
Immune Checkpoint Inhibitors for Vaccine Improvements: Current Status and New Approaches

 , , ,
, , ,  and
and
Abstract
:1. Introduction
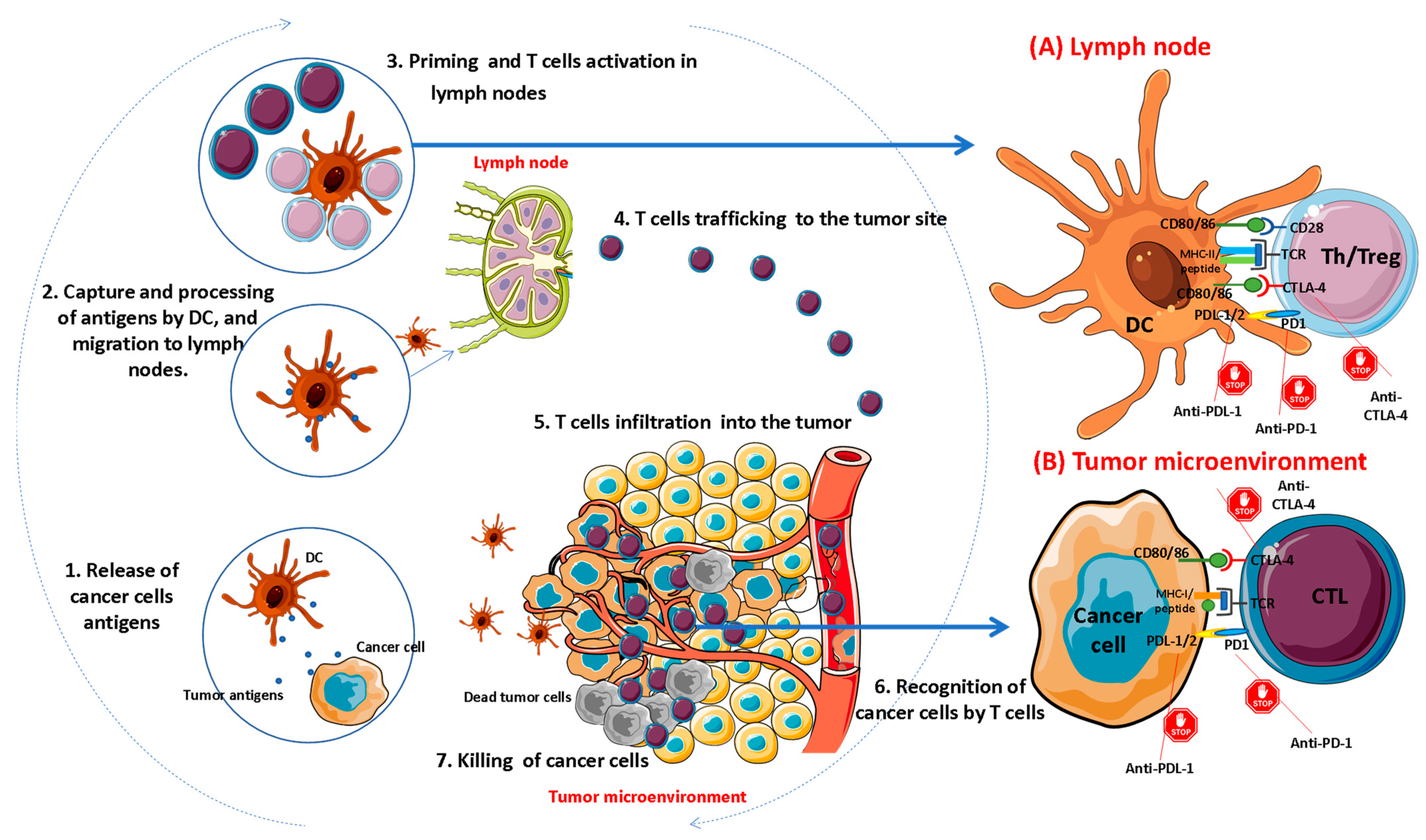
2. Overview of Immune Checkpoints
3. Pharmacological Inhibition of Immune Checkpoints
4. Combining ICIs and Anti-Tumor Vaccines
5. ICIs and Anti-Infectious Vaccines
6. Newly Emerging ICIs (Second Generation) for Combination Therapy
7. New Alternatives of Immune Checkpoint Inhibition
7.1. ASOs
7.2. Small Non-Coding RNAs
7.3. Aptamers
7.4. Peptides and Other Small-Molecule ICIs
8. Future Directions and Closing Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pollard, A.J.; Bijker, E.M. A guide to vaccinology: From basic principles to new developments. Nat. Rev. Immunol. 2021, 21, 83–100. [Google Scholar] [CrossRef] [PubMed]
- Heaton, P.M. Challenges of Developing Novel Vaccines with Particular Global Health Importance. Front. Immunol. 2020, 11, 517290. [Google Scholar] [CrossRef] [PubMed]
- Mazzola, P.; Radhi, S.; Mirandola, L.; Annoni, G.; Jenkins, M.; Cobos, E.; Chiriva-Internati, M. Aging, cancer, and cancer vaccines. Immun Ageing. 2012, 9, 4. [Google Scholar] [CrossRef] [PubMed]
- Pulendran, B.; SArunachalam, P.; O’Hagan, D.T. Emerging concepts in the science of vaccine adjuvants. Nat. Rev. Drug Discov. 2021, 20, 454–475. [Google Scholar] [CrossRef]
- Pogostin, B.H.; McHugh, K.J. Novel Vaccine Adjuvants as Key Tools for Improving Pandemic Preparedness. Bioengineering 2021, 8, 155. [Google Scholar] [CrossRef] [PubMed]
- Petrovsky, N. Comparative Safety of Vaccine Adjuvants: A Summary of Current Evidence and Future Needs. Drug Saf. 2015, 38, 1059–1074. [Google Scholar] [CrossRef] [PubMed]
- Batista-Duharte, A.; Martínez, D.T.; Carlos, I.Z. Efficacy and safety of immunological adjuvants. Where is the cut-off? Biomed. Pharmacother. 2018, 105, 616–624. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, D.T.; Fox, C.B. New generation adjuvants—From empiricism to rational design. Vaccine 2015, 33 (Suppl. 2), B14–B20. [Google Scholar] [CrossRef] [PubMed]
- Tom, J.K.; Albin, T.J.; Manna, S.; Moser, B.A.; Steinhardt, R.C.; Esser-Kahn, A.P. Applications of Immunomodulatory Immune Synergies to Adjuvant Discovery and Vaccine Development. Trends Biotechnol. 2019, 37, 373–388. [Google Scholar] [CrossRef]
- Sabbaghi, A.; Ghaemi, A. Molecular Adjuvants for DNA Vaccines: Application, Design, Preparation, and Formulation. Methods Mol. Biol. 2021, 2197, 87–112. [Google Scholar] [CrossRef]
- Li, L.; Petrovsky, N. Molecular mechanisms for enhanced DNA vaccine immunogenicity. Expert. Rev. Vaccines 2016, 15, 313–329. [Google Scholar] [CrossRef] [PubMed]
- Batista-Duharte, A.; Téllez-Martínez, D.; Fuentes, D.L.P.; Carlos, I.Z. Molecular adjuvants that modulate regulatory T cell function in vaccination: A critical appraisal. Pharmacol. Res. 2018, 129, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Baksh, K.; Weber, J. Immune checkpoint protein inhibition for cancer: Preclinical justification for CTLA-4 and PD-1 blockade and new combinations. Semin. Oncol. 2015, 42, 363–377. [Google Scholar] [CrossRef] [PubMed]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef]
- Tundo, G.R.; Sbardella, D.; Lacal, P.M.; Graziani, G.; Marini, S. On the Horizon: Targeting Next-Generation Immune Checkpoints for Cancer Treatment. Chemotherapy 2019, 64, 62–80. [Google Scholar] [CrossRef]
- Collins, J.M.; Redman, J.M.; Gulley, J.L. Combining vaccines and immune checkpoint inhibitors to prime, expand, and facilitate effective tumor immunotherapy. Expert Rev. Vaccines 2018, 17, 697–705. [Google Scholar] [CrossRef]
- Cavalcante, L.; Chowdhary, A.; Sosman, J.A.; Chandra, S. Combining Tumor Vaccination and Oncolytic Viral Approaches with Checkpoint Inhibitors: Rationale, Pre-Clinical Experience, and Current Clinical Trials in Malignant Melanoma. Am. J. Clin. Dermatol. 2018, 19, 657–670. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, Y.; Ding, Z.Y.; Liu, J.Y. Safety and Efficacy of Therapeutic Cancer Vaccines Alone or in Combination with Immune Checkpoint Inhibitors in Cancer Treatment. Front. Pharmacol. 2019, 10, 1184. [Google Scholar] [CrossRef]
- Vafaei, S.; Zekiy, A.O.; Khanamir, R.A.; Zaman, B.A.; Ghayourvahdat, A.; Azimizonuzi, H.; Zamani, M. Combination therapy with immune checkpoint inhibitors (ICIs); a new frontier. Cancer Cell Int. 2022, 22, 2. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer immunity Cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef]
- Mehdizadeh, S.; Bayatipoor, H.; Pashangzadeh, S.; Jafarpour, R.; Shojaei, Z.; Motallebnezhad, M. Immune checkpoints and cancer development: Therapeutic implications and future directions. Pathol. Res. Pract. 2021, 223, 153485. [Google Scholar] [CrossRef] [PubMed]
- Pisibon, C.; Ouertani, A.; Bertolotto, C.; Ballotti, R.; Cheli, Y. Immune Checkpoints in Cancers: From Signaling to the Clinic. Cancers 2021, 13, 4573. [Google Scholar] [CrossRef] [PubMed]
- Andrews, L.P.; Marciscano, A.E.; Drake, C.G.; Vignali, D.A. LAG3 (CD223) as a cancer immunotherapy target. Immunol. Rev. 2017, 276, 80–96. [Google Scholar] [CrossRef] [PubMed]
- Monney, L.; Sabatos, C.A.; Gaglia, J.L.; Ryu, A.; Waldner, H.; Chernova, T.; Manning, S.; Greenfield, E.A.; Coyle, A.J.; Sobel, R.A.; et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature 2002, 415, 536–541. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Harden, K.; Gonzalez, L.C.; Francesco, M.; Chiang, E.; Irving, B.; Tom, I.; Ivelja, S.; Refino, C.J.; Clark, H.; et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2009, 10, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Rubinstein, R.; Lines, J.L.; Wasiuk, A.; Ahonen, C.; Guo, Y.; Lu, L.F.; Gondek, D.; Wang, Y.; Fava, R.A.; et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J. Exp. Med. 2011, 208, 577–592. [Google Scholar] [CrossRef]
- Chapoval, A.I.; Ni, J.; Lau, J.S.; Wilcox, R.A.; Flies, D.B.; Liu, D.; Dong, H.; Sica, G.L.; Zhu, G.; Tamada, K.; et al. B7-H3: A costimulatory molecule for T cell activation and IFN-gamma production. Nat. Immunol. 2001, 2, 269–274. [Google Scholar] [CrossRef]
- Yu, X.; Zheng, Y.; Mao, R.; Su, Z.; Zhang, J. BTLA/HVEM Signaling: Milestones in Research and Role in Chronic Hepatitis B Virus Infection. Front. Immunol. 2019, 10, 617. [Google Scholar] [CrossRef]
- Wang, J.; Sun, J.; Liu, L.N.; Flies, D.B.; Nie, X.; Toki, M.; Zhang, J.; Song, C.; Zarr, M.; Zhou, X.; et al. Siglec-15 as an immune suppressor and potential target for normalization cancer immunotherapy. Nat. Med. 2019, 25, 656–666. [Google Scholar] [CrossRef]
- Leone, R.D.; Lo, Y.C.; Powell, J.D. A2aR antagonists: Next generation checkpoint blockade for cancer immunotherapy. Comput. Struct. Biotechnol. J. 2015, 13, 265–272. [Google Scholar] [CrossRef]
- Allard, B.; Longhi, M.S.; Robson, S.C.; Stagg, J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol. Rev. 2017, 276, 121–144. [Google Scholar] [CrossRef] [PubMed]
- Perrot, I.; Michaud, H.A.; Giraudon-Paoli, M.; Augier, S.; Docquier, A.; Gros, L.; Courtois, R.; Déjou, C.; Jecko, D.; Becquart, O.; et al. Blocking Antibodies Targeting the CD39/CD73 Immunosuppressive Pathway Unleash Immune Responses in Combination Cancer Therapies. Cell Rep. 2019, 27, 2411–2425.e9. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Kadlecek, T.A.; Au-Yeung, B.B.; Goodfellow, H.E.; Hsu, L.Y.; Freedman, T.S.; Weiss, A. ZAP-70: An essential kinase in T-cell signaling. Cold Spring Harb. Perspect. Biol. 2010, 2, a002279. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, 328rv4. [Google Scholar] [CrossRef]
- Youngnak, P.; Kozono, Y.; Kozono, H.; Iwai, H.; Otsuki, N.; Jin, H.; Omura, K.; Yagita, H.; Pardoll, D.M.; Chen, L.; et al. Differential binding properties of B7-H1 and B7-DC to programmed death-1. Biochem. Biophys. Res. Commun. 2003, 307, 672–677. [Google Scholar] [CrossRef]
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H.; et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858–17863. [Google Scholar] [CrossRef]
- Fu, Z.; Liu, H.; Wang, L.; Yu, C.; Yang, Y.; Feng, M.; Wang, J. A reporter gene assay for determining the biological activity of therapeutic antibodies targeting TIGIT. Acta Pharm. Sin. B 2021, 11, 3925–3934. [Google Scholar] [CrossRef]
- Zhang, Q.; Bi, J.; Zheng, X.; Chen, Y.; Wang, H.; Wu, W.; Wang, Z.; Wu, Q.; Peng, H.; Wei, H.; et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 2018, 19, 723–732. [Google Scholar] [CrossRef]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef]
- Baixeras, E.; Huard, B.; Miossec, C.; Jitsukawa, S.; Martin, M.; Hercend, T.; Auffray, C.; Triebel, F.; Piatier-Tonneau, D. Characterization of the lymphocyte activation gene 3-encoded protein. A new ligand for human leukocyte antigen class II antigens. J. Exp. Med. 1992, 176, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Weber, S.; Karjalainen, K. Mouse CD4 binds MHC class II with extremely low affinity. Int. Immunol. 1993, 5, 695–698. [Google Scholar] [CrossRef] [PubMed]
- Ruiter, D.J.; Mattijssen, V.; Broecker, E.B.; Ferrone, S. MHC antigens in human melanomas. Semin. Cancer Biol. 1991, 2, 35–45. [Google Scholar] [PubMed]
- Bruniquel, D.; Borie, N.; Hannier, S.; Triebel, F. Regulation of expression of the human lymphocyte activation gene-3 (LAG-3) molecule, a ligand for MHC class II. Immunogenetics 1998, 48, 116–124. [Google Scholar] [CrossRef]
- Hannier, S.; Tournier, M.; Bismuth, G.; Triebel, F. CD3/TCR complex-associated lymphocyte activation gene-3 molecules inhibit CD3/TCR signaling. J. Immunol. 1998, 161, 4058–4065. [Google Scholar]
- Golden-Mason, L.; Palmer, B.E.; Kassam, N.; Townshend-Bulson, L.; Livingston, S.; McMahon, B.J.; Castelblanco, N.; Kuchroo, V.; Gretch, D.R.; Rosen, H.R. Negative immune regulator Tim-3 is overexpressed on T cells in hepatitis C virus infection and its blockade rescues dysfunctional CD4+ and CD8+ T cells. J. Virol. 2009, 83, 9122–9130. [Google Scholar] [CrossRef]
- Jones, R.B.; Ndhlovu, L.C.; Barbour, J.D.; Sheth, P.M.; Jha, A.R.; Long, B.R.; Wong, J.C.; Satkunarajah, M.; Schweneker, M.; Chapman, J.M.; et al. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J. Exp. Med. 2008, 205, 2763–2779. [Google Scholar] [CrossRef]
- Takamura, S.; Tsuji-Kawahara, S.; Yagita, H.; Akiba, H.; Sakamoto, M.; Chikaishi, T.; Kato, M.; Miyazawa, M. Premature terminal exhaustion of Friend virus-specific effector CD8+ T cells by rapid induction of multiple inhibitory receptors. J. Immunol. 2010, 184, 4696–4707. [Google Scholar] [CrossRef]
- Zhu, C.; Anderson, A.C.; Schubart, A.; Xiong, H.; Imitola, J.; Khoury, S.J.; Zheng, X.X.; Strom, T.B.; Kuchroo, V.K. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat. Immunol. 2005, 6, 1245–1252. [Google Scholar] [CrossRef]
- Sabatos, C.A.; Chakravarti, S.; Cha, E.; Schubart, A.; Sánchez-Fueyo, A.; Zheng, X.X.; Coyle, A.J.; Strom, T.B.; Freeman, G.J.; Kuchroo, V.K. Interaction of Tim-3 and Tim-3 ligand regulates T helper type 1 responses and induction of peripheral tolerance. Nat. Immunol. 2003, 4, 1102–1110. [Google Scholar] [CrossRef]
- Sánchez-Fueyo, A.; Tian, J.; Picarella, D.; Domenig, C.; Zheng, X.X.; Sabatos, C.A.; Manlongat, N.; Bender, O.; Kamradt, T.; Kuchroo, V.K.; et al. Tim-3 inhibits T helper type 1–mediated auto- and alloimmune responses and promotes immunological tolerance. Nat. Immunol. 2003, 4, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Zhu, C.; Kondo, Y.; Anderson, A.C.; Gandhi, A.; Russell, A.; Dougan, S.K.; Petersen, B.S.; Melum, E.; Pertel, T.; et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature 2015, 517, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Su, E.W.; Zhu, C.; Hainline, S.; Phuah, J.; Moroco, J.A.; Smithgall, T.E.; Kuchroo, V.K.; Kane, L.P. Phosphotyrosine-Dependent Coupling of Tim-3 to T-Cell Receptor Signaling Pathways. Mol. Cell. Biol. 2011, 31, 3963–3974. [Google Scholar] [CrossRef] [PubMed]
- Hosseinkhani, N.; Derakhshani, A.; Shadbad, M.A.; Argentiero, A.; Racanelli, V.; Kazemi, T.; Mokhtarzadeh, A.; Brunetti, O.; Silvestris, N.; Baradaran, B. The Role of V-Domain Ig Suppressor of T Cell Activation (VISTA) in Cancer Therapy: Lessons Learned and the Road Ahead. Front. Immunol. 2021, 12, 676181. [Google Scholar] [CrossRef] [PubMed]
- ElTanbouly, M.A.; Schaafsma, E.; Noelle, R.J.; Lines, J.L. VISTA: Coming of age as a multi-lineage immune checkpoint. Clin. Exp. Immunol. 2020, 200, 120–130. [Google Scholar] [CrossRef]
- Flem-Karlsen, K.; Fodstad, Ø.; Nunes-Xavier, C.E. B7-H3 Immune Checkpoint Protein in Human Cancer. Curr. Med. Chem. 2020, 27, 4062–4086. [Google Scholar] [CrossRef]
- Kontos, F.; Michelakos, T.; Kurokawa, T.; Sadagopan, A.; Schwab, J.H.; Ferrone, C.R.; Ferrone, S. B7-H3: An Attractive Target for Antibody-based Immunotherapy. Clin. Cancer Res. 2021, 27, 1227–1235. [Google Scholar] [CrossRef]
- Johnson, D.B.; Nebhan, C.A.; Moslehi, J.J.; Balko, J.M. Immune-checkpoint inhibitors: Long-term implications of toxicity. Nat. Rev. Clin. Oncol. 2022, 19, 254–267. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, H.; Chen, X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist. 2019, 2, 141–160. [Google Scholar] [CrossRef]
- Crosby, E.J.; Lyerly, H.K.; Hartman, Z.C. Cancer vaccines: The importance of targeting oncogenic drivers and the utility of combinations with immune checkpoint inhibitors. Oncotarget 2021, 12, 1–3. [Google Scholar] [CrossRef]
- Liao, J.Y.; Zhang, S. Safety and Efficacy of Personalized Cancer Vaccines in Combination with Immune Checkpoint Inhibitors in Cancer Treatment. Front. Oncol. 2021, 11, 663264. [Google Scholar] [CrossRef]
- Puzanov, I.; Milhem, M.M.; Minor, D.; Hamid, O.; Li, A.; Chen, L.; Chastain, M.; Gorski, K.S.; Anderson, A.; Chou, J.; et al. Talimogene laherparepvec in combination with ipilimumab in previously untreated, unresectable stage IIIB-IV melanoma. J. Clin. Oncol. 2016, 34, 2619–2626. [Google Scholar] [CrossRef] [PubMed]
- Chesney, J.; Puzanov, I.; Collichio, F.; Singh, P.; Milhem, M.M.; Glaspy, J.; Hamid, O.; Ross, M.; Friedlander, P.; Garbe, C.; et al. Randomized, open-label phase II study evaluating the efficacy and safety of talimogene laherparepvec in combination with ipilimumab versus ipilimumab alone in patients with advanced, unresectable melanoma. J. Clin. Oncol. 2018, 36, 1658–1667. [Google Scholar] [CrossRef] [PubMed]
- Sinha, M.; Zhang, L.; Subudhi, S.; Chen, B.; Marquez, J.; Liu, E.V.; Allaire, K.; Cheung, A.; Ng, S.; Nguyen, C.; et al. Pre-existing immune status associated with response to combination of sipuleucel-T and ipilimumab in patients with metastatic castration-resistant prostate cancer. J. Immunother. Cancer 2021, 9, e002254. [Google Scholar] [CrossRef] [PubMed]
- Madan, R.A.; Mohebtash, M.; Arlen, P.M.; Vergati, M.; Rauckhorst, M.; Steinberg, S.M.; Tsang, K.Y.; Poole, D.J.; Parnes, H.L.; Wright, J.J.; et al. Ipilimumab and a poxviral vaccine targeting prostate-specific antigen in metastatic castration-resistant prostate cancer: A phase 1 dose-escalation trial. Lancet Oncol. 2012, 13, 501–508. [Google Scholar] [CrossRef]
- Singh, H.; Madan, R.A.; Dahut, W.L.; Strauss, J.; Rauckhorst, M.; McMahon, S.; Heery, C.R.; Schlom, J.; Gulley, J.L. Combining active immunotherapy and immune checkpoint inhibitors in prostate cancer. J. Clin. Oncol. 2015, 33 (Suppl. 7), e14008. [Google Scholar] [CrossRef]
- Gerritsen, W.; Eertwegh, A.J.V.D.; De Gruijl, T.; Berg, H.P.V.D.; Scheper, R.J.; Sacks, N.; Lowy, I.; Stankevich, E.; Hege, K. Expanded phase I combination trial of GVAX immunotherapy for prostate cancer and ipilimumab in patients with metastatic hormone-refractory prostate cancer (mHPRC). J. Clin. Oncol. 2008, 26 (Suppl. 15), 5146. [Google Scholar] [CrossRef]
- van den Eertwegh, A.J.; Versluis, J.; van den Berg, H.P.; Santegoets, S.J.; van Moorselaar, R.J.; van der Sluis, T.M.; Gall, H.E.; Harding, T.C.; Jooss, K.; Lowy, I.; et al. Combined immunotherapy with granulocyte-macrophage colony-stimulating factor-transduced allogeneic prostate cancer cells and ipilimumab in patients with metastatic castration-resistant prostate cancer: A phase 1 dose-escalation trial. Lancet Oncol. 2012, 13, 509–517. [Google Scholar] [CrossRef]
- Le, D.T.; Lutz, E.; Uram, J.N.; Sugar, E.A.; Onners, B.; Solt, S.; Zheng, L.; Diaz, L.A., Jr.; Donehower, R.C.; Jaffee, E.M.; et al. Evaluation of ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer. J. Immunother. 2013, 36, 382–389. [Google Scholar] [CrossRef]
- Attia, P.; Phan, G.Q.; Maker, A.V.; Robinson, M.R.; Quezado, M.M.; Yang, J.C.; Sherry, R.M.; Topalian, S.L.; Kammula, U.S.; Royal, R.E.; et al. Autoimmunity correlates with tumor regression in patients with metastatic melanoma treated with anti-cytotoxic T-lymphocyte antigen-4. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 6043–6053. [Google Scholar] [CrossRef]
- Wilgenhof, S.; Corthals, J.; Heirman, C.; Van Baren, N.; Lucas, S.; Kvistborg, P.; Thielemans, K.; Neyns, B. Phase II study of autologous monocyte-derived mRNA electroporated dendritic cells (TriMixDC-MEL) plus ipilimumab in patients with pretreated advanced melanoma. J. Clin. Oncol. 2016, 34, 1330–1338. [Google Scholar] [CrossRef] [PubMed]
- Aamdal, E.; Inderberg, E.M.; Ellingsen, E.B.; Rasch, W.; Brunsvig, P.F.; Aamdal, S.; Heintz, K.M.; Vodák, D.; Nakken, S.; Hovig, E.; et al. Combining a Universal Telomerase Based Cancer Vaccine With Ipilimumab in Patients with Metastatic Melanoma—Five-Year Follow Up of a Phase I/IIa Trial. Front. Immunol. 2021, 12, 663865. [Google Scholar] [CrossRef] [PubMed]
- Rosser, C.J.; Hirasawa, Y.; Acoba, J.D.; Tamura, D.J.; Pal, S.K.; Huang, J.; Scholz, M.C.; Dorffet, T.B. Phase Ib study assessing different sequencing regimens of atezolizumab (anti-PD-L1) and sipuleucel-T (SipT)in patients who have asymptomatic or minimally symptomatic metastatic castrate resistant prostate cancer. J. Clin. Oncol. 2020, 38, e17564. [Google Scholar] [CrossRef]
- Long, G.V.; Dummer, R.; Ribas, A.; Puzanov, I.; Michielin, O.; VanderWalde, A.; Andtbacka, R.H.; Cebon, J.; Fernandez, E.; Malvehy, J.; et al. A phase I/III, multicenter, open-label trial of talimogene laherparepvec (T-VEC) in combination with pembrolizumab for the treatment of unresected, stage IIIB-IV melanoma (MASTERKEY-265). J. ImmunoTher. Cancer 2015, 3 (Suppl. 2), P181. [Google Scholar] [CrossRef]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic virotherapy promotes intratumoral t cell infiltration and improves anti–PD-1 immunotherapy. Cell 2017, 170, 1109–1119. [Google Scholar] [CrossRef]
- Harrington, K.; Kong, A.; Mach, N.; Rordorf, T.; Corral, J.; Espeli, V.; Treichel, S.; Cheng, J.; Kim, J.; Chesney, J. Early safety from phase 1b/3, multicenter, open-label, randomized trial of talimogene laherparepvec (T-VEC) + pembrolizumab (pembro) for recurrent or metastatic squamous cell carcinoma of the head and neck (R/M SCCHN): MASTERKEY-232. Ann. Oncol. 2017, 28 (Suppl. 5), v394. [Google Scholar] [CrossRef]
- Harrington, K.J.; Kong, A.H.; Mach, N.; Rordorf, T.; Jaime, J.C.; Espeli, V.; Treichel, S.; Gumuscu, B.; Kim, J.J.; Chesney, J.A. Safety and preliminary efficacy of talimogene laherparepvec (T-VEC) in combination (combo) with pembrobrolizumab (Pembro) in patients (pts) with recurrent or metastatic squamous cell carcinoma of the head and neck (R/M HNSCC): A multicenter, phase 1b study (MASTERKEY-232). J. Clin. Oncol. 2018, 36 (Suppl. 15), 6036. [Google Scholar]
- Kudchadkar, R.R.; Gallenstein, D.; Martinez, A.J.; Yu, B.; Weber, J.S. Phase I trial of extended-dose anti–PD-1 antibody BMS-936558 with a multipeptide vaccine for previously treated stage IV melanoma. J. Clin. Oncol. 2012, 30 (Suppl. 15), 8582. [Google Scholar] [CrossRef]
- Weber, J.S.; Kudchadkar, R.R.; Yu, B.; Gallenstein, D.; Horak, C.E.; Inzunza, H.D.; Zhao, X.; Martinez, A.J.; Wang, W.; Gibney, G.; et al. Safety, efficacy, and biomarkers of nivolumab with vaccine in ipilimumab-refractory or -naive melanoma. J. Clin. Oncol. 2013, 31, 4311–4318. [Google Scholar] [CrossRef]
- Gibney, G.T.; Kudchadkar, R.R.; DeConti, R.C.; Thebeau, M.S.; Czupryn, M.P.; Tetteh, L.; Eysmans, C.; Richards, A.; Schell, M.J.; Fisher, K.J.; et al. Safety, correlative markers, and clinical results of adjuvant nivolumab in combination with vaccine in resected high-risk metastatic melanoma. Clin. Cancer Res. 2015, 21, 712–720. [Google Scholar] [CrossRef]
- Hsueh, E.C.; Essner, R.; Foshag, L.J.; Ollila, D.W.; Gammon, G.; O’Day, S.J.; Boasberg, P.D.; Stern, S.L.; Ye, X.; Morton, D.L. Prolonged survival after complete resection of disseminated melanoma and active immunotherapy with a therapeutic cancer vaccine. J. Clin. Oncol. 2002, 20, 4549–4554. [Google Scholar] [CrossRef] [PubMed]
- Sosman, J.A.; Moon, J.; Tuthill, R.J.; Warneke, J.A.; Vetto, J.T.; Redman, B.G.; Liu, P.Y.; Unger, J.M.; Flaherty, L.E.; Sondak, V.K. A phase 2 trial of complete resection for stage IV melanoma: Results of Southwest Oncology Group Clinical Trial S9430. Cancer 2011, 117, 4740–4746. [Google Scholar] [CrossRef] [PubMed]
- Läubli, H.; Balmelli, C.; Kaufmann, L.; Stanczak, M.; Syedbasha, M.; Vogt, D.; Hertig, A.; Müller, B.; Gautschi, O.; Stenner, F.; et al. Influenza vaccination of cancer patients during PD-1 blockade induces serological protection but may raise the risk for immune-related adverse events. J. Immunother. Cancer 2018, 6, 40. [Google Scholar] [CrossRef] [PubMed]
- Finnefrock, A.C.; Tang, A.; Li, F.; Freed, D.C.; Feng, M.; Cox, K.S.; Sykes, K.J.; Guare, J.P.; Miller, M.D.; Olsen, D.B.; et al. PD-1 blockade in rhesus macaques: Impact on chronic infection and prophylactic vaccination. J. Immunol. 2009, 182, 980–987. [Google Scholar] [CrossRef] [PubMed]
- Kanaloupitis, D.K.; Chandran, A.; Ralph, A.; Thompson, R.; Hallmeyer, S. Safety and efficacy of concurrent administration of influenza vaccine in patients undergoing anti-PD-1 immunotherapy. J. Clin. Oncol. 2017, 35 (Suppl. 15), e14607. [Google Scholar] [CrossRef]
- Weber, J.S.; Hamid, O.; Chasalow, S.D.; Wu, D.Y.; Parker, S.M.; Galbraith, S.; Gnjatic, S.; Berman, D. Ipilimumab increases activated T cells and enhances humoral immunity in patients with advanced melanoma. J. Immunother. 2012, 35, 89–97. [Google Scholar] [CrossRef]
- Desage, A.L.; Bouleftour, W.; Rivoirard, R.; Magne, N.; Collard, O.; Fournel, P.; Tissot, C. Vaccination and Immune Checkpoint Inhibitors: Does Vaccination Increase the Risk of Immune-related Adverse Events? A Systematic Review of Literature. Am. J. Clin. Oncol. 2021, 44, 109–113. [Google Scholar] [CrossRef]
- Spagnolo, F.; Boutros, A.; Croce, E.; Cecchi, F.; Arecco, L.; Tanda, E.; Pronzato, P.; Lambertini, M. Influenza vaccination in cancer patients receiving immune checkpoint inhibitors: A systematic review. Eur. J. Clin. Investig. 2021, 51, e13604. [Google Scholar] [CrossRef]
- Bersanelli, M.; Giannarelli, D.; De Giorgi, U.; Pignata, S.; Di Maio, M.; Clemente, A.; Verzoni, E.; Giusti, R.; Di Napoli, M.; Aprile, G.; et al. INfluenza Vaccine Indication During therapy with Immune checkpoint inhibitors: A multicenter prospective observational study (INVIDIa-2). J. Immunother. Cancer 2021, 9, e002619. [Google Scholar] [CrossRef]
- Bersanelli, M.; Scala, S.; Affanni, P.; Veronesi, L.; Colucci, M.E.; Banna, G.L.; Cortellini, A.; Liotta, F. Immunological insights on influenza infection and vaccination during immune checkpoint blockade in cancer patients. Immunotherapy 2020, 12, 105–110. [Google Scholar] [CrossRef]
- Niewolik, J.; Mikuteit, M.; Cossmann, A.; Vahldiek, K.; Gutzmer, R.; Müller, F.; Schröder, D.; Heinemann, S.; Behrens, G.M.N.; Dopfer-Jablonka, A.; et al. Immunogenicity of COVID-19 vaccination in melanoma patients under immune checkpoint blockade. Oncology 2022, 100, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Xu, L.; Yi, M.; Yu, S.; Wu, K.; Luo, S. Novel immune checkpoint targets: Moving beyond PD-1 and CTLA-4. Mol Cancer. 2019, 18, 155. [Google Scholar] [CrossRef] [PubMed]
- Zahm, C.D.; Moseman, J.E.; Delmastro, L.E.; Mcneel, D.G. PD-1 and LAG-3 blockade improve anti-tumor vaccine efficacy. Oncoimmunology 2021, 10, 1912892. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Coulon, P.G.; Srivastava, R.; Vahed, H.; Kim, G.J.; Walia, S.S.; Yamada, T.; Fouladi, M.A.; Ly, V.T.; BenMohamed, L. Blockade of LAG-3 Immune Checkpoint Combined with Therapeutic Vaccination Restore the Function of Tissue-Resident Anti-viral CD8+ T Cells and Protect Against Recurrent Ocular Herpes Simplex Infection and Disease. Front. Immunol. 2018, 9, 2922. [Google Scholar] [CrossRef] [PubMed]
- Potaczek, D.P.; Garn, H.; Unger, S.D.; Renz, H. Antisense molecules: A new class of drugs. J. Allergy Clin. Immunol. 2016, 137, 1334–1346. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Delgado, M.; Sendra, L.; Herrero, M.J.; Olivera-Pasquini, G.G.; Batista-Duharte, A.; Aliño, S.F. Study of Oligonucleotides Access and Distribution in Human Peripheral Blood Mononuclear Cells. Int. J. Mol. Sci. 2022, 23, 5839. [Google Scholar] [CrossRef]
- Mansoor, M.; Melendez, A.J. Advances in antisense oligonucleotide development for target identification, validation, and as novel therapeutics. Gene Regul. Syst. Biol. 2008, 2, 275–295. [Google Scholar] [CrossRef]
- Khvorova, A.; Watts, J.K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 2017, 35, 238–248. [Google Scholar] [CrossRef]
- Quemener, A.M.; Bachelot, L.; Forestier, A.; Donnou-Fournet, E.; Gilot, D.; Galibert, M.D. The powerful world of antisense oligonucleotides: From bench to bedside. Wiley Interdiscip. Rev. RNA 2020, 11, e1594. [Google Scholar] [CrossRef]
- Batista-Duharte, A.; Sendra, L.; Herrero, M.J.; Téllez-Martínez, D.; Carlos, I.Z.; Aliño, S.F. Progress in the Use of Antisense Oligonucleotides for Vaccine Improvement. Biomolecules 2020, 10, 316. [Google Scholar] [CrossRef]
- Miguel, A.; Sendra, L.; Noé, V.; Ciudad, C.J.; Dasí, F.; Hervas, D.; Herrero, M.J.; Aliño, S.F. Silencing of Foxp3 enhances the antitumor efficacy of GM-CSF genetically modified tumor cell vaccine against B16 melanoma. Onco Targets Ther. 2017, 10, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Batista-Duharte, A.; Sendra, L.; Herrero, M.J.; Portuondo, D.L.; Téllez-Martínez, D.; Olivera, G.; Fernández-Delgado, M.; Javega, B.; Herrera, G.; Martínez, A.; et al. Foxp3 Silencing with Antisense Oligonucleotide Improves Immunogenicity of an Adjuvanted Recombinant Vaccine against Sporothrix schenckii. Int. J. Mol. Sci. 2021, 22, 3470. [Google Scholar] [CrossRef] [PubMed]
- Ripple, M.J.; You, D.; Honnegowda, S.; Giaimo, J.D.; Sewell, A.B.; Becnel, D.M.; Cormier, S.A. Immunomodulation with IL-4Rα antisense oligonucleotide prevents respiratory syncytial virus-mediated pulmonary disease. J. Immunol. 2010, 185, 4804–4811. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, N.; Lu, Y.; Huang, Z.; Zang, Y.; Chen, J.; Zhang, J.; Ding, Z. Phosphorothioated antisense oligodeoxynucleotide suppressing interleukin-10 is a safe and potent vaccine adjuvant. Vaccine 2019, 37, 4081–4088. [Google Scholar] [CrossRef]
- Zhou Tu, L.; Sun, X.; Yang, L.; Zhang, T.; Zhang, X.; Li, X.; Dong, B.; Liu, Y.; Yang, M.; Wang, L.; et al. TGF-β2 interfering oligonucleotides used as adjuvants for microbial vaccines. J. Leukoc. Biol. 2020, 108, 1673–1692. [Google Scholar] [CrossRef]
- Li, X.; Yang, L.; Zhao, P.; Yao, Y.; Lu, F.; Tu, L.; Liu, J.; Li, Z.; Yu, Y.; Wang, L. Adjuvanticity of a CTLA-4 3′ UTR complementary oligonucleotide for emulsion formulated recombinant subunit and inactivated vaccines. Vaccine 2017, 35, 2379–2389. [Google Scholar] [CrossRef]
- Li, Z.; Song, Y.; Cui, C.; Lan, Y.; Li, X.; Liu, Y.; Lu, F.; Zhang, Y.; Yu, Y.; Wang, L. A LAG3-interfering oligonucleotide acts as an adjuvant to enhance the antibody responses induced by recombinant protein vaccines and inactivated influenza virus vaccines. Appl. Microbiol. Biotechnol. 2019, 103, 6543–6557. [Google Scholar] [CrossRef]
- Zhou, T. Small non-coding RNAs as epigenetic regulators. In Nutritional Epigenomics; Academic Press: Cambridge, MA, USA, 2019; pp. 37–47. [Google Scholar]
- Dana, H.; Chalbatani, G.M.; Mahmoodzadeh, H.; Karimloo, R.; Rezaiean, O.; Moradzadeh, A.; Mehmandoost, N.; Moazzen, F.; Mazraeh, A.; Marmari, V.; et al. Molecular Mechanisms and Biological Functions of siRNA. Int. J. Biomed. Sci. 2017, 13, 48–57. [Google Scholar]
- Hobo, W.; Novobrantseva, T.I.; Fredrix, H.; Wong, J.; Milstein, S.; Epstein-Barash, H.; Liu, J.; Schaap, N.; van der Voort, R.; Dolstra, H. Improving dendritic cell vaccine immunogenicity by silencing PD-1 ligands using siRNA-lipid nanoparticles combined with antigen mRNA electroporation. Cancer Immunol. Immunother. 2013, 62, 285–297. [Google Scholar] [CrossRef]
- Roeven, M.W.; Hobo, W.; van der Voort, R.; Fredrix, H.; Norde, W.J.; Teijgeler, K.; Ruiters, M.H.; Schaap, N.; Dolstra, H. Efficient nontoxic delivery of PD-L1 and PD-L2 siRNA into dendritic cell vaccines using the cationic lipid SAINT-18. J. Immunother. 2015, 38, 145–154. [Google Scholar] [CrossRef]
- van der Waart, A.B.; Fredrix, H.; van der Voort, R.; Schaap, N.; Hobo, W.; Dolstra, H. siRNA silencing of PD-1 ligands on dendritic cell vaccines boosts the expansion of minor histocompatibility antigen-specific CD8(+) T cells in NOD/SCID/IL2Rg(null) mice. Cancer Immunol. Immunother. 2015, 64, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Jadidi-Niaragh, F.; Atyabi, F.; Rastegari, A.; Kheshtchin, N.; Arab, S.; Hassannia, H.; Ajami, M.; Mirsanei, Z.; Habibi, S.; Masoumi, F.; et al. CD73 specific siRNA loaded chitosan lactate nanoparticles potentiate the antitumor effect of a dendritic cell vaccine in 4T1 breast cancer bearing mice. J. Control. Release 2017, 246, 46–59. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, L.; Xu, Z.; Miao, L.; Huang, L. mRNA Vaccine with Antigen-Specific Checkpoint Blockade Induces an Enhanced Immune Response against Established Melanoma. Mol. Ther. 2018, 26, 420–434. [Google Scholar] [CrossRef] [PubMed]
- Esmaily, M.; Masjedi, A.; Hallaj, S.; Nabi Afjadi, M.; Malakotikhah, F.; Ghani, S.; Ahmadi, A.; Sojoodi, M.; Hassannia, H.; Atyabi, F.; et al. Blockade of CTLA-4 increases anti-tumor response inducing potential of dendritic cell vaccine. J. Control. Release 2020, 326, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Hassannia, H.; Ghasemi Chaleshtari, M.; Atyabi, F.; Nosouhian, M.; Masjedi, A.; Hojjat-Farsangi, M.; Namdar, A.; Azizi, G.; Mohammadi, H.; Ghalamfarsa, G.; et al. Blockage of immune checkpoint molecules increases T-cell priming potential of dendritic cell vaccine. Immunology 2020, 159, 75–87. [Google Scholar] [CrossRef]
- Sugii, N.; Matsuda, M.; Okumura, G.; Shibuya, A.; Ishikawa, E.; Kaneda, Y.; Matsumura, A. Hemagglutinating virus of Japan-envelope containing programmed cell death-ligand 1 siRNA inhibits immunosuppressive activities and elicits antitumor immune responses in glioma. Cancer Sci. 2021, 112, 81–90. [Google Scholar] [CrossRef]
- Won, J.E.; Byeon, Y.; Wi, T.I.; Lee, C.M.; Lee, J.H.; Kang, T.H.; Lee, J.W.; Lee, Y.; Park, Y.M.; Han, H.D. Immune checkpoint silencing using RNAi-incorporated nanoparticles enhances antitumor immunity and therapeutic efficacy compared with antibody-based approaches. J. Immunother. Cancer 2022, 10, e003928. [Google Scholar] [CrossRef]
- Wan, W.J.; Huang, G.; Wang, Y.; Tang, Y.; Li, H.; Jia, C.H.; Liu, Y.; You, B.G.; Zhang, X.N. Coadministration of iRGD peptide with ROS-sensitive nanoparticles co-delivering siFGL1 and siPD-L1 enhanced tumor immunotherapy. Acta Biomater. 2021, 136, 473–484. [Google Scholar] [CrossRef]
- Ni, X.; Castanares, M.; Mukherjee, A.; Lupold, S.E. Nucleic acid aptamers: Clinical applications and promising new horizons. Curr. Med. Chem. 2011, 18, 4206–4214. [Google Scholar] [CrossRef]
- Santulli-Marotto, S.; Nair, S.K.; Rusconi, C.; Sullenger, B.; Gilboa, E. Multivalent RNA aptamers that inhibit CTLA-4 and enhance tumor immunity. Cancer Res. 2003, 63, 7483–7489. [Google Scholar]
- McNamara, J.O., 2nd; Andrechek, E.R.; Wang, Y.; Viles, K.D.; Rempel, R.E.; Gilboa, E.; Sullenger, B.A.; Giangrande, P.H. Cell type-specific delivery of siRNAs with aptamer-siRNA chimeras. Nat. Biotechnol. 2006, 24, 1005–1015. [Google Scholar] [CrossRef] [PubMed]
- Prodeus, A.; Abdul-Wahid, A.; Fischer, N.W.; Huang, E.H.; Cydzik, M.; Gariépy, J. Targeting the PD-1/PD-L1 Immune Evasion Axis With DNA Aptamers as a Novel Therapeutic Strategy for the Treatment of Disseminated Cancers. Mol. Ther. Nucleic Acids 2015, 4, e237. [Google Scholar] [CrossRef] [PubMed]
- Lai, W.Y.; Huang, B.T.; Wang, J.W.; Lin, P.Y.; Yang, P.C. A Novel PD-L1-targeting Antagonistic DNA Aptamer with Antitumor Effects. Mol. Therapy. Nucleic Acids 2016, 5, e397. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Mao, Z.; Li, W.; Pei, R. Anti-PD-L1 DNA aptamer antagonizes the interaction of PD-1/PD-L1 with antitumor effect. J. Mater. Chem. B 2021, 9, 746–756. [Google Scholar] [CrossRef]
- Hervas-Stubbs, S.; Soldevilla, M.M.; Villanueva, H.; Mancheño, U.; Bendandi, M.; Pastor, F. Identification of TIM3 2′-fluoro oligonucleotide aptamer by HT-SELEX for cancer immunotherapy. Oncotarget 2016, 7, 4522–4530. [Google Scholar] [CrossRef]
- Gefen, T.; Castro, I.; Muharemagic, D.; Puplampu-Dove, Y.; Patel, S.; Gilboa, E. A TIM-3 Oligonucleotide Aptamer Enhances T Cell Functions and Potentiates Tumor Immunity in Mice. Mol. Ther. 2017, 25, 2280–2288. [Google Scholar] [CrossRef]
- Soldevilla, M.M.; Hervas, S.; Villanueva, H.; Lozano, T.; Rabal, O.; Oyarzabal, J.; Lasarte, J.J.; Bendandi, M.; Inoges, S.; López-Díaz de Cerio, A.; et al. Identification of LAG3 high affinity aptamers by HT-SELEX and Conserved Motif Accumulation (CMA). PLoS ONE 2017, 12, e0185169. [Google Scholar] [CrossRef]
- Wu, Q.; Jiang, L.; Li, S.C.; He, Q.J.; Yang, B.; Cao, J. Small molecule inhibitors targeting the PD-1/PD-L1 signaling pathway. Acta Pharmacol. Sin. 2021, 42, 1–9. [Google Scholar] [CrossRef]
- Zarganes-Tzitzikas, T.; Konstantinidou, M.; Gao, Y.; Krzemien, D.; Zak, K.; Dubin, G.; Holak, T.A.; Dömling, A. Inhibitors of programmed cell death 1 (PD-1): A patent review (2010–2015). Expert Opin. Ther. Pat. 2016, 26, 973–977. [Google Scholar] [CrossRef]
- Guzik, K.; Tomala, M.; Muszak, D.; Konieczny, M.; Hec, A.; Błaszkiewicz, U.; Pustuła, M.; Butera, R.; Dömling, A.; Holak, T.A. Development of the Inhibitors that Target the PD-1/PD-L1 Interaction-A Brief Look at Progress on Small Molecules, Peptides and Macrocycles. Molecules 2019, 24, 2071. [Google Scholar] [CrossRef]
- Chen, T.; Li, Q.; Liu, Z.; Chen, Y.; Feng, F.; Sun, H. Peptide-based and small synthetic molecule inhibitors on PD-1/PD-L1 pathway: A new choice for immunotherapy? Eur. J. Med. Chem. 2019, 161, 378–398. [Google Scholar] [CrossRef]
- Miao, Q.; Zhang, W.; Zhang, K.; Li, H.; Zhu, J.; Jiang, S. Rational design of a potent macrocyclic peptide inhibitor targeting the PD-1/PD-L1 protein-protein interaction. RSC Adv. 2021, 11, 23270–23279. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Qi, Z.; Wang, T.; Zhang, X.; Zhang, K.; Wang, K.; Cheng, Y.; Xiao, Y.; Li, Z.; Jiang, S. Design, Synthesis, and Evaluation of PD-1/PD-L1 Antagonists Bearing a Benzamide Scaffold. ACS Med. Chem. Lett. 2022, 13, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Orafaie, A.; Sadeghian, H.; Bahrami, A.R.; Rafatpanah, H.; Matin, M.M. Design, synthesis and evaluation of PD-L1 peptide antagonists as new anticancer agents for immunotherapy. Bioorg. Med. Chem. 2021, 30, 115951. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xia, Y.; Yu, C.; Du, H.; Liu, J.; Li, H.; Huang, S.; Zhu, Q.; Xu, Y.; Zou, Y. Discovery of Novel Small-Molecule Inhibitors of PD-1/PD-L1 Interaction via Structural Simplification Strategy. Molecules 2021, 26, 3347. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Lu, X.; Luo, G.; Xiang, H. Progress in PD-1/PD-L1 pathway inhibitors: From biomacromolecules to small molecules. Eur. J. Med. Chem. 2020, 186, 111876. [Google Scholar] [CrossRef]
- Arab, S.; Kheshtchin, N.; Ajami, M.; Ashurpoor, M.; Safvati, A.; Namdar, A.; Mirzaei, R.; Mousavi Niri, N.; Jadidi-Niaragh, F.; Ghahremani, M.H.; et al. Increased efficacy of a dendritic cell-based therapeutic cancer vaccine with adenosine receptor antagonist and CD73 inhibitor. Tumour Biol. 2017, 39, 1010428317695021. [Google Scholar] [CrossRef]
- Yan, P.; Luo, Y.; Li, X.; Li, Y.; Wang, Y.; Wu, J.; Zhou, S. A Redox-Responsive Nanovaccine Combined with A2A Receptor Antagonist for Cancer Immunotherapy. Adv. Healthc. Mater. 2021, 10, e2101222. [Google Scholar] [CrossRef]


{kind=link}
| Target; Format | 1st Indication Approved/Reviewed | 1st EU Approval Year | 1st US Approval Year | |
|---|---|---|---|---|
| Ipilimumab | CTLA-4; Human IgG1 | Metastatic melanoma | 2011 | 2011 |
| Pembrolizumab | PD1; Humanized IgG4 | Melanoma | 2015 | 2014 |
| Nivolumab | PD1; Human IgG4 | Melanoma, non-small cell lung cancer | 2015 | 2014 |
| Atezolizumab | PD-L1; Humanized IgG1 | Bladder cancer | 2017 | 2016 |
| Avelumab | PD-L1; Human IgG1 | Merkel cell carcinoma | 2017 | 2017 |
| Durvalumab | PD-L1; Human IgG1 | Bladder cancer | 2018 | 2017 |
| Cemiplimab | PD-1; Human IgG4 | Cutaneous squamous cell carcinoma | 2019 | 2018 |
| Dostarlimab | PD-1; Humanized IgG4 | Endometrial cancer | 2021 | 2021 |
| Relatlimab | LAG-3; Human IgG4 | Melanoma | Review | 2022 |
| Tremelimumab | CTLA-4; Human IgG2A | Antineoplastic; liver cancer | Review | Review |
| Tislelizumab | PD-1; Humanized IgG4 | Esophageal squamous cell carcinoma | Review | Review |
| Sintilimab | PD-1; Human IgG4 | Non-small cell lung cancer | NA | Review |
| Retifanlimab | PD-1; Humanized IgG4 | Squamous cell carcinoma of the anal canal | MAA withdrawn | Review |
| Penpulimab | PD-1; Humanized IgG1 | Metastatic nasopharyngeal carcinoma | NA | Review |
| Omburtamab | B7-H3; Murine IgG1 | CNS/leptomeningeal metastasis from neuroblastoma | Review | Review |
| Other regions | ||||
| Sintilimab (Tyvyt): human anti-PD-1 mAb approved in China in December 2018 for Hodgkin’s lymphoma; Toripalimab (Tuoyi): humanized anti-PD-1 mAb approved in China in December 2018 for melanoma; Camrelizumab: humanized anti-PD-1 mAb approved in China in 2019 for Hodgkin’s lymphoma; Tislelizumab: humanized anti-PD-1 mAb, approved in China in December 2019 as a treatment for classical Hodgkin’s lymphoma; Disitamab vedotin (Aidixi): anti-HER2 humanized ADC approved in China in June 2021 as a treatment for gastric cancer; Penpulimab: ant-PD-1 humanized mAb approved in China in August 2021 for Hodgkin’s lymphoma; Zimberelimab: anti-PD-1 human mAb approved in China in August 2021 for Hodgkin’s lymphoma; Prolgolimab (Forteca), anti-PD-1 mAb approved in Russia in 2020 for melanoma. | ||||
| ICI/Target | Vaccine | Disease | n | ICI-Vaccine Combination Strategy | Results | irAEs | References |
|---|---|---|---|---|---|---|---|
| Ipilimumab/CTLA-4 | T-VEC | Unresectable stage IIIB-IV melanoma | 198 | T-VEC was administered intratumorally at the first dose ≤4 mL × 106 pfu/mL, after 3 weeks at subsequent doses ≤4 mL × 108 pfu/mL every 2 weeks; four doses of ipilimumab 3 mg/kg were given intravenously every 3 weeks. | Combination therapy promoted a significantly higher objective therapeutic response rate than ipilimumab alone (39% vs. 18%). A decrease in visceral lesions was observed in 52% of patients treated with the combination and 23% of patients with ipilimumab alone. | Fatigue (59%), chills (53%), diarrhea (42%), pruritus (40%), rash (39%). The incidence rate of grade ≥3 irAEs was 45% in the combination and 35% with ipilimumab alone. | [62,63] |
| Sipuleucel-T | mCRPC | 50 | All patients received 3 doses of intravenous Sipuleucel-T infusion once every 2 weeks. Patients received the first dose of ipilimumab either immediately following their last Sipuleucel-T infusion, or 3 weeks after their last vaccine infusion. Additional 3 doses of ipilimumab, 3 mg/kg was given to all patients every 3 weeks, for a total of 4 ipilimumab doses | The combination treatment induced CD4(+) and CD8(+) T lymphocytes activation that was most pronounced with the immediate schedule. Lower frequencies of CTLA-4(+) circulating T lymphocytes, were associated with better clinical outcomes. However, combining Ipilimumab with Sipuleucel-T resulted in modest clinical activity. | The treatment was well tolerated. One patient underwent a grade 4 event (colitis with colonic perforation) and nine grade 3 events in seven patients. Interestingly, patients with an irAE were more likely to have a significant PSA response (any grade, p = 0.001, grade 3/4, p = 0.037). | [64] | |
| PROSTVAC | mCRPC | 30 | PROSTVAC was administered subcutaneously at prime doses of 2 × 108 pfu/mL, with subsequent monthly doses of 1 × 109 pfu/mL. Intravenous ipilimumab was given (1, 3, 5, and 10 mg/kg) on the same day as the vaccine. | For patients receiving ipilimumab 10 mg/kg, overall survival was 37.2 months, very longer than historical controls of treatment with PROSTVAC or ipilimumab alone. | irAEs mostly occurred in patients treated with ipilimumab 10 mg/kg. Grades 1 to 2 injection-site reactions were the most common adverse events. Grades 3 to 4 irAEs, including rash, diarrhea, colitis, and endocrine events, were observed in 27% of patients, requiring replacement hormones or supportive measures | [65,66] | |
| GVAX | mCRPC | 28 | All patients received GVAX intradermally. Priming dose of 5 × 108 cells with additional injections of 3 × 108 cells every 2 weeks for 24 weeks plus intravenous ipilimumab at doses of 0.3, 1, 3, and 5 mg/kg every 4 weeks. | 25% of patients showed >50% PSA reduction from baseline, and four patients obtained stable disease measured by bone scan. | Adverse events (>30%) were grades 1 to 2 injection-site reactions, fatigue, influenza-like symptoms, and rash. At, one patient receiving ipilimumab at 5 mg/kg, had grade 4 sarcoid alveolitis. Other irAEs related to ipilimumab included hypophysitis and hepatitis. Both responded to hormone replacement therapy. | [67,68] | |
| Advanced pancreatic adenocarcinoma | 30 | Patients received either intravenous ipilimumab 10 mg/kg alone or intradermal GVAX at doses of 5 × 108 cells with subsequent ipilimumab 10 mg/kg. | The combination promoted prolonged disease stabilization, improved 1-year survival (27% vs. 7%), and a trend of favorable median overall survival (5.7 vs. 3.6 months; p = 0.072) compared with ipilimumab alone. CA19-9 responses were observed in 47% of patients that received combination therapy, whereas none in ipilimumab alone. | Grades 1 to 2 injection-site reactions, rash, fatigue, fever, and influenza-like illness. 20% of patients experienced grades 3 to 4 irAEs including rash, colitis, pneumonitis, and nephritis. All irAEs responded to steroids except for nephritis requiring hemodialysis | [69] | ||
| Peptide Vaccine (gp100:209-217 and gp100:280-288 from gp100, a melanoma-associated antigen. | Progressive stage IV melanoma | 56 | Twenty-nine patients received 3 mg/kg Ipilimumab every 3 weeks, whereas 27 received 3 mg/kg as their initial dose with subsequent doses reduced to 1 mg/kg every 3 weeks. The patients received concomitant vaccination with peptide vaccine. | Two patients had a complete response (at 30 and 31 months, respectively) while five patients achieved a partial response, for an overall objective response rate of 13%. Tumor regression was seen in lung, liver, brain, lymph nodes, and subcutaneous sites, and it was correlated with autoimmune reactions. | Of 14 patients with grade 3/4 autoimmune reactions, 36% experienced favorable clinical response. Only two favorable responses were observed in the 42 patients (5%) with no autoimmune reactions (p = 0.008). There were no significant differences in response rate or toxicity between the two-dose schedules. | [70] | |
| TriMixDC-MEL | Pretreated advanced melanoma | 39 | TriMixDC-MEL was given subcutaneously and intravenously plus ipilimumab (10 mg/kg) every 3 weeks for four doses, followed by nivolumab (anti-PD1) maintenance every 3 months | The disease control rate was 51% at 6 months, and tumor objective response rate with the combination was 38%, which was higher than ipilimumab alone (10–15%). Tumor responses included eight complete and seven partial responses. | The most common adverse events (>30%) were injection-site reactions, influenza-like illness, dermatitis, and chills. 14 patients (36%) underwent grades 3 to 4 events, but most of them were reversible by using established treatment. | [71] | |
| UV1 | Unresectable metastatic melanoma | 12 | Ipilimumab (3 mg/kg) was administered every 3 weeks for a total of 4 doses. Intradermal abdominal injections of UV1 vaccines (300 µg doses) were administered as before and between treatments of ipilimumab. Thereafter every fourth week up to 28 weeks, and at weeks 36 and 48. GM-CSF (sargramostim 75 µg) was injected at the same site 10–15 min prior to UV1. | Ten patients showed a Th1 immune response to UV1, occurring early and after a few vaccinations. Three patients obtained a partial response. One patient had a complete response. Overall survival was 50% at 5 years. | The adverse events observed were injection site reaction, pruritus, rash, nausea, diarrhea, and fatigue. | [72] | |
| Atezolizumab/PDL-1 | Sipuleucel-T | mCRPC | Patients received either atezolizumab 1200 mg intravenously every 3 weeks for 2 doses followed by Sipuleucel-T three infusions every 2 weeks, or Sipuleucel-T every 2 weeks for a total of three infusions followed by atezolizumab as described (Phase Ib study) | The primary endpoint of this study was safety. There were no grade 5 adverse events attributed to the study drugs. Two patients underwent grade 4 toxicities, while eight grade 3 toxicities and four grade 3 toxicities were observed | None of the grade 3 or 4 adverse events were irAEs. | [73] | |
| Pembrolizumab/PD1 | T-VEC | Unresectable stages IIIB-IV melanoma | 21 | Patients received T-VEC at an initial dose of 4 mL × 106 pfu/mL, followed 3 weeks later at a full dose of 4 mL × 108 pfu/mL every two weeks. Pembrolizumab 200 mg was injected intravenously coinciding with subsequent doses of T-VEC. | Combination therapy induced an objective response rate of 62%, almost twice as shown in the phase III study of pembrolizumab (34%) and T-VEC (26%). The complete response rate for per immune-related response criteria was 33%. An increase in lymphocyte infiltration, and IFN-γ gene expression was observed in patients who responded to combination therapy. | The most common adverse events observed were. fatigue (62%), chills (48%), fever (43%), rash (33%), and arthralgia (33%). One grade 1 reaction associated with the combination resulted in hospitalization, while other grades 3 to 4 AEs were due to pembrolizumab. In general, combination therapy did not increase the toxicity of monotherapy. | [74,75] |
| Advanced squamous cell carcinoma of the head and neck | 36 | T-VEC was injected intralesionally at a first dose of 8 mL × 106 pfu/mL, and subsequent doses of 8 mL × 108 pfu/mL every 3 weeks. Intravenous pembrolizumab 200 mg was administered every 3 weeks | The objective response rate was 16.7% (six patients with five subjects PDL-1 positive), and the disease control rate was 38.9% (14 patients with 11 subjects PD-L1 positive). | The detected adverse events included pyrexia (36.1%), dyspnea (33.3%), and fatigue (25.0%). Grades 3 to 4 reactions were observed in 24 patients (66.7%). | [76,77] | ||
| Nivolumab/PD1 | Peptide Vaccine (MART-1/NY-ESO-1/gp100 with Montanide ISA 51 VG) | Unresectable stages III to IV melanoma | 90 | In a phase I trial, patients were treated with an extended dose of nivolumab (1, 3, or 10 mg/kg) with or without vaccines. | For both ipilimumab-refractory and -naive subjects, the RECIST (Response Evaluation Criteria in Solid Tumors) response rates were 25%, and nivolumab-induced durable responses for up to 140 weeks | Fatigue and injection-site reaction were the most common adverse events, most of which were mild to moderate. Grade 3 irAEs (optic neuritis, fever, pneumonitis, and rash) were also observed and were successfully treated with prednisone as described previously for nivolumab monotherapy. | [78,79] |
| Resected stages IIIC to IV melanoma | 33 | Patients were treated with an extended dose of nivolumab (1, 3, or 10 mg/kg) plus peptide vaccine every 2 weeks for 24 weeks, followed by nivolumab alone every 3 months for up to 2 years | The estimated median relapse-free survival (RFS) was 47.1 months compared with the historical median RFS (12–21 months). | Injection-site reaction, fatigue, rash, pruritus, nausea, and arthralgia were the most common reactions registered (>40%). Grade 3 reactions included hypokalemia, rash, enteritis, and colitis. All of them responded to systemic management of steroids and supportive care | [80,81,82] |
| mAbs | Alternative Molecular ICIs | |
|---|---|---|
| Specificity | Highly specific but cross-reactivity can be observed | Highly specific but off-target interaction can be observed |
| Diversity | mAbs can be obtained against a very wide range of target structures | |
| Mechanisms of action | Specific blockade of the checkpoint’s interaction with their natural ligand on the cell surface | Blocking direct interaction between checkpoint’s interaction with their natural ligand, inhibiting transcription and translation of checkpoint; promoting checkpoint degradation |
| Purity | High purity | High purity |
| Molecular weight | High molecular weight (~150 kDa) | Low molecular weight (~6 to10 kDa) |
| Structure | Glycoproteins with complex structure | Short, single-stranded DNA or RNA with chemical modifications, short linear peptides, cyclopeptides, or small synthetic molecules |
| Thermal Stability | Low stability. Cold chain through the storage, handling, and transportation is necessary | Highly stable. Lyophilization and freezing do not modify their biological activity |
| Bioavailability | No oral bioavailability and inability to penetrate the cells. The Fc domain of IgG antibody can interact with diverse cell receptors which hinder reaching the targets | They can penetrate the cells and act on intracellular targets |
| Secreted target | Secreted targets (e.g from tumor cells), can interrupt antibody-mediated immune reactions in the tumor microenvironment. | Their targets are mainly intracellular |
| Immunogenicity | Highly immunogenic by xenogeneic differences, e.g., between mice and humans | They are not properly immunogenic |
| Toxicity | Different grades of toxicity have been described | Relatively low toxicity associated with off-target effects |
| Development and Manufacturing | Immortal hybridomas cell lines produce unlimited quantities of antibodies, but industrial production is technologically complex | They are obtained synthetically. The use of vehicles can add complexity to the manufacturing process |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Batista-Duharte, A.; Hassouneh, F.; Alvarez-Heredia, P.; Pera, A.; Solana, R. Immune Checkpoint Inhibitors for Vaccine Improvements: Current Status and New Approaches. Pharmaceutics 2022, 14, 1721. https://doi.org/10.3390/pharmaceutics14081721
Batista-Duharte A, Hassouneh F, Alvarez-Heredia P, Pera A, Solana R. Immune Checkpoint Inhibitors for Vaccine Improvements: Current Status and New Approaches. Pharmaceutics. 2022; 14(8):1721. https://doi.org/10.3390/pharmaceutics14081721
Chicago/Turabian StyleBatista-Duharte, Alexander, Fakhri Hassouneh, Pablo Alvarez-Heredia, Alejandra Pera, and Rafael Solana. 2022. "Immune Checkpoint Inhibitors for Vaccine Improvements: Current Status and New Approaches" Pharmaceutics 14, no. 8: 1721. https://doi.org/10.3390/pharmaceutics14081721