Yang, X.; Huang, G. Preparation and Evaluation of Stearylamine-Bearing Pemetrexed Disodium-Loaded Cationic Liposomes In Vitro and In Vivo. AAPS PharmSciTech 2020, 21, 193. [Google Scholar] [CrossRef]
- Adams, D.H.; Joyce, G.; Richardson, V.J.; Ryman, B.E.; Wisniewski, H.M. Liposome toxicity in the mouse central nervous system. J. Neurol. Sci. 1977, 31, 173–179. [Google Scholar] [CrossRef]
- Inglut, C.T.; Sorrin, A.J.; Kuruppu, T.; Vig, S.; Cicalo, J.; Ahmad, H.; Huang, H.C. Immunological and toxicological considerations for the design of liposomes. Nanomaterials 2020, 10, 190. [Google Scholar] [CrossRef]
- Mehanna, M.M.; El-Kader, N.A.; Samaha, M.W. Liposomes as potential carriers for ketorolac ophthalmic delivery: Formulation and stability issues. Braz. J. Pharm. Sci. 2017, 53, 1–10. [Google Scholar] [CrossRef]
- Salem, H.F.; Ahmed, S.M.; Omar, M.M. Liposomal flucytosine capped with gold nanoparticle formulations for improved ocular delivery. Drug Des. Devel. Ther. 2016, 10, 277–295. [Google Scholar] [CrossRef] [PubMed]
- Ternullo, S.; Werning, L.V.S.; Holsæter, A.M.; Škalko-Basnet, N. Curcumin-in-deformable liposomes-in-chitosan-hydrogel as a novel wound dressing. Pharmaceutics 2020, 12, 8. [Google Scholar] [CrossRef] [PubMed]
- Ternullo, S.; Gagnat, E.; Julin, K.; Johannessen, M.; Basnet, P.; Vanić, Ž.; Škalko-Basnet, N. Liposomes augment biological benefits of curcumin for multitargeted skin therapy. Eur. J. Pharm. Biopharm. 2019, 144, 154–164. [Google Scholar] [CrossRef]
- Mura, P.; Maestrelli, F.; Cirri, M.; Nerli, G.; Di Cesare Mannelli, L.; Ghelardini, C.; Mennini, N. Improvement of butamben anesthetic efficacy by the development of deformable liposomes bearing the drug as cyclodextrin complex. Pharmaceutics 2021, 13, 872. [Google Scholar] [CrossRef]
- Soni, A.; Jain, V.; Jain, S.K.; Khangar, P.K. Preparation and characterization of amphotericin B mannosylated liposomes for effective management of visceral leishmaniasis. J. Drug Deliv. Ther. 2021, 11, 113–118. [Google Scholar] [CrossRef]
- De, M.; Ghosh, S.; Asad, M.; Banerjee, I.; Ali, N. Combining doxorubicin with stearylamine-bearing liposomes elicits Th1 cytokine responses and cures metastasis in a mouse model. Cancer Immunol. Immunother. 2020, 69, 1725–1735. [Google Scholar] [CrossRef]
- Verma, A.; Jain, A.; Tiwari, A.; Saraf, S.; Panda, P.K.; Agrawal, G.P.; Jain, S.K. Folate Conjugated Double Liposomes Bearing Prednisolone and Methotrexate for Targeting Rheumatoid Arthritis. Pharm. Res. 2019, 36, 123. [Google Scholar] [CrossRef]
- Narayan, R.; Singh, M.; Ranjan, O.P.; Nayak, Y.; Garg, S.; Shavi, G.V.; Nayak, U.Y. Development of risperidone liposomes for brain targeting through intranasal route. Life Sci. 2016, 163, 38–45. [Google Scholar] [CrossRef]
- Rajendran, V.; Rohra, S.; Raza, M.; Hasan, G.M.; Dutt, S.; Ghosh, P.C. Stearylamine liposomal delivery of monensin in combination with free artemisinin eliminates blood stages of Plasmodium falciparum in culture and P. berghei infection in murine malaria. Antimicrob. Agents Chemother. 2016, 60, 1304–1318. [Google Scholar] [CrossRef]
- Migliore, M.M.; Ortiz, R.; Dye, S.; Campbell, R.B.; Amiji, M.M.; Waszczak, B.L. Neurotrophic and neuroprotective efficacy of intranasal GDNF in a rat model of Parkinson’s disease. Neuroscience 2014, 274, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Migliore, M.M.; Vyas, T.K.; Campbell, R.B.; Amiji, M.M.; Waszczak, B.L. Brain Delivery of Proteins by the Intranasal Route of Administration: A Comparison of Cationic Liposomes Versus Aqueous Solution Formulations. J. Pharm. Sci. 2010, 99, 1745–1761. [Google Scholar] [CrossRef] [PubMed]
- Tran, B.H.; Yu, Y.; Chang, L.; Tan, B.; Jia, W.; Xiong, Y.; Dai, T.; Zhong, R.; Zhang, W.; Le, V.M.; et al. A novel liposomal S-propargyl-cysteine: A sustained release of hydrogen sulfide reducing myocardial fibrosis via TGF-β1/smad pathway. Int. J. Nanomed. 2019, 14, 10061–10077. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Rajendran, V.; Kulshreshtha, R.; Ghosh, P.C. Enhanced efficacy of anti-miR-191 delivery through stearylamine liposome formulation for the treatment of breast cancer cells. Int. J. Pharm. 2017, 530, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Caddeo, C.; Gabriele, M.; Fernàndez-Busquets, X.; Valenti, D.; Fadda, A.M.; Pucci, L.; Manconi, M. Antioxidant activity of quercetin in Eudragit-coated liposomes for intestinal delivery. Int. J. Pharm. 2019, 565, 64–69. [Google Scholar] [CrossRef]
- De, M.; Ghosh, S.; Sen, T.; Shadab, M.; Banerjee, I.; Basu, S.; Ali, N. A Novel Therapeutic Strategy for Cancer Using Phosphatidylserine Targeting Stearylamine-Bearing Cationic Liposomes. Mol. Ther.—Nucleic Acids 2018, 10, 9–27. [Google Scholar] [CrossRef]
- Lotosh, N.Y.; Alyaseva, S.O.; Vasilov, R.G.; Selishcheva, A.A. Stearylamine Causes the Formation of Neutrophil Extracellular Traps Independently of Reactive Oxygen Species. Cell Tissue Biol. 2019, 13, 366–375. [Google Scholar] [CrossRef]
- Lotosh, N.Y.; Aliaseva, S.O.; Malashenkova, I.K.; Sorokoumova, G.M.; Vasilov, R.G.; Selischeva, A.A. Cationic Liposomes Cause ROS Generation and Release of Neutrophil Extracellular Traps. Biochem. Suppl. Ser. A Membr. Cell Biol. 2019, 13, 40–49. [Google Scholar] [CrossRef]
- Rahman, S.A.; Abdelmalak, N.S.; Badawi, A.; Elbayoumy, T.; Sabry, N.; El Ramly, A. Tretinoin-loaded liposomal formulations: From lab to comparative clinical study in acne patients. Drug Deliv. 2016, 23, 1184–1193. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Rai, A.; Reddy, N.D.; Shenoy, R.R.; Mudgal, J.; Bansal, P.; Mudgal, P.P.; Arumugam, K.; Udupa, N.; Sharma, N.; et al. Improved in vitro and in vivo hepatoprotective effects of liposomal silymarin in alcohol-induced hepatotoxicity in Wistar rats. Pharmacol. Rep. 2019, 71, 703–712. [Google Scholar] [CrossRef]
- Alomrani, A.; Badran, M.; Harisa, G.I.; ALshehry, M.; Alhariri, M.; Alshamsan, A.; Alkholief, M. The use of chitosan-coated flexible liposomes as a remarkable carrier to enhance the antitumor efficacy of 5-fluorouracil against colorectal cancer. Saudi Pharm. J. 2019, 27, 603–611. [Google Scholar] [CrossRef]
- Togami, K.; Daisho, T.; Yumita, Y.; Kitayama, A.; Tada, H.; Chono, S. Evaluation of various tissue-clearing techniques for the three-dimensional visualization of liposome distribution in mouse lungs at the alveolar scale. Int. J. Pharm. 2019, 562, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Calvo, A.; Moreno, E.; Larrea, E.; Sanmartín, C.; Irache, J.M.; Espuelas, S. Berberine-loaded liposomes for the treatment of leishmania infantum-infected balb/c mice. Pharmaceutics 2020, 12, 858. [Google Scholar] [CrossRef] [PubMed]
- Ethemoglu, M.S.; Seker, F.B.; Akkaya, H.; Kilic, E.; Aslan, I.; Erdogan, C.S.; Yilmaz, B. Anticonvulsant activity of resveratrol-loaded liposomes in vivo. Neuroscience 2017, 357, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Togami, K.; Maruta, Y.; Nanbu, M.; Tada, H.; Chono, S. Prolonged distribution of aerosolized PEGylated liposomes in the lungs of mice with bleomycin-induced pulmonary fibrosis. Drug Dev. Ind. Pharm. 2020, 46, 1873–1880. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency (EMA). ICH Topic Q1A (R2) Stability Testing of New Drug Substances and Products. 2003; 20p. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-1-r2-stability-testing-new-drug-substances-products-step-5_en.pdf (accessed on 30 March 2022).
- Danaei, M.; Dehghankhold, M.; Ataei, S.; Hasanzadeh Davarani, F.; Javanmard, R.; Dokhani, A.; Khorasani, S.; Mozafari, M.R. Impact of particle size and polydispersity index on the clinical applications of lipidic nanocarrier systems. Pharmaceutics 2018, 10, 57. [Google Scholar] [CrossRef]
- Porfire, A.; Achim, M.; Barbalata, C.; Rus, I.; Tomuta, I.; Cristea, C. Pharmaceutical Development of Liposomes Using the QbD Approach. In Liposomes—Advances and Perspectives; IntechOpen: London, UK, 2019; pp. 1–20. [Google Scholar] [CrossRef]
- Ingvarsson, P.T.; Yang, M.; Nielsen, H.M.; Rantanen, J.; Foged, C. Stabilization of liposomes during drying. Expert Opin. Drug Deliv. 2011, 8, 375–388. [Google Scholar] [CrossRef]
- Demetzos, C. Differential Scanning Calorimetry (DSC): A tool to study the thermal behavior of lipid bilayers and liposomal stability. J. Liposome Res. 2008, 18, 159–173. [Google Scholar] [CrossRef]
- Taylor, K.M.G.; Morris, R.M. Thermal analysis of phase transition behaviour in liposomes. Thermochim. Acta 1995, 248, 289–301. [Google Scholar] [CrossRef]
- Pinilla, C.M.B.; Reque, P.M.; Brandelli, A. Effect of Oleic Acid, Cholesterol, and Octadecylamine on Membrane Stability of Freeze-Dried Liposomes Encapsulating Natural Antimicrobials. Food Bioprocess Technol. 2020, 13, 599–610. [Google Scholar] [CrossRef]
- Derenne, A.; Claessens, T.; Conus, C.; Goormaghtigh, E. Infrared Spectroscopy of Membrane Lipids. In Encyclopedia of Biophysics; Springer: Berlin/Heidelberg, Germany, 2013; pp. 1074–1081. ISBN 978-3-642-16711-9. [Google Scholar]
- Sharma, A.; Sharma, U.S. Liposomes in drug delivery: Progress and limitations. Int. J. Pharm. 1997, 154, 123–140. [Google Scholar] [CrossRef]
- Czamara, K.; Majzner, K.; Pacia, M.Z.; Kochan, K.; Kaczor, A.; Baranska, M. Raman spectroscopy of lipids: A review. J. Raman Spectrosc. 2015, 46, 4–20. [Google Scholar] [CrossRef]
- European Medicines Agency. ICH Guideline Q3C (R6) on Impurities: Guideline for Residual Solvents. 2019; p. 24. Available online: https://database.ich.org/sites/default/files/Q3C-R6_Guideline_ErrorCorrection_2019_0410_0.pdf (accessed on 29 March 2022).











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Composition | Molar Ratio | Drug | Size (nm) | PdI | Zeta Potential (mV) | Source |
|---|---|---|---|---|---|---|
| EPC:CH:SA | 1:1:3.85 | ketorolac tromethamine | 7060 ± - | 0.43 ± - | - | Mehanna et al. [48] |
| SPC:CH:SA:Span 60 | 1:1:0.15:1 | flucytosine | 135 ± 12 | 0.27 ± - | +42.5 ± 2.1 | Salem et al. [49] |
| SPC:SA:Tween 20 | 20:6.3:2.4 | curcumin | 252 ± 52 | 0.17 ± 0.01 | +34.0 ± 0.6 | Ternullo et al. [50] |
| SPC:SA:Tween 20 | 20:6.3:2.4 | curcumin | 232 ± 68 | 0.22 ± 0.04 | +33.7 ± 1.1 | Ternullo et al. [51] |
| EPC:CH:SA | 5.5:1.0: 1.5 | butamben | 240 ± 65 | 0.22 ± - | +30.2 ± 3.9 | Mura et al. [52] |
| EPC:CH:SA | 6.6:10.3:11.13 | sumatriptan | 349 ± 100 | 0.28 ± 0.24 | +37.9 ± 3.7 | Villasmil-Sánchez et al. [35] |
| SPC:CH:SA | 7:3:1.5 | amphotericin B | 940 ± 40 | - | +28.4 ± 0.3 | Soni et al. [53] |
| SPC:SA | 2:0.5 | amphotericin B | 140 ± 4 | 0.24 ± 0.04 | +32.0 ± 0.2 | Mishra et al. [39] |
| SPC:SA | 1:0.5 | amphotericin B | 202 ± 6 | 0.39 ± 0.03 | +63.0 ± 0.4 | Mishra et al. [39] |
| SPC:CH; SA | 9.13:1; 5.18 mg | paclitaxel | 193 ± 2 | 0.17 ± 0.03 | +38.2 ± 3.5 | Ingle et al. [37] |
| SPC:CH; SA | 7:2; 5.00 mg | resveratrol | 146 ± 10 | - | +38.0 ±9.1 | Jagwani et al. [41] |
| PC:SA | 7:2 | doxorubicin | 148 ± - | - | +43.1 ± - | De et al. [54] |
| DSPC:CH:SA | 7.5:2.5:0.5 | prednisolone, methotrexate | 159 ± 2 | 0.09 ± - | +6.3 ± 0.4 | Verma et al. [55] |
| EPC:CH:SA | 7.8:2.6:2.9 | pemetrexed disodium | 220 ± 5 | 0.23 ± 0.02 | +22.2 ± 0.5 | He et al. [45] |
| SPC:CH:SA | 8:1:2 | risperidone | 209 ± 16 | - | +22.4 ± 1.5 | Narayan et al. [56] |
| SPC:CH:SA | 8:1:0.25 | risperidone | 99 ± 7 | - | +15.6 ± 1.4 | Narayan et al. [56] |
| SPC:CH:SA | 7:3:1.1 | monensin | 121 ± 20 | 0.25 ± 0.01 | +43.9 ± 0.9 | Rajendran et al. [57] |
| DOPC:CH:SA | 10:6:1 | GDNF | 149 ±11 | - | +30.0 ± 3.0 | Migliore et al. [58] |
| DOPC:CH:SA | 10:6:1 | ovalbumin | 299 ± 26 | - | +19.0 ± 1.5 | Migliore et al. [59] |
| DOPE:SPC:CH:SA | 10:45:29:16 | - | 95 ± 9 | 0.24 ± 0.03 | +52.8 ± 3.7 | Vhora et al. [44] |
| SPC:CH:SA:DSPE-PEG2000 | 11:7:0.6:1.4 | - | 209 ± 2 | - | +48.7 ± 4.3 | Tran et al. [60] |
| SPC:CH:SA | 7:3:1.1 | - | 77 ± 2 | 0.21 ± - | +32.9 ± 2.1 | Sharma et al. [61] |
| SPC:SA | 7.3:1 | - | 81 ± 6 | 0.24 ± 0.02 | +17.5 ± 1.8 | Caddeo et al. [62] |
| PC:SA | 7:2 | - | 146 ± - | 0.20 ± - | +52.0 ± - | De et al. [63] |
| SPC:SA | 3:1 | - | 140 ± 49 | - | +11.4 ± 0.4 | Lotosh et al. [64,65] |
| EPC:CH:SA | 12:5:5 | - | 108 ± 15 | 0.20 ± 0.04 | +30.1 ± 1.2 | SA-PBS pH 5.6 |
| Composition | Molar Ratio | Drug | Size (nm) | PdI | Zeta Potential (mV) | Source |
|---|---|---|---|---|---|---|
| EPC:CH:DCP | 1:1:3.85 | ketorolac tromethamine | 8350 ± - | 0.45 ± - | - | Mehanna et al. [48] |
| PC:CH:DCP | 1:1:0.7 | tretinoin | 318 ± 3 | 0.43 ± - | −41.2 ± 1.2 | Rahman et al. [66] |
| SPC:CH:DCP | 6:1:1.5 | silymarin | 756 ± - | 0.61 ± - | −77.3 ± - | Kumar et al. [67] |
| EPC:CH:DCP | 6.6:10.3:5.49 | sumatriptan | 549 ± 10 | 0.37 ± 0.09 | −68.1 ± 0.4 | Villasmil-Sánchez et al. [35] |
| SPC:CH:DCP:Span 60 | 1:1:0.1:1 | flucytosine | 159 ± 5 | 0.26 ± - | −59.1 ± 1.7 | Salem et al. [49] |
| SPC:CH:DCP:Tween 80 | 9:3:1:1 | 5-fluorouracil | 108 ± 11 | 0.31 ± 0.05 | −16.3 ± 1.5 | Alomrani et al. [68] |
| EPC:CH:DCP:DSPE-PEG2000:DPPE | 7:2:1:1:0.025 | FITC-dextran | 116 ± - | 0.12 ± - | −29.0 ± - | Togami et al. [69] |
| EPC:CH:DCP:DSPE-PEG2000:DPPE | 7:2:1:1:0.025 | rhodamine B | 125 ± - | 0.09 ± - | −32.0 ± - | Togami et al. [69] |
| SPC:CH:DCP: DSPE-PEG2000: | 11:7:1.4:0.6 | - | 191 ± 4 | −45.1 ± 2.5 | Tran et al. [60] | |
| SPC:CH:DCP | 15:8:1 | - | 195 ± 5 | 0.28 ± - | −47.0 ± 1.0 | Calvo et al. [70] |
| SPC:CH:DCP | 10:4:1 | - | 146 ± 6 | −18.6 ± 0.5 | Ethemoglu et al. [71] | |
| EPC:CH:DCP | 7:2:1 | - | 134 ± 4 | 0.12 ± 0.03 | −49.4 ± 3.5 | Togami et al. [72] |
| EPC:CH:DCP | 8.5:4.5:6.5 | - | 88 ±1 | 0.21 ± 0.02 | −36.7 ± 3.3 | DCP-PBS pH 5.6 |
| Run | Composition (Molar Ratio) | ||
|---|---|---|---|
| PC | CH | SA/DCP | |
| 1 | 7.5 | 3.5 | 3.0 |
| 2 | 7.5 | 4.5 | 9.0 |
| 3 | 7.5 | 5.5 | 6.0 |
| 4 | 10.0 | 3.5 | 9.0 |
| 5 | 10.0 | 4.5 | 6.0 |
| 6 | 10.0 | 5.5 | 3.0 |
| 7 | 12.5 | 3.5 | 6.0 |
| 8 | 12.5 | 4.5 | 3.0 |
| 9 | 12.5 | 5.5 | 9.0 |
| Run | Composition (Molar Ratio) | Responses | ||||
|---|---|---|---|---|---|---|
| PC | CH | SA | Vesicle Size (nm) | Polydispersity Index | Zeta Potential (mV) | |
| 1 | 7.5 | 3.5 | 3.0 | 121 ± 28 | 0.22 ± 0.02 | +22.0 ± 7.8 |
| 2 | 7.5 | 4.5 | 9.0 | 106 ± 21 | 0.23 ± 0.03 | +17.6 ± 3.4 |
| 3 | 7.5 | 5.5 | 6.0 | 116 ± 14 | 0.23 ± 0.02 | +24.6 ± 1.4 |
| 4 | 10.0 | 3.5 | 9.0 | 93 ± 6 | 0.22 ± 0.03 | +25.0 ± 3.5 |
| 5 | 10.0 | 4.5 | 6.0 | 113 ± 16 | 0.23 ± 0.06 | +25.8 ± 3.7 |
| 6 | 10.0 | 5.5 | 3.0 | 112 ± 7 | 0.16 ± 0.01 | +26.6. ± 2.7 |
| 7 | 12.5 | 3.5 | 6.0 | 111 ± 6 | 0.19 ± 0.03 | +26.3 ± 1.2 |
| 8 | 12.5 | 4.5 | 3.0 | 109 ± 7 | 0.17 ± 0.03 | +26.6 ± 0.8 |
| 9 | 12.5 | 5.5 | 9.0 | 100 ± 17 | 0.17 ± 0.01 | +27.1 ± 2.8 |
| Run | Composition (molar ratio) | Responses | ||||
| PC | CH | DCP | Vesicle Size (nm) | Polydispersity Index | Zeta Potential (mV) | |
| 1 | 7.5 | 3.5 | 3.0 | 98 ± 11 | 0.20 ± 0.03 | −29.9 ± 1.6 |
| 2 | 7.5 | 4.5 | 9.0 | 82 ± 16 | 0.24 ± 0.02 | −29.6 ± 3.4 |
| 3 | 7.5 | 5.5 | 6.0 | 108 ± 9 | 0.21 ± 0.04 | −32.5 ± 6,5 |
| 4 | 10.0 | 3.5 | 9.0 | 87 ± 16 | 0.24 ± 0.03 | −32.6 ± 2.7 |
| 5 | 10.0 | 4.5 | 6.0 | 93 ± 23 | 0.23 ± 0.03 | −29.7 ± 6.2 |
| 6 | 10.0 | 5.5 | 3.0 | 119 ± 25 | 0.21 ± 0.07 | −29.7 ± 3.3 |
| 7 | 12.5 | 3.5 | 6.0 | 95 ± 8 | 0.18 ± 0.03 | −29.2 ± 3.2 |
| 8 | 12.5 | 4.5 | 3.0 | 104 ± 25 | 0.18 ± 0.02 | −27.6 ± 1.3 |
| 9 | 12.5 | 5.5 | 9.0 | 105 ± 2 | 0.18 ± 0.02 | −17.7 ± 3.1 |
| Sample | Composition (Molar Ratio) | Responses | ||||
|---|---|---|---|---|---|---|
| PC | CH | SA/DCP | Vesicle Size (nm) | Polydispersity Index | Zeta Potential (mV) | |
| SA-PBS pH 5.6 | 12.0 | 5.0 | 5.0 | 108 ± 15 | 0.20 ± 0.04 | +30.1 ± 1.2 |
| SA-PBS pH 7.4 | 134 ± 24 | 0.22 ± 0.07 | +5.2 ± 2.2 | |||
| SA-saline sol. | 105 ± 18 | 0.20 ± 0.10 | +13.6 ± 5.3 | |||
| DCP-PBS pH 5.6 | 8.5 | 4.5 | 6.5 | 88 ± 14 | 0.21 ± 0.02 | −36.7 ± 3.3 |
| DCP-PBS pH 7.4 | 112 ± 11 | 0.22 ± 0.02 | −23.5 ± 1.6 | |||
| DCP-saline sol. | 120 ± 10 | 0.21 ± 0.05 | −19.8 ± 2.0 | |||
| SA-PBS pH 5.6 | DCP-PBS pH 5.6 | PC-CH-60-40 | PC-CH-80-20 | |
|---|---|---|---|---|
| PC:CH:SA | PC:CH:DCP | PC:CH | PC:CH | |
| Molar ratio | 12.0:5.0:5.0 | 8.5:4.5:6.5 | 1:1.32 | 2.01:1 |
| Molar % (n/n%) | 54.5:22.75:22.75 | 43.6:23.1:33.3 | 43.1:56.9 | 66.8:33.2 |
| Weight ratio | 59.9:12.5:8.8 | 60.3:16.1:32.8 | 60.0:40.0 | 80.0:20.0 |
| Weight % (w/w%) | 73.8:15.4:10.8 | 55.2:14.7:30.1 | 60.0:40.0 | 80.0:20.0 |
| PC:CH weight ratio | 82.7:17.3 | 78.9:21.1 | 60.0:40.0 | 80.0:20.0 |
| Vesicle size (nm) | 108 ± 15 | 88 ± 14 | 151 ± 28 | 172 ± 44 |
| Polydispersity index | 0.20 ± 0.04 | 0.21 ± 0.02 | 0.18 ± 0.07 | 0.24 ± 0.03 |
| Zeta potential (mV) | +30.1 ± 1.2 | −36.7 ± 3.3 | −9.0 ± 2,4 | −8.9 ± 1.3 |
| 2-Week Stability | SA-PBS pH 5.6 | DCP-PBS pH 5.6 |
|---|---|---|
| Vesicle size (nm) | 118 ± 17 | 113 ± 9 |
| Polydispersity index | 0.23 ± 0.07 | 0.30 ± 0.09 |
| Zeta potential (mV) | +26.7 ± 3.1 | −39.0 ± 3.2 |
| Composition | Starting Point of Weight Loss (°C) | Maximal Weight Loss at 300 °C (%) |
|---|---|---|
| SA-PBS pH 5.6 | 75 | 4 |
| DCP-PBS pH 5.6 | 80 | 4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Németh, Z.; Csóka, I.; Semnani Jazani, R.; Sipos, B.; Haspel, H.; Kozma, G.; Kónya, Z.; Dobó, D.G. Quality by Design-Driven Zeta Potential Optimisation Study of Liposomes with Charge Imparting Membrane Additives. Pharmaceutics 2022, 14, 1798. https://doi.org/10.3390/pharmaceutics14091798
Németh Z, Csóka I, Semnani Jazani R, Sipos B, Haspel H, Kozma G, Kónya Z, Dobó DG. Quality by Design-Driven Zeta Potential Optimisation Study of Liposomes with Charge Imparting Membrane Additives. Pharmaceutics. 2022; 14(9):1798. https://doi.org/10.3390/pharmaceutics14091798
Chicago/Turabian StyleNémeth, Zsófia, Ildikó Csóka, Reza Semnani Jazani, Bence Sipos, Henrik Haspel, Gábor Kozma, Zoltán Kónya, and Dorina Gabriella Dobó. 2022. "Quality by Design-Driven Zeta Potential Optimisation Study of Liposomes with Charge Imparting Membrane Additives" Pharmaceutics 14, no. 9: 1798. https://doi.org/10.3390/pharmaceutics14091798