
Insights into the Pharmacogenetics of Tacrolimus Pharmacokinetics and Pharmacodynamics


{kind=link}
Abstract
:1. Introduction
2. CYP3A Pharmacogenetics and Its Influence on Tacrolimus Pharmacokinetics, IPV, C0/D Ratio, and Clinical Outcome
2.1. CYP3A5 Genotype and Tacrolimus IPV
2.2. The CYP3A5 Genotype and Tacrolimus C0/D Ratio
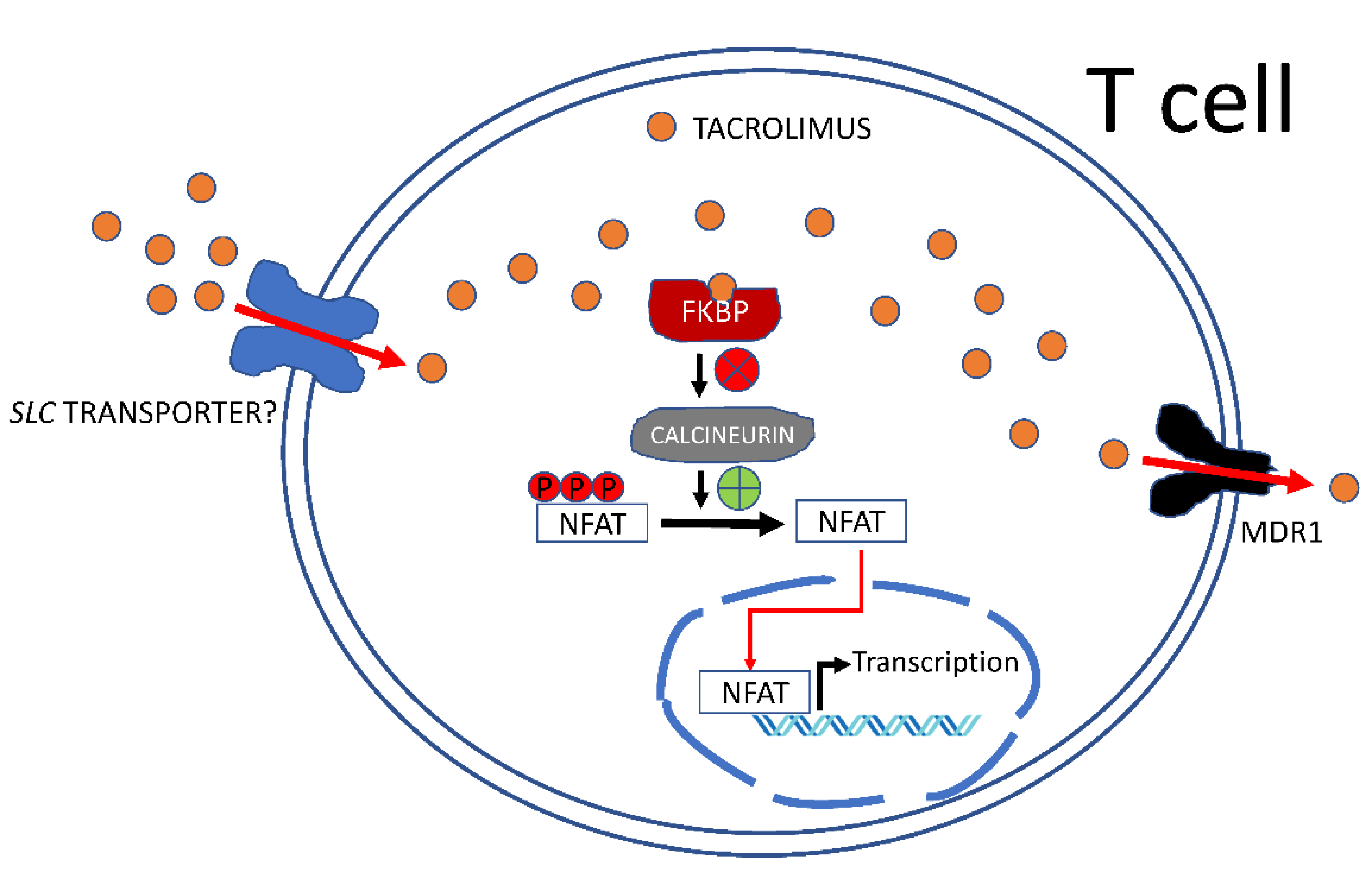
3. Will Membrane Transporters Ever Become Pharmacogenetic Biomarkers of Tacrolimus Pharmacokinetics and Pharmacodynamics?
3.1. The Role of ABC Pumps in Tacrolimus Handling
3.2. Tacrolimus Influx Transporters
4. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Birdwell, K.A.; Decker, B.; Barbarino, J.M.; Peterson, J.F.; Stein, C.M.; Sadee, W.; Wang, D.; Vinks, A.A.; He, Y.; Swen, J.J.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines for CYP3A5 genotype and tacrolimus dosing. Clin. Pharmacol. Ther. 2015, 98, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Brunet, M.; Van Gelder, T.; Åsberg, A.; Haufroid, V.; Hesselink, D.A.; Langman, L.; Lemaitre, F.; Marquet, P.; Seger, C.; Shipkova, M.; et al. Therapeutic Drug Monitoring of Tacrolimus-Personalized Therapy: Second Consensus Report. Ther. Drug Monit. 2019, 41, 261–307. [Google Scholar] [CrossRef] [PubMed]
- Degraeve, A.L.; Moudio, S.; Haufroid, V.; Chaib Eddour, D.; Mourad, M.; Bindels, L.B.; Elens, L. Predictors of tacrolimus pharmacokinetic variability: Current evidences and future perspectives. Expert Opin. Drug Metab. Toxicol. 2020, 16, 769–782. [Google Scholar] [CrossRef] [PubMed]
- Neuberger, J.M.; Bechstein, W.O.; Kuypers, D.R.J.; Burra, P.; Citterio, F.; De Geest, S.; Duvoux, C.; Jardine, A.G.; Kamar, N.; Krämer, B.K.; et al. Practical recommendations for long-term management of modifiable risks in kidney and liver transplant recipients: A guidance report and clinical checklist by the consensus on managing modifiable risk in transplantation (COMMIT) group. Transplantation 2017, 101, S1–S56. [Google Scholar] [CrossRef]
- Yu, M.; Liu, M.; Zhang, W.; Ming, Y. Pharmacokinetics, Pharmacodynamics and Pharmacogenetics of tacrolimus in Kidney Transplantation. Curr. Drug Metab. 2018, 19, 513–522. [Google Scholar] [CrossRef]
- Tron, C.; Florian, L.; Verstuyft, C.; Petitcollin, A.; Verdier, M.C.; Bellissant, E. Pharmacogenetics of membrane transporters of tacrolimus in solid organ transplantation. Clin. Pharmacokinet. 2019, 58, 593–613. [Google Scholar] [CrossRef]
- Mendoza Rojas, A.; Hesselink, D.A.; van Besouw, N.M.; Baan, C.C.; van Gelder, T. Impact of low tacrolimus exposure and high tacrolimus intra-patient variability on the development of de novo anti-HLA donor-specific antibodies in kidney transplant recipients. Expert Rev. Clin. Immunol. 2019, 15, 1323–1331. [Google Scholar] [CrossRef]
- Gonzales, H.M.; McGillicuddy, J.W.; Rohan, V.; Chandler, J.L.; Nadig, S.N.; Dubay, D.A.; Taber, D.J. A comprehensive review of the impact of tacrolimus intrapatient variability on clinical outcomes in kidney transplantation. Am. J. Transplant. 2020, 20, 1969–1983. [Google Scholar] [CrossRef]
- Lemaitre, F.; Monchaud, C.; Woillard, J.B.; Picard, N.; Marquet, P. Synthèse des recommandations de l’International Association of Therapeutic Drug Monitoring and Clinical Toxicology (IATDMCT) sur le suivi thérapeutique pharmacologique du tacrolimus. Therapies 2020, 75, 681–685. [Google Scholar] [CrossRef]
- Shuker, N.; van Gelder, T.; and Hesselink, D.A. Intra-patient variability in tacrolimus exposure: Causes, consequences for clinical management. Transplant. Rev. 2015, 29, 78–84. [Google Scholar] [CrossRef]
- Vanhov, T.; Vermeulen, T.; Annaert, P.; Lerut, E.; Kuypers, D.R.J. High intrapatient variability of tacrolimus concentrations predicts accelerated progression of chonic histologic lesions in renal recipients. Am. J. Transplant. 2016, 16, 2954–2963. [Google Scholar] [CrossRef]
- Shuker, N.; Shuker, L.; van Rosmalen, J.; Roodnat, J.I.; Borra, L.C.; Weimar, W.; Hesselink, D.A.; van Gelder, T. A high intrapatient variability in tacrolimus exposure is associated with poor long-term outcome of kidney transplantation. Transpl. Int. 2016, 29, 1158–1167. [Google Scholar] [CrossRef]
- Rodrigo, E.; Segundo, D.S.; Fernández-Fresnedo, G.; López-Hoyos, M.; Benito, A.; Ruiz, J.C.; de Cos, M.A.; Arias, M. Within-Patient Variability in Tacrolimus Blood Levels Predicts Kidney Graft Loss and Donor-Specific Antibody Development. Transplantation 2016, 100, 2479–2485. [Google Scholar] [CrossRef]
- Kuypers, D.R.J. Intrapatient variability of tacrolimus exposure in solid organ transplantation: A novel marker for clinical outcome. Clin. Pharmacol. Ther. 2020, 107, 347–358. [Google Scholar] [CrossRef]
- Thölking, G.; Fortmann, C.; Koch, R.; Gerth, H.U.; Pabst, D.; Pavenstädt, H.; Kabar, I.; Hüsing, A.; Wolters, H.; Reuter, S.; et al. The tacrolimus metabolism rate influences renal function after kidney transplantation. PLoS ONE 2014, 9, e111128. [Google Scholar] [CrossRef]
- Schütte-Nütgen, K.; Thölking, G.; Steinke, J.; Pavenstädt, H.; Schmidt, R.; Suwelack, B.; Reuter, S. Fast Tac Metabolizers at Risk—It is Time for a C/D Ratio Calculation. J. Clin. Med. 2019, 8, 587. [Google Scholar] [CrossRef]
- Oberbauer, R.; Bestard, O.; Furian, L.; Maggiore, U.; Pascual, J.; Rostaing, L.; Budde, K. Optimization of tacrolimus in kidney transplantation: New pharmacokinetic perspectives. Transplant. Rev. 2020, 34, 100531. [Google Scholar] [CrossRef]
- Rong, H.; Barratt, D.T.; Coller, J.K.; Sallustio, B.C.; Somogyi, A.A. CYP3A5*3 and ABCB1 61A>G significantly influence dose-adjusted trough blood tacrolimus concentrations in the first three months post-kidney transplantation. Basic Clin. Pharmacol. Toxicol. 2018, 123, 320–326. [Google Scholar]
- Hesselink, D.A.; Bouamar, R.; Elens, L.; van Schaik, R.H.; van Gelder, T. The role of pharmacogenetics in the disposition of and response to tacrolimus in solid organ transplantation. Clin. Pharmacokinet. 2014, 53, 123–139. [Google Scholar] [CrossRef]
- Kamdem, L.K.; Streit, F.; Zanger, U.M.; Brockmöller, J.; Oellerich, M.; Armstrong, V.W.; Wojnowski, L. Contribution of CYP3A5 to the in vitro hepatic clearance of tacrolimus. Clin. Chem. 2005, 51, 1374–1381. [Google Scholar] [CrossRef]
- Thervet, E.; Anglicheau, D.; King, B.; Schlageter, M.H.; Cassinat, B.; Beaune, P.; Legendre, C.; Daly, A.K. Impact of cytochrome P450 3A5 genetic polymorphism on tacrolimus doses and concentration-to-dose ratio in renal transplant recipients. Transplantation 2003, 76, 1233–1235. [Google Scholar] [CrossRef]
- Jacobson, P.A.; Oetting, W.S.; Brearley, A.M.; Leduc, R.; Guan, W.; Schladt, D.; Matas, A.J.; Lamba, V.; Julian, B.A.; Mannon, R.B.; et al. Novel polymorphisms associated with tacrolimus trough concentrations: Results from a multicenter kidney transplant consortium. Transplantation 2011, 91, 300–308. [Google Scholar] [CrossRef]
- Haufroid, V.; Mourad, M.; Van Kerckhove, V.; Wawrzyniak, J.; De Meyer, M.; Eddour, D.C.; Malaise, J.; Lison, D.; Squifflet, J.; Wallemacq, P. The effect of CYP3A5 and MDR1 (ABCB1) polymorphisms on cyclosporine and tacrolimus dose requirements and trough blood levels in stable renal transplant patients. Pharmacogenetics 2004, 14, 147–154. [Google Scholar] [CrossRef]
- Gómez-Bravo, M.A.; Salcedo, M.; Fondevila, C.; Suarez, F.; Castellote, J.; Rufian, S.; Pons, J.A.; Alamo, J.M.; Millán, O.; Brunet, M. Impact of donor and recipient CYP3A5 and ABCB1 genetic polymorphisms on tacrolimus dosage requirements and rejection in Caucasian Spanish liver transplant patients. J. Clin. Pharmacol. 2013, 53, 1146–1154. [Google Scholar] [CrossRef]
- Wang, D.; Guo, Y.; Wrighton, S.A.; Cooke, G.E.; Sadee, W. Intronic polymorphism in CYP3A4 affects hepatic expression and response to statin drugs. Pharm. J. 2011, 11, 274–286. [Google Scholar] [CrossRef]
- Elens, L.; Bouamar, R.; Hesselink, D.A.; Haufroid, V.; van der Heiden, I.P.; van Gelder, T.; van Schaik, R.H. A new functional CYP3A4 intron 6 polymorphism significantly affects tacrolimus pharmacokinetics in kidney transplant recipients. Clin. Chem. 2011, 57, 1574–1583. [Google Scholar] [CrossRef]
- Gómez-Bravo, M.A.; Apellaniz-Ruiz, M.; Salcedo, M.; Fondevila, C.; Suarez, F.; Castellote, J.; Rufian, S.; Pons, J.A.; Bilbao, I.; Alamo, J.M.; et al. Influence of donor liver CYP3A4*20 loss-of-function genotype on tacrolimus pharmacokinetics in transplanted patients. Pharm. Genom. 2018, 28, 41–48. [Google Scholar] [CrossRef]
- Elens, L.; Capron, A.; van Schaik, R.H.; De Meyer, M.; De Pauw, L.; Eddour, D.C.; Latinne, D.; Wallemacq, P.; Mourad, M.; Haufroid, V. Impact of CYP3A4*22 allele on tacrolimus pharmacokinetics in early period after renal transplantation: Toward updated genotype-based dosage guidelines. Ther. Drug Monit. 2013, 35, 608–616. [Google Scholar] [CrossRef]
- Elens, L.; van Schaik, R.H.; Panin, N.; de Meyer, M.; Wallemacq, P.; Lison, D.; Mourad, M.; Haufroid, V. Effect of a new functional CYP3A4 polymorphism on calcineurin inhibitors’ dose requirements and trough blood levels in stable renal transplant patients. Pharmacogenomics 2011, 12, 1383–1396. [Google Scholar] [CrossRef]
- Lunde, I.; Bremer, S.; Midtvedt, K.; Mohebi, B.; Dahl, M.; Bergan, S.; Åsberg, A.; Christensen, H. The influence of CYP3A, PPARA, and POR genetic variants on the pharmacokinetics of tacrolimus and cyclosporine in renal transplant recipients. Eur. J. Clin. Pharmacol. 2014, 70, 685–693. [Google Scholar] [CrossRef]
- Picard, N.; Bergan, S.; Marquet, P.; van Gelder, T.; Wallemac, P.; Hesselink, D.A.; Haufroid, V. Pharmacogenetic biomarkers predictive of the pharmacokinetics and pharmacodynamics of immunosuppressive drugs. Ther. Drug. Monit. 2016, 38, S57–S69. [Google Scholar] [CrossRef] [PubMed]
- From 1000 Genomes Project Data. Available online: http:www.internationalgenome.org (accessed on 30 May 2022).
- Mohamed, M.E.; Schladt, D.P.; Guan, W.; Wu, B.; van Setten, J.; Keating, B.J.; Iklé, D.; Remmel, R.P.; Dorr, C.R.; Mannon, R.B.; et al. DeKAF Genomics and GEN03 Investigators. Tacrolimus troughs and genetic determinants of metabolism in kidney transplant recipients: A comparison of four ancestry groups. Am. J. Transplant. 2019, 19, 2795–2804. [Google Scholar] [CrossRef] [PubMed]
- de Jonge, H.; de Loor, H.; Verbeke, K.; Vanrenterghem, Y.; Kuypers, D.R. In vivo CYP3A4 activity, CYP3A5 genotype, and hematocrit predict tacrolimus dose requirements and clearance in renal transplant patients. Clin. Pharmacol. Ther. 2012, 92, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Kurzawski, M.; Malinowski, D.; Dziewanowski, K.; Drozdzik, M. Impact of PPARA and POR polymorphisms on tacrolimus pharmacokinetics and new-onset diabetes in kidney transplant recipients. Pharmacogenet. Genom. 2014, 24, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Sallustio, B.C.; Noll, B.D.; Hu, R.; Barratt, D.T.; Tuke, J.; Coller, J.K.; Russ, G.R.; Somogyi, A.A. Tacrolimus dose, blood concentrations and acute nephrotoxicity, but not CYP3A5/ABCB1 genetics, are associated with allograft tacrolimus concentrations in renal transplant recipients. Br. J. Clin. Pharmacol. 2021, 87, 3901–3909. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Huan, G.; Wang, M. Influence of CYP3A and ABCB1 Single Nucleotide Polymorphisms on the Pharmacokinetics/Pharmacodynamics of Tacrolimus in Pediatric Patients. Curr. Drug Metab. 2018, 19, 1141–1151. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Men, T.; Liu, H.; Li, X.; Wang, J.; Lv, J. Genetic risk factors for post-transplantation diabetes mellitus in Chinese Han renal allograft recipients treated with tacrolimus. Transpl. Immunol. 2018, 49, 39–42. [Google Scholar] [CrossRef]
- Lancia, P.; Adam de Beaumais, T.; Elie, V.; Garaix, F.; Fila, M.; Nobili, F.; Ranchin, B.; Testevuide, P.; Ulinski, T.; Zhao, W.; et al. Pharmacogenetics of post-transplant diabetes mellitus in children with renal transplantation treated with tacrolimus. Pediatr. Nephrol. 2018, 33, 1045–1055. [Google Scholar] [CrossRef]
- Xi, B.; Wang, C.; Liu, L.; Zeng, T.; Liang, Y.; Li, J.; Mi, J. Association of the CYP3A5 polymorphism (6986G>A) with blood pressure and hypertension. Hypertens. Res. 2011, 34, 1216–1220. [Google Scholar] [CrossRef]
- Swen, J.J.; Nijenhuis, M.; de Boer, A.; Grandia, L.; Maitland-van der Zee, A.H.; Mulder, H.; Rongen, G.A.; van Schaik, R.H.; Schalekamp, T.; Touw, D.J.; et al. Pharmacogenetics: From bench to byte—An update of guidelines. Clin. Pharmacol. Ther. 2011, 89, 662–673. [Google Scholar] [CrossRef]
- Andrews, L.M.; Hesselink, D.A.; van Schaik, R.H.N.; van Gelder, T.; de Fijter, J.W.; Lloberas, N.; Elens, L.; Moes, D.A.R.; de Winter, B.C.M. A population pharmacokinetic model to predict the individual starting dose of tacrolimus in adult renal transplant recipients. Br. J. Clin. Pharmacol. 2019, 85, 601–615. [Google Scholar] [CrossRef]
- Francke, M.I.; Hesselink, D.A.; Li, Y.; Koch, B.C.P.; de Wit, L.E.A.; van Schaik, R.H.N.; Yang, L.; Baan, C.C.; van Gelder, T.; de Winter, B.C.M. Avoiding Tacrolimus Underexposure and Overexposure with a Dosing Algorithm for Renal Transplant Recipients: A Single Arm Prospective Intervention Trial. Br. J. Clin. Pharmacol. 2021, 87, 1918–1929. [Google Scholar] [CrossRef]
- Thervet, E.; Loriot, M.A.; Barbier, S.; Buchler, M.; Ficheux, M.; Choukroun, G.; Toupance, O.; Touchard, G.; Alberti, C.; Le Pogamp, P.; et al. Optimization of initial tacrolimus dose using pharmacogenetic testing. Clin. Pharmacol. Ther. 2010, 87, 721–726. [Google Scholar] [CrossRef]
- Venkat, V.L.; Nick, T.G.; Wang, Y.; Bucuvalas, J.C. An objective measure to identify pediatric liver transplant recipients at risk for late allograft rejection related to non-adherence. Pediatr. Transplant. 2008, 12, 67–72. [Google Scholar] [CrossRef]
- Shemesh, E.; Bucuvalas, J.C.; Anand, R.; Mazariegos, G.V.; Alonso, E.M.; Venick, R.S.; Reyes-Mugica, M.; Annunziato, R.A.; Shneider, B.L. The Medication Level Variability Index (MLVI) Predicts Poor Liver Transplant Outcomes: A Prospective Multi-Site Study. Am. J. Transplant. 2017, 17, 2668–2678. [Google Scholar] [CrossRef]
- Kim, E.; Kim, B.; Cho, J.; Lee, J.H.; Lee, E.; Yu, Y.M.; Cho, J.Y.; Lee, E.; Choi, Y. The effects of intrapatient variability in tacrolimus concentration on clinical outcomes immediately after liver transplantation. Korean J. Clin. Pharm. 2020, 30, 36–43. [Google Scholar] [CrossRef]
- Rayar, M.; Tron, C.; Jézéquel, C.; Beaurepaire, J.M.; Petitcollin, A.; Houssel-Debry, P.; Camus, C.; Verdier, M.C.; Dehlawi, A.; Lakéhal, M.; et al. High Intrapatient Variability of Tacrolimus Exposure in the Early Period After Liver Transplantation Is Associated with Poorer Outcomes. Transplantation 2018, 102, e108–e114. [Google Scholar] [CrossRef]
- Del Bello, A.; Congy-Jolivet, N.; Danjoux, M.; Muscari, F.; Lavayssière, L.; Esposito, L.; Hebral, A.L.; Bellière, J.; Kamar, N. High tacrolimus intra-patient variability is associated with graft rejection, and de novo donor-specific antibodies occurrence after liver transplantation. World J. Gastroenterol. 2018, 24, 1795–1802. [Google Scholar] [CrossRef]
- Van der Veer, M.A.A.; Nangrahary, N.; Hesselink, D.A.; Erler, N.S.; Metselaar, H.J.; van Gelder, T.; Murad, S.D. High Intrapatient Variability in Tacrolimus Exposure Is Not Associated with Immune-mediated Graft Injury After Liver Transplantation. Transplantation 2019, 103, 2329–2337. [Google Scholar] [CrossRef]
- Dopazo, C.; Bilbao, I.; García, S.; Gómez-Gavara, C.; Caralt, M.; Campos-Varela, I.; Lluis Castells, L.; Hidalgo, E.; Moreso, F.; Montoro, B.; et al. High intrapatient variability of tacrolimus exposure associated with poorer outcomes in liver transplantation. Clin. Transl. Sci. 2022, 15, 1544–1555. [Google Scholar] [CrossRef]
- Coste, G.; Lemaitre, F. The Role of Intra-Patient Variability of Tacrolimus Drug Concentrations in Solid Organ Transplantation: A Focus on Liver, Heart, Lung and Pancreas. Pharmaceutics 2022, 14, 379. [Google Scholar] [CrossRef]
- Schumacher, L.; Leino, A.D.; Park, J.M. Tacrolimus intrapatient variability in solid organ transplantation: A multiorgan perspective. Pharmacotherapy 2021, 41, 103–118. [Google Scholar] [CrossRef]
- Kim, E.J.; Kim, S.J.; Huh, K.H.; Kim, B.S.; Kim, M.S.; Kim, S.; Kim, S.I.; Kim, Y.S.; Lee, J. Clinical significance of tacrolimus intra-patient variability on kidney transplant outcomes according to pre-transplant immunological risk. Sci. Rep. 2021, 9, 12114. [Google Scholar] [CrossRef]
- Pashaee, N.; Bouamar, R.; Hesselink, D.A.; Roodnat, J.I.; van Schaik, R.H.N.; Weimar, W.; van Gelder, T. CYP3A5 genotype is not related to the intrapatient variability of tacrolimus clearance. Ther. Drug Monit. 2011, 33, 369–371. [Google Scholar] [CrossRef]
- Spierings, N.; Holt, D.W.; MacPhee, I.A.M. CYP3A5 genotype had no impact on intrapatient variability of tacrolimus clearance in renal transplant recipients. Ther. Drug Monit. 2013, 35, 328–331. [Google Scholar] [CrossRef]
- Ro, H.; Min, S.I.; Yang, J.; Moon, K.C.; Kim, Y.S.; Kim, S.J.; Ahn, C.; Ha, J. Impact of tacrolimus intraindividual variability and CYP3A5 genetic polymorphism on acute rejection in kidney transplantation. Ther. Drug Monit. 2012, 34, 680–685. [Google Scholar] [CrossRef]
- Choi, J.S.; Ko, H.; Kim, H.K.; Chung, C.; Han, A.; Min, S.K.; Ha, J.; Kang, H.G.; Ha, I.S.; Min, S. Effects of tacrolimus intrapatient variability and CYP3A5 polymorphism on the outcomes of pediatric kidney transplantation. Pediatr. Transplant. 2022, 26, e14297. [Google Scholar] [CrossRef]
- Seibert, S.R.; Schladt, D.P.; Wu, B.; Guan, W.; Dorr, C.; Remmel, R.P.; Matas, A.J.; Mannon, R.B.; Israni, A.K.; Oetting, W.S.; et al. Tacrolimus trough and dose intra-patient variability and CYP3A5 genotype: Effects on acute rejction and graft failure in European American and African American Kidney transplant recipients. Clin. Transplant. 2018, 32, e13424. [Google Scholar] [CrossRef]
- Thölking, G.; Schmidt, C.; Koch, R.; Schuette-Nuetgen, K.; Pabst, D.; Wolters, H.; Kabar, I.; Hüsing, A.; Pavenstädt, H.; Reuter, S.; et al. Influence of tacrolimus metabolism rate on BKV infection after kidney transplantation. Sci. Rep. 2016, 6, 32273. [Google Scholar] [CrossRef]
- van Gelder, T.; Meziyerh, S.; Swen, J.J.; de Vries, A.P.J.; Moes, D.J.A.R. The Clinical Impact of the C0/D Ratio and the CYP3A5 Genotype on Outcome in Tacrolimus Treated Kidney Transplant Recipients. Front. Pharmacol. 2020, 11, 1142. [Google Scholar] [CrossRef]
- Stefanovic, N.Z.; Velickovic-Radovanovic, R.M.; Dankovic, K.S.; Mitic, B.P.; Paunovic, G.J.; Cvetkovic, M.B.; Cvetkovic, T.P. Combined effect of inter- and intrapatient variability in tacrolimus exposure on graft impairment within a 3-year period following kidney transplantation: A single-center experience. Eur. J. Drug Metab. Pharmacokinet. 2020, 45, 749–760. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Nigam, S.K. What do drug transporters really do? Nat. Rev. Drug Discov. 2015, 14, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Kell, D.B. Implications of endogenous roles of transporters for drug discovery: Hitchhiking and metabolite-likeness. Nat. Rev. Drug Discov. 2016, 15, 143. [Google Scholar] [CrossRef] [PubMed]
- Kell, D.B. The Transporter-Mediated Cellular Uptake and Efflux of Pharmaceutical Drugs and Biotechnology Products: How and Why Phospholipid Bilayer Transport Is Negligible in Real Biomembranes. Molecules 2021, 26, 5629. [Google Scholar] [CrossRef] [PubMed]
- International Transporter Consortium; Giacomini, K.M.; Huang, S.M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar] [PubMed]
- Chu, X.; Liao, M.; Shen, H.; Yoshida, K.; Zur, A.A.; Arya, V.; Galetin, A.; Giacomini, K.M.; Hanna, I.; Kusuhara, H.; et al. International Transporter Consortium. Clinical Probes and Endogenous Biomarkers as Substrates for Transporter Drug-Drug Interaction Evaluation: Perspectives from the International Transporter Consortium. Clin. Pharmacol. Ther. 2018, 104, 836–864. [Google Scholar] [CrossRef] [PubMed]
- Zamek-Gliszczynski, M.J.; Taub, M.E.; Chothe, P.P.; Chu, X.; Giacomini, K.M.; Kim, R.B.; Ray, A.S.; Stocker, S.L.; Unadkat, J.D.; Wittwer, M.B.; et al. International Transporter Consortium. Transporters in Drug Development: 2018 ITC Recommendations for Transporters of Emerging Clinical Importance. Clin. Pharmacol. Ther. 2018, 104, 890–899. [Google Scholar] [CrossRef]
- Zamek-Gliszczynski, M.J.; Sangha, V.; Shen, H.; Feng, B.; Wittwer, M.B.; Varma, M.V.S.; Liang, X.; Sugiyama, Y.; Zhang, L.; Bendayan, R.; et al. Transporters in drug development: International transporter consortium update on emerging transporters of clinical importance. Clin. Pharmacol. Ther. 2022. [Google Scholar] [CrossRef]
- Dvorak, V.; Wiedmer, T.; Ingles-Prieto, A.; Altermatt, P.; Batoulis, H.; Bärenz, F.; Bender, E.; Digles, D.; Dürrenberger, F.; Heitman, L.H.; et al. An Overview of Cell-Based Assay Platforms for the Solute Carrier Family of Transporters. Front. Pharmacol. 2021, 12, 722889. [Google Scholar] [CrossRef]
- Saeki, T.; Ueda, K.; Tanigawara, Y.; Hori, R.; Komano, T. Human P-glycoprotein transports cyclosporin A and FK506. J. Biol. Chem. 1993, 268, 6077–6080. [Google Scholar] [CrossRef]
- Fromm, M.F. Importance of P-glycoprotein at blood-tissue barriers. Trends Pharmacol. Sci. 2004, 25, 423–429. [Google Scholar] [CrossRef]
- Liu, X. ABC Family Transporters. Adv. Exp. Med. Biol. 2019, 1141, 13–100. [Google Scholar]
- Eichelbaum, M.; Fromm, M.F.; Schwab, M. Clinical aspects of the MDR1 (ABCB1) gene polymorphism. Ther. Drug Monit. 2004, 26, 180–185. [Google Scholar] [CrossRef]
- Hodges, L.M.; Markova, S.M.; Chinn, L.W.; Gow, J.M.; Kroetz, D.L.; Klein, T.E.; Altman, R.B. Very important pharmacogene summary: ABCB1 (MDR1, P-glycoprotein). Pharm. Genom. 2011, 21, 152–161. [Google Scholar] [CrossRef]
- Hoffmeyer, S.; Burk, O.; von Richter, O.; Arnold, H.P.; Brockmöller, J.; Johne, A.; Cascorbi, I.; Gerloff, T.; Roots, I.; Eichelbaum, M.; et al. Functional polymorphisms of the human multidrug-resistance gene: Multiple sequence variations and correlation of one allele with P-glycoprotein expression and activity in vivo. Proc. Natl. Acad. Sci. USA 2000, 97, 3473–3478. [Google Scholar] [CrossRef]
- Elens, L.; Capron, A.; Kerckhove, V.V.; Lerut, J.; Mourad, M.; Lison, D.; Wallemacq, P.; Haufroid, V. 1199G>A and 2677G>T/A polymorphisms of ABCB1 independently affect tacrolimus concentration in hepatic tissue after liver transplantation. Pharm. Genom. 2007, 17, 873–883. [Google Scholar] [CrossRef]
- Tron, C.; Woillard, J.B.; Houssel-Debry, P.; David, V.; Jezequel, C.; Rayar, M.; Balakirouchenane, D.; Blanchet, B.; Debord, J.; Petitcollin, A.; et al. Pharmacogenetic-Whole blood and intracellular pharmacokinetic-Pharmacodynamic (PG-PK2-PD) relationship of tacrolimus in liver transplant recipients. PLoS ONE 2020, 15, e0230195. [Google Scholar] [CrossRef]
- Sallustio, B.C. Monitoring Intra-cellular Tacrolimus Concentrations in Solid Organ Transplantation: Use of Peripheral Blood Mononuclear Cells and Graft Biopsy Tissue. Front. Pharmacol. 2021, 12, 733285. [Google Scholar] [CrossRef]
- De Nicolòf, A.; Pinon, M.; Palermiti, A.; Nonnato, A.; Manca, A.; Mula, J.; Catalano, S.; Tandoi, F.; Romagnoli, R.; D’Avolio, A.; et al. Monitoring Tacrolimus Concentrations in Whole Blood and Peripheral Blood Mononuclear Cells: Inter- and Intra-Patient Variability in a Cohort of Pediatric Patients. Front. Pharmacol. 2021, 12, 750433. [Google Scholar] [CrossRef]
- Dessilly, G.; Elens, L.; Panin, N.; Capron, A.; Decottignies, A.; Demoulin, J.B.; Haufroid, V. ABCB1 1199G>A genetic polymorphism (Rs2229109) influences the intracellular accumulation of tacrolimus in HEK293 and K562 recombinant cell lines. PLoS ONE 2014, 9, e91555. [Google Scholar]
- Minuesa, G.; Arimany-Nardi, C.; Erkizia, I.; Cedeño, S.; Moltó, J.; Clotet, B.; Pastor-Anglada, M.; Martinez-Picado, J. P-glycoprotein (ABCB1) activity decreases raltegravir disposition in primary CD4+P-gp high cells and correlates with HIV-1 viral load. J. Antimicrob. Chemother. 2016, 71, 2782–2792. [Google Scholar] [CrossRef] [PubMed]
- Lemaitre, F.; Vethe, N.T.; D’Avolio, A.; Tron, C.; Robertsen, I.; De Winter, B.; Denicolo, A.; Koch, B.C.P.; Venkataramanan, R.; Van Gelder, T.; et al. Measuring Intracellular Concentrations of Calcineurin Inhibitors: Expert Consensus from the International Association of Therapeutic Drug Monitoring and Clinical Toxicology Expert Panel. Ther. Drug Monit. 2020, 42, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Fehrenbach, T.; Cui, Y.; Faulstich, H.; Keppler, D. Characterization of the transport of the bicyclic peptide phalloidin by human hepatic transport proteins. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2003, 368, 415–420. [Google Scholar] [CrossRef]
- Shitara, Y.; Takeuchi, K.; Nagamatsu, Y.; Wada, S.; Sugiyama, Y.; Horie, T. Long-lasting inhibitory effects of cyclosporin A, but not tacrolimus, on OATP1B1- and OATP1B3-mediated uptake. Drug Metab. Pharmacokinet. 2012, 27, 368–378. [Google Scholar] [CrossRef]
- de Graaf, W.; Häusler, S.; Heger, M.; van Ginhoven, T.M.; van Cappellen, G.; Bennink, R.J.; Kullak-Ublick, G.A.; Hesselmann, R.; van Gulik, T.M.; Stieger, B. Transporters involved in the hepatic uptake of (99m)Tc-mebrofenin and indocyanine green. J. Hepatol. 2011, 54, 738–745. [Google Scholar] [CrossRef]
- Hagenbuch, B.; Stieger, B. The SLCO (former SLC21) superfamily of transporters. Mol. Aspects Med. 2013, 34, 396–412. [Google Scholar] [CrossRef]
- Niemi, M.; Pasanen, M.K.; Neuvonen, P.J. Organic anion transporting polypeptide 1B1: A genetically polymorphic transporter of major importance for hepatic drug uptake. Pharmacol. Rev. 2011, 63, 157–181. [Google Scholar] [CrossRef]
- Turner, R.M.; Pirmohamed, M. Statin-Related Myotoxicity: A Comprehensive Review of Pharmacokinetic, Pharmacogenomic and Muscle Components. J. Clin. Med. 2019, 9, 22. [Google Scholar] [CrossRef]
- Cooper-DeHoff, R.M.; Niemi, M.; Ramsey, L.B.; Luzum, J.A.; Tarkiainen, E.K.; Straka, R.J.; Gong, L.; Tuteja, S.; Wilke, R.A.; Wadelius, M.; et al. The Clinical Pharmacogenetics Implementation Consortium Guideline for SLCO1B1, ABCG2, and CYP2C9 genotypes and Statin-Associated Musculoskeletal Symptoms. Clin. Pharmacol. Ther. 2022, 111, 1007–1021. [Google Scholar] [CrossRef]
- Nies, A.T.; Niemi, M.; Burk, O.; Winter, S.; Zanger, U.M.; Stieger, B.; Schwab, M.; Schaeffeler, E. Genetics is a major determinant of expression of the human hepatic uptake transporter OATP1B1, but not of OATP1B3 and OATP2B1. Genome Med. 2013, 5, 1. [Google Scholar] [CrossRef]
- Lemahieu, W.P.; Hermann, M.; Asberg, A.; Verbeke, K.; Holdaas, H.; Vanrenterghem, Y.; Maes, B.D. Combined therapy with atorvastatin and calcineurin inhibitors: No interactions with tacrolimus. Am. J. Transplant. 2005, 5, 2236–2243. [Google Scholar] [CrossRef]
- Neuvonen, M.; Hirvensalo, P.; Tornio, A.; Rago, B.; West, M.; Lazzaro, S.; Mathialagan, S.; Varma, M.; Cerny, M.A.; Costales, C.; et al. Identification of Glycochenodeoxycholate 3-O-Glucuronide and Glycodeoxycholate 3-O-Glucuronide as Highly Sensitive and Specific OATP1B1 Biomarkers. Clin. Pharmacol. Ther. 2021, 109, 646–657. [Google Scholar] [CrossRef]
- Neuvonen, M.; Tornio, A.; Hirvensalo, P.; Backman, J.T.; Niemi, M. Performance of Plasma Coproporphyrin I and III as OATP1B1 Biomarkers in Humans. Clin. Pharmacol. Ther. 2021, 110, 1622–1632. [Google Scholar] [CrossRef]
- Damon, C.; Luck, M.; Toullec, L.; Etienne, I.; Buchler, M.; Hurault de Ligny, B.; Choukroun, G.; Thierry, A.; Vigneau, C.; Moulin, B.; et al. Predictive Modeling of Tacrolimus Dose Requirement Based on High-Throughput Genetic Screening. Am. J. Transplant. 2017, 17, 1008–1019. [Google Scholar] [CrossRef]
- Pastor-Anglada, M.; Pérez-Torras, S. Nucleoside transporter proteins as biomarkers of drug responsiveness and drug targets. Front. Pharmacol. 2015, 6, 13. [Google Scholar] [CrossRef]
- Pastor-Anglada, M.; Urtasun, N.; Pérez-Torras, S. Intestinal Nucleoside Transporters: Function, Expression, and Regulation. Compr. Physiol. 2018, 8, 1003–1017. [Google Scholar]
- Pastor-Anglada, M.; Pérez-Torras, S. Who Is Who in Adenosine Transport. Front. Pharmacol. 2018, 9, 627. [Google Scholar] [CrossRef]
- Kim, D.G.; Bynoe, M.S. A2A adenosine receptor modulates drug efflux transporter P-glycoprotein at the blood-brain barrier. J. Clin. Investig. 2016, 126, 1717–1733. [Google Scholar] [CrossRef]
- Zheng, X.; Huai, C.; Xu, Q.; Xu, L.; Zhang, M.; Zhong, M.; Qiu, X. FKBP-CaN-NFAT pathway polymorphisms selected by in silico biological function prediction are associated with tacrolimus efficacy in renal transplant patients. Eur. J. Pharm. Sci. 2021, 160, 105694. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brunet, M.; Pastor-Anglada, M. Insights into the Pharmacogenetics of Tacrolimus Pharmacokinetics and Pharmacodynamics. Pharmaceutics 2022, 14, 1755. https://doi.org/10.3390/pharmaceutics14091755
Brunet M, Pastor-Anglada M. Insights into the Pharmacogenetics of Tacrolimus Pharmacokinetics and Pharmacodynamics. Pharmaceutics. 2022; 14(9):1755. https://doi.org/10.3390/pharmaceutics14091755
Chicago/Turabian StyleBrunet, Mercè, and Marçal Pastor-Anglada. 2022. "Insights into the Pharmacogenetics of Tacrolimus Pharmacokinetics and Pharmacodynamics" Pharmaceutics 14, no. 9: 1755. https://doi.org/10.3390/pharmaceutics14091755