
Beyond Formulation: Contributions of Nanotechnology for Translation of Anticancer Natural Products into New Drugs

 , ,
, ,
Abstract
:1. Introduction
2. Doxorubicin (DOX) and Paclitaxel (PTX): Discovery, Mechanism of Action, and Conventional Formulations
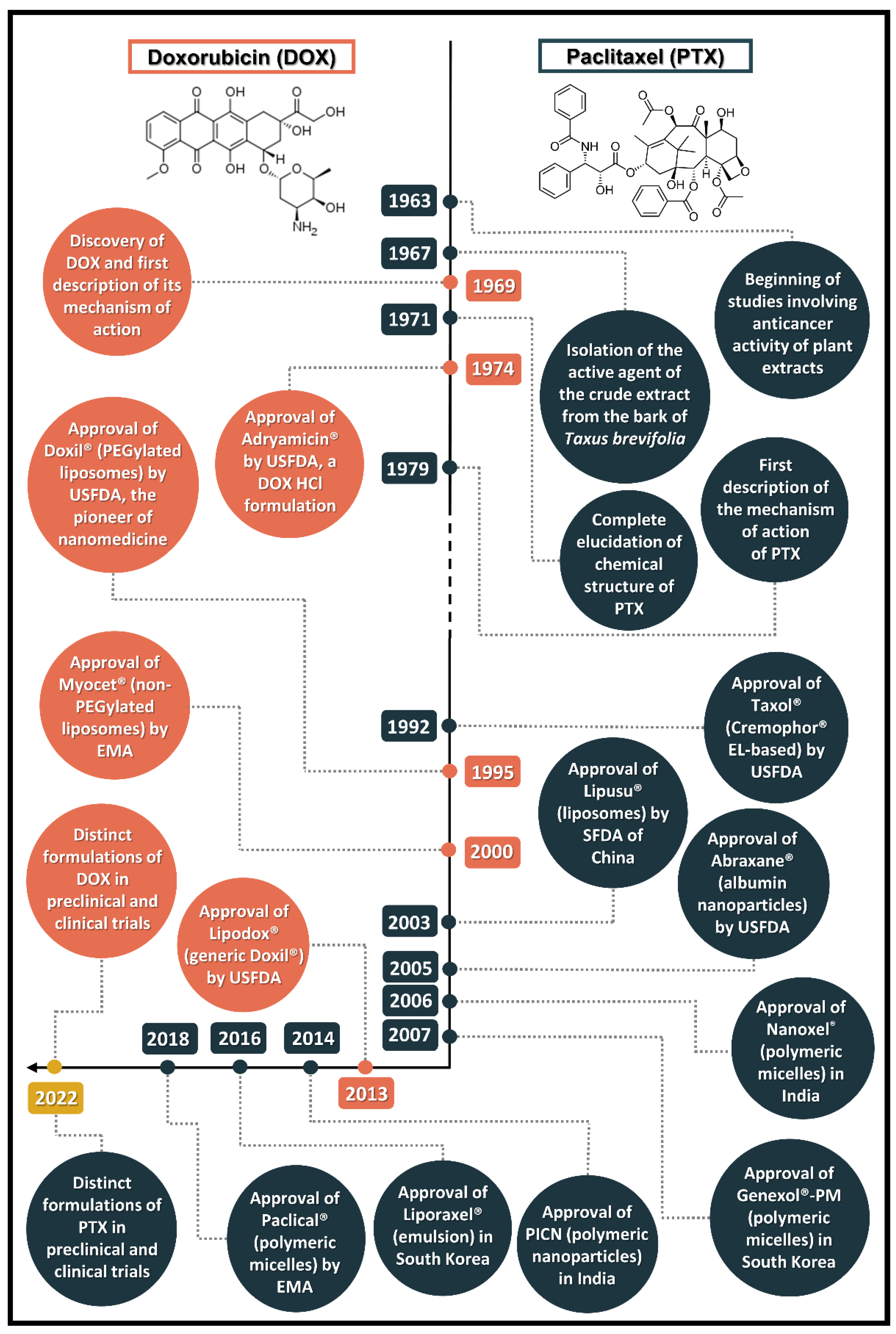
2.1. Discovery of the Prototypes
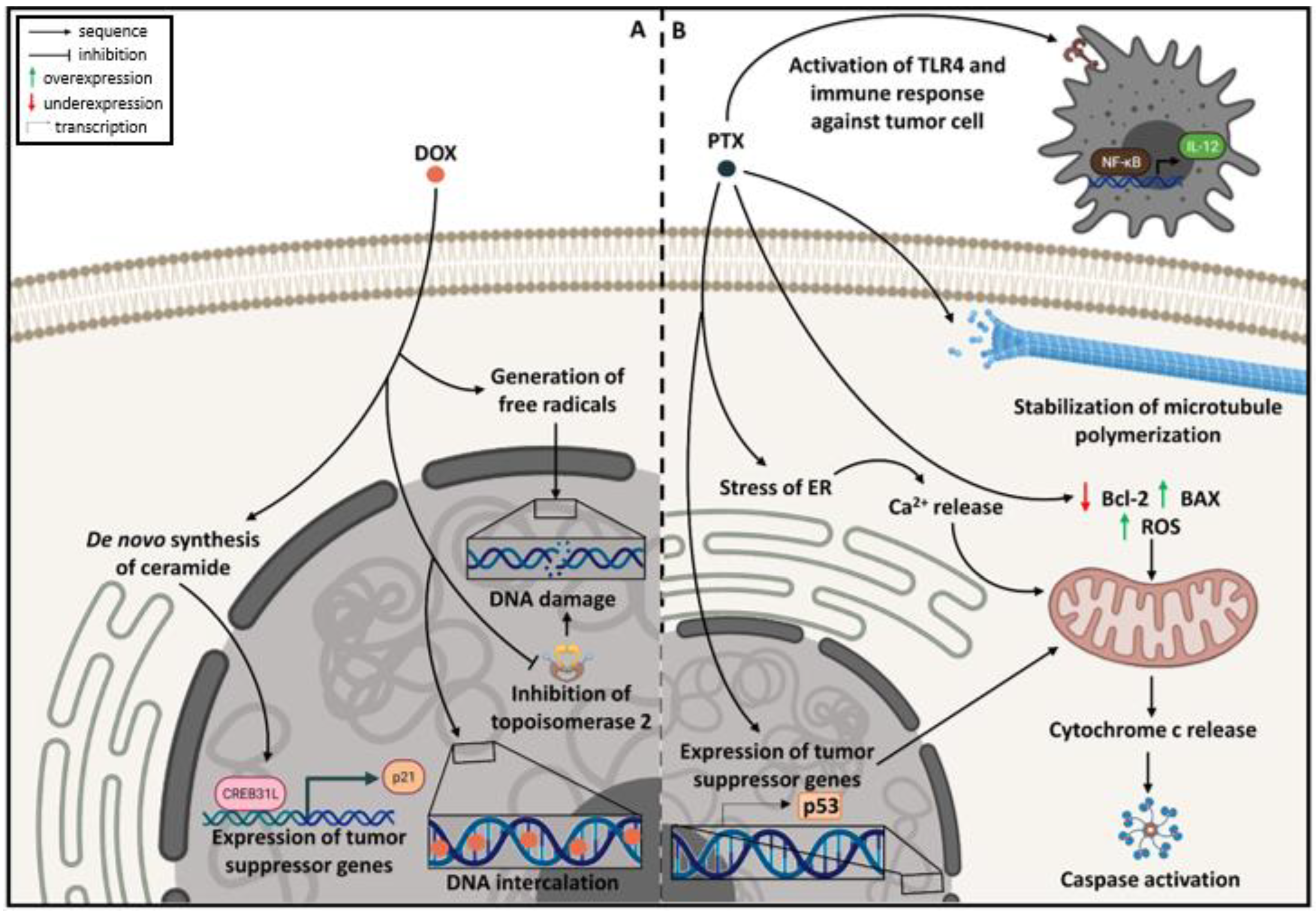
2.2. Mechanism of Action
2.3. Conventional Formulations
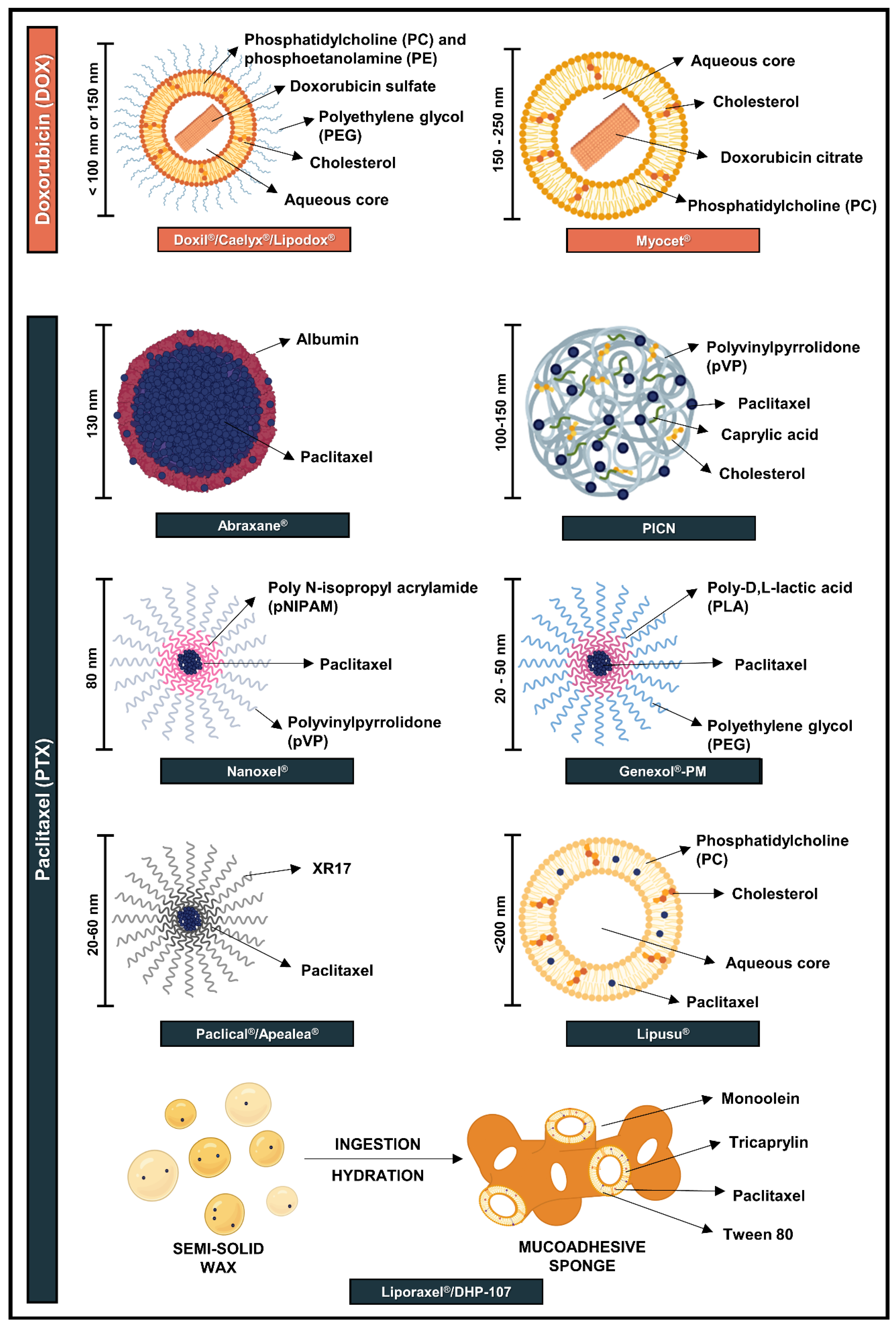
3. Approved Nanotechnology-Based Formulations for Doxorubicin (DOX) and Paclitaxel (PTX)
3.1. Nanoformulations Approved for DOX
3.2. Nanoformulations Approved for PTX
3.2.1. Polymeric Nanoparticles
3.2.2. Polymeric Micelles
3.2.3. Lipid-Based Formulations
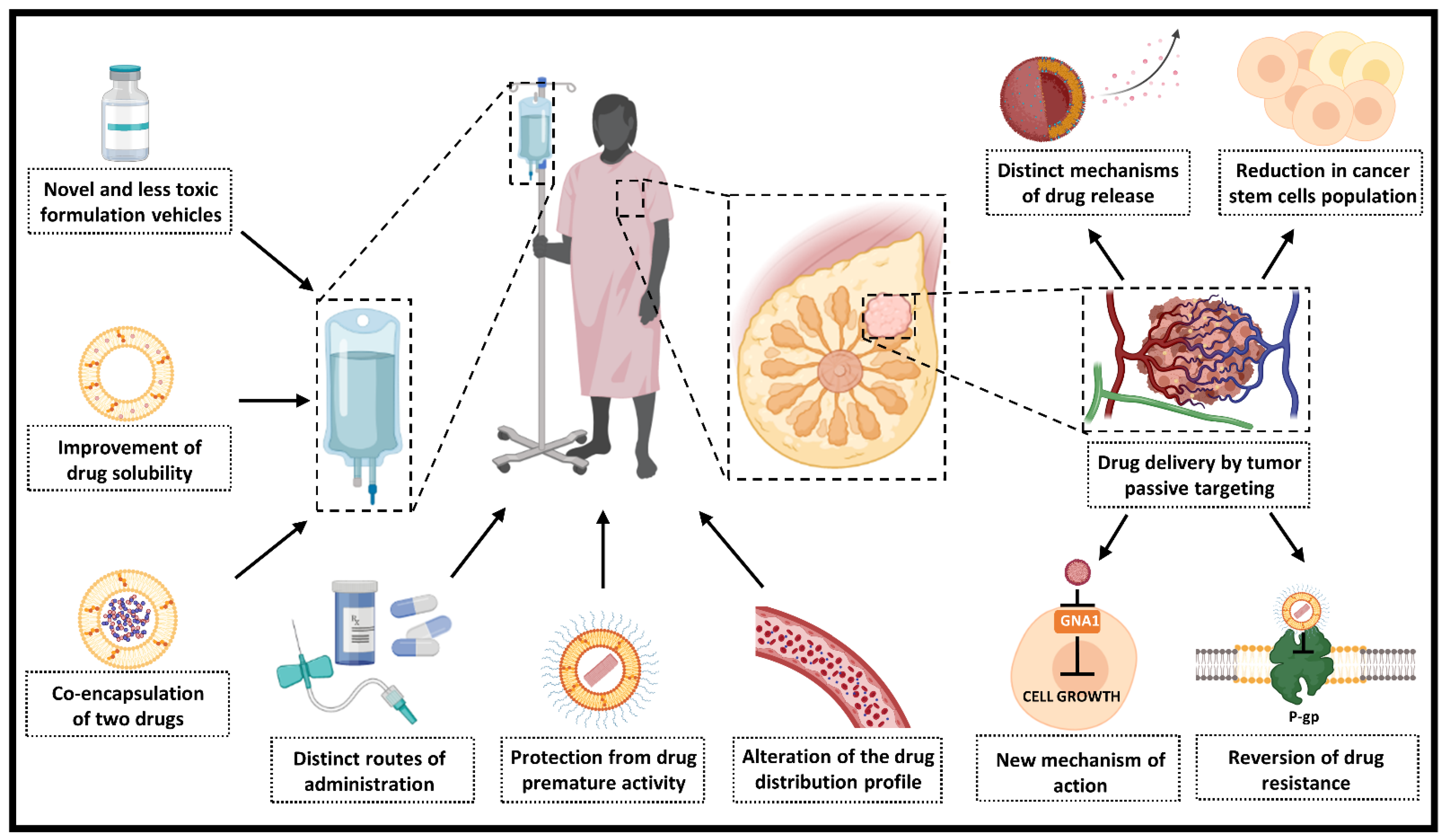
4. Contribution of Nanotechnology for Doxorubicin (DOX) and Paclitaxel (PTX) Therapeutic Use
4.1. Novel and Less Toxic Formulation Vehicles
4.2. Reduction of Drug-Related Toxic Effects and Improvement of Safety Profile
4.3. Protection from Premature Drug Activity and Alteration in the Distribution Profile
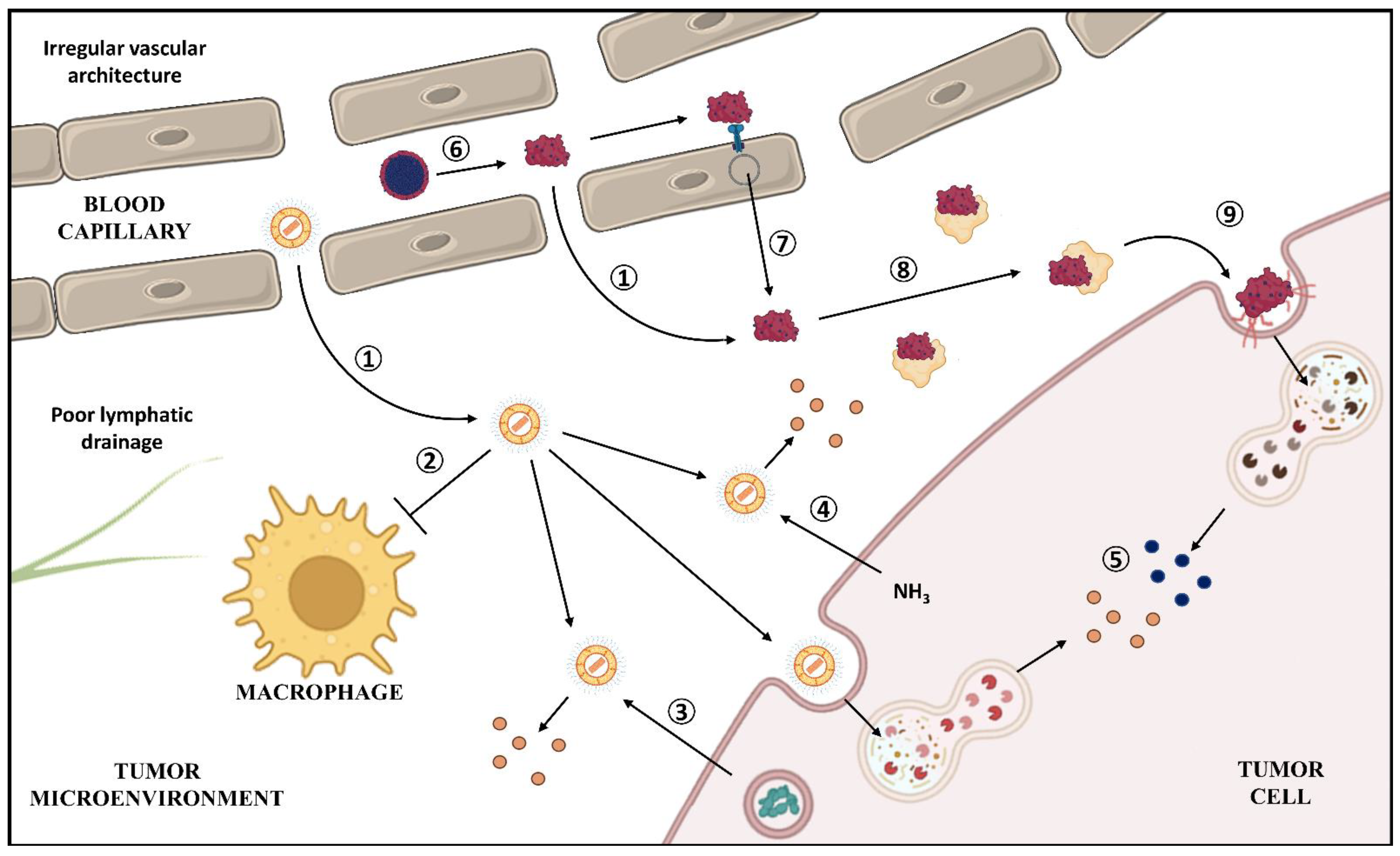
4.4. Tumor Passive Targeting
4.5. Distinct Routes of Administration
4.6. Influence on Drug Release and Mechanisms of Uptake
4.7. Reversion of Tumor Resistance to Chemotherapy
4.8. Influence on the Mechanism of Action
5. Other Possible Contributions of Nanotechnology for DOX and PTX
5.1. Drug Release by Thermal Stimuli
5.2. Tumor Active Targeting
5.3. Increase in Solubility
5.4. Co-Encapsulation of Drugs
6. Cost–Benefits
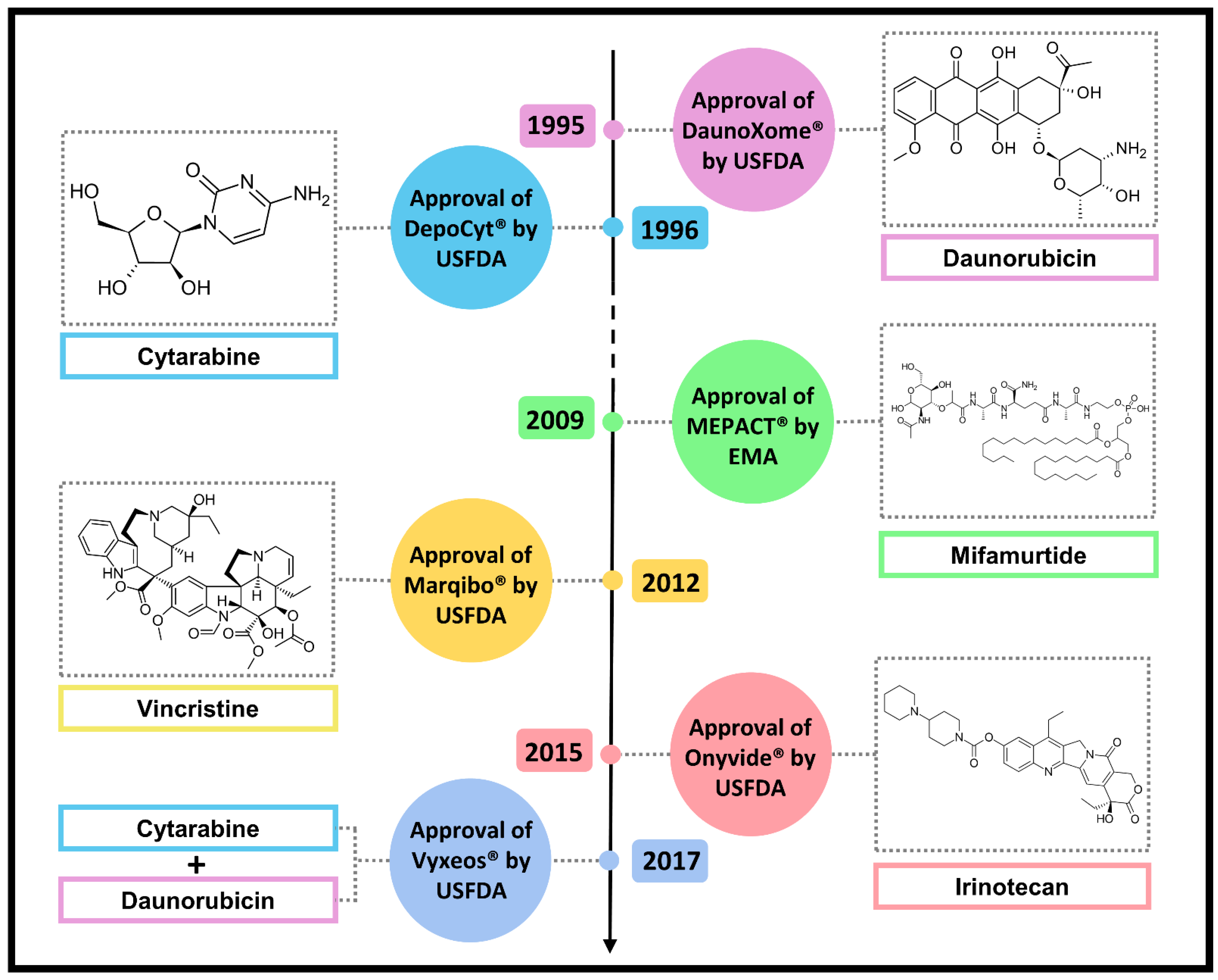
7. Other Approved Natural Anticancer Nanodrugs
7.1. DaunoXome®
7.2. DepoCyt®
7.3. MEPACT®
7.4. Marqibo®
7.5. Onivyde®
7.6. Vyxeos®
8. Nanotechnology Controversies
9. Conclusions and Further Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
References
- Chopra, B.; Dhingra, A.K. Natural products: A lead for drug discovery and development. Phytother. Res. 2021, 35, 4660–4702. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the nearly four decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, L.C.; Calil, F.A.; Machado-Neto, J.A.; Costa-Lotufo, L.V. DNA damaging agents and DNA repair: From carcinogenesis to cancer therapy. Cancer Genet. 2021, 252, 6–24. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Chen, L. Progress in research on paclitaxel and tumor immunotherapy. Cell. Mol. Biol. Lett. 2019, 24, 40. [Google Scholar] [CrossRef] [PubMed]
- Tomuleasa, C.; Braicu, C.; Irimie, A.; Craciun, L.; Berindan-Neagoe, I. Nanopharmacology in translational hematology and oncology. Int. J. Nanomed. 2014, 9, 3465–3479. [Google Scholar] [CrossRef]
- Schütz, C.A.; Juillerat-Jeanneret, L.; Mueller, H.; Lynch, I.; Riediker, M.; Consortium, N. Therapeutic nanoparticles in clinics and under clinical evaluation. Nanomedicine 2013, 8, 449–467. [Google Scholar] [CrossRef]
- US Food and Drug Administration. Considering Whether an FDA-Regulated Product Involves the Application of Nanotechnology. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/considering-whether-fda-regulated-product-involves-application-nanotechnology (accessed on 11 June 2022).
- Apolinário, A.C.; Hirata, A.S.; Anjos Miguel, R.D.; Costa-Lotufo, L.V.; Pessoa, A.; La Clair, J.J.; Fenical, W.; Lopes, L.B. Exploring the benefits of nanotechnology for cancer drugs in different stages of the drug development pipeline. Nanomedicine 2020, 15, 2539–2542. [Google Scholar] [CrossRef]
- Di Marco, A.; Cassinelli, G.; Arcamone, F. The discovery of daunorubicin. Cancer Treat. Rep. 1981, 65 (Suppl. S4), 3–8. [Google Scholar]
- Arcamone, F.; Cassinelli, G.; Fantini, G.; Grein, A.; Orezzi, P.; Pol, C.; Spalla, C. Adriamycin, 14-hydroxydaunomycin, a new antitumor antibiotic from S. peucetius var. caesius. Biotechnol. Bioeng. 1969, 11, 1101–1110. [Google Scholar] [CrossRef]
- Wani, M.C.; Taylor, H.L.; Wall, M.E.; Coggon, P.; McPhail, A.T. Plant antitumor agents. VI. The isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J. Am. Chem Soc. 1971, 93, 2325–2327. [Google Scholar] [CrossRef]
- Donehower, R.C. The clinical development of paclitaxel: A successful collaboration of academia, industry and the national cancer institute. Oncologist 1996, 1, 240–243. [Google Scholar] [CrossRef] [PubMed]
- Henry, D.W. Adriamycin. In Cancer Chemotherapy; ACS Publications: Washington, DC, USA, 1976; Volume 30, pp. 15–57. [Google Scholar] [CrossRef]
- Marco, A.; Arcamone, F. DNA complexing antibiotics: Daunomycin, adriamycin and their derivatives. Arzneimittelforschung 1975, 25, 368–374. [Google Scholar] [PubMed]
- Varela-López, A.; Battino, M.; Navarro-Hortal, M.D.; Giampieri, F.; Forbes-Hernández, T.Y.; Romero-Márquez, J.M.; Collado, R.; Quiles, J.L. An update on the mechanisms related to cell death and toxicity of doxorubicin and the protective role of nutrients. Food Chem. Toxicol. 2019, 134, 110834. [Google Scholar] [CrossRef] [PubMed]
- Denard, B.; Pavia-Jimenez, A.; Chen, W.; Williams, N.S.; Naina, H.; Collins, R.; Brugarolas, J.; Ye, J. Identification of CREB3L1 as a Biomarker Predicting Doxorubicin Treatment Outcome. PLoS ONE 2015, 10, e0129233. [Google Scholar] [CrossRef]
- Schiff, P.B.; Fant, J.; Horwitz, S.B. Promotion of microtubule assembly in vitro by taxol. Nature 1979, 277, 665–667. [Google Scholar] [CrossRef]
- Parness, J.; Horwitz, S.B. Taxol binds to polymerized tubulin in vitro. J. Cell Biol. 1981, 91, 479–487. [Google Scholar] [CrossRef]
- Horwitz, S.B. Mechanism of action of taxol. Trends Pharmacol. Sci. 1992, 13, 134–136. [Google Scholar] [CrossRef]
- Zasadil, L.M.; Andersen, K.A.; Yeum, D.; Rocque, G.B.; Wilke, L.G.; Tevaarwerk, A.J.; Raines, R.T.; Burkard, M.E.; Weaver, B.A. Cytotoxicity of paclitaxel in breast cancer is due to chromosome missegregation on multipolar spindles. Sci. Transl. Med. 2014, 6, 229ra43. [Google Scholar] [CrossRef]
- Komlodi-Pasztor, E.; Sackett, D.; Wilkerson, J.; Fojo, T. Mitosis is not a key target of microtubule agents in patient tumors. Nat. Rev. Clin. Oncol. 2011, 8, 244–250. [Google Scholar] [CrossRef]
- Weaver, B.A. How Taxol/paclitaxel kills cancer cells. Mol. Biol. Cell 2014, 25, 2677–2681. [Google Scholar] [CrossRef]
- Strobel, T.; Swanson, L.; Korsmeyer, S.; Cannistra, S.A. BAX enhances paclitaxel-induced apoptosis through a p53-independent pathway. Proc. Natl. Acad. Sci. USA 1996, 93, 14094–14099. [Google Scholar] [CrossRef] [PubMed]
- Li, M. Paclitaxel inhibits proliferation and promotes apoptosis through regulation ROS and endoplasmic reticulum stress in osteosarcoma cell. Mol. Cell. Toxicol. 2020, 16, 377–384. [Google Scholar] [CrossRef]
- Ren, X.; Zhao, B.; Chang, H.; Xiao, M.; Wu, Y.; Liu, Y. Paclitaxel suppresses proliferation and induces apoptosis through regulation of ROS and the AKT/MAPK signaling pathway in canine mammary gland tumor cells. Mol. Med. Rep. 2018, 17, 8289–8299. [Google Scholar] [CrossRef]
- Gallego-Jara, J.; Lozano-Terol, G.; Sola-Martínez, R.A.; Cánovas-Díaz, M.; de Diego Puente, T. A compressive review about taxol. Molecules 2020, 25, 5986. [Google Scholar] [CrossRef] [PubMed]
- Wanderley, C.W.; Colón, D.F.; Luiz, J.P.M.; Oliveira, F.F.; Viacava, P.R.; Leite, C.A.; Pereira, J.A.; Silva, C.M.; Silva, C.R.; Silva, R.L.; et al. Paclitaxel Reduces tumor growth by reprogramming tumor-associated macrophages to an M1 profile in a TLR4-dependent manner. Cancer Res. 2018, 78, 5891–5900. [Google Scholar] [CrossRef]
- US Food and Drug Administration. Doxorubicin Hydrochloride Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/050467s073lbl.pdf (accessed on 27 July 2022).
- Renu, K.; Abilash, V.G.; Pichhia, P.B.T.; Arunachalam, S. Molecular mechanism of doxorubicin-induced cardiomyopathy—An update. Eur. J. Pharmacol. 2018, 818, 241–253. [Google Scholar] [CrossRef]
- US Food and Drug Administration. Taxol (Paclitaxel Injection) Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/020262s049lbl.pdf (accessed on 27 July 2022).
- Wani, M.C.; Horwitz, S.B. Nature as a remarkable chemist: A personal story of the discovery and development of Taxol. Anticancer. Drugs 2014, 25, 482–487. [Google Scholar] [CrossRef]
- Gradishar, W.J. Albumin-bound paclitaxel: A next-generation taxane. Expert Opin. Pharmacother. 2006, 7, 1041–1053. [Google Scholar] [CrossRef]
- Stinchcombe, T.E. Nanoparticle albumin-bound paclitaxel: A novel Cremphor-EL-free formulation of paclitaxel. Nanomedicine 2007, 2, 415–423. [Google Scholar] [CrossRef]
- Gelderblom, H.; Verweij, J.; Nooter, K.; Sparreboom, A. Cremophor EL: The drawbacks and advantages of vehicle selection for drug formulation. Eur. J. Cancer 2001, 37, 1590–1598. [Google Scholar] [CrossRef]
- Desai, N.; Trieu, V.; Yao, Z.; Louie, L.; Ci, S.; Yang, A.; Tao, C.; De, T.; Beals, B.; Dykes, D.; et al. Increased antitumor activity, intratumor paclitaxel concentrations, and endothelial cell transport of cremophor-free, albumin-bound paclitaxel, ABI-007, compared with cremophor-based paclitaxel. Clin. Cancer Res. 2006, 12, 1317–1324. [Google Scholar] [CrossRef]
- Apolinário, A.C.; Hauschke, L.; Nunes, J.R.; Lopes, L.B. Lipid nanovesicles for biomedical applications: ‘What is in a name’? Prog. Lipid Res. 2021, 82, 101096. [Google Scholar] [CrossRef] [PubMed]
- Farokhzad, O.C.; Langer, R. Impact of nanotechnology on drug delivery. ACS Nano 2009, 3, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Lasic, D.D. Doxorubicin in sterically stabilized liposomes. Nature 1996, 380, 561–562. [Google Scholar] [CrossRef]
- US Food and Drug Administration. Doxil Label. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2007/050718s029lbl.pdf (accessed on 27 July 2022).
- European Medicines Agency. Caelyx, INN-Doxorubicin Hydrochloride. Available online: https://www.ema.europa.eu/en/documents/product-information/caelyx-pegylated-liposomal-epar-product-information_en.pdf (accessed on 27 July 2022).
- Gabizon, A.; Peretz, T.; Sulkes, A.; Amselem, S.; Ben-Yosef, R.; Ben-Baruch, N.; Catane, R.; Biran, S.; Barenholz, Y. Systemic administration of doxorubicin-containing liposomes in cancer patients: A phase I study. Eur. J. Cancer Clin. Oncol. 1989, 25, 1795–1803. [Google Scholar] [CrossRef]
- Cagel, M.; Grotz, E.; Bernabeu, E.; Moretton, M.A.; Chiappetta, D.A. Doxorubicin: Nanotechnological overviews from bench to bedside. Drug Discov. Today 2017, 22, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Barenholz, Y. Doxil®—The first FDA-approved nano-drug: Lessons learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef]
- Bulbake, U.; Doppalapudi, S.; Kommineni, N.; Khan, W. Liposomal formulations in clinical use: An updated review. Pharmaceutics 2017, 9, 12. [Google Scholar] [CrossRef]
- European Medicines Agency. Myocet, INN-Doxorubicin. Available online: https://www.ema.europa.eu/en/documents/product-information/myocet-liposomal-previously-myocet-epar-product-information_en.pdf (accessed on 27 July 2022).
- Balazsovits, J.A.; Mayer, L.D.; Bally, M.B.; Cullis, P.R.; McDonell, M.; Ginsberg, R.S.; Falk, R.E. Analysis of the effect of liposome encapsulation on the vesicant properties, acute and cardiac toxicities, and antitumor efficacy of doxorubicin. Cancer Chemother. Pharmacol. 1989, 23, 81–86. [Google Scholar] [CrossRef]
- Maiti, K.; Bhowmick, S.; Jain, P.; Zope, M.; Doshi, K.; Rajamannar, T. Comparison of physicochemical properties of generic doxorubicin HCl liposome injection with the reference listed drug. Anticancer Agents Med. Chem. 2018, 18, 597–609. [Google Scholar] [CrossRef]
- Burade, V.; Bhowmick, S.; Maiti, K.; Zalawadia, R.; Ruan, H.; Thennati, R. Lipodox® (generic doxorubicin hydrochloride liposome injection): In vivo efficacy and bioequivalence versus Caelyx® (doxorubicin hydrochloride liposome injection) in human mammary carcinoma (MX-1) xenograft and syngeneic fibrosarcoma (WEHI 164) mouse models. BMC Cancer 2017, 17, 405. [Google Scholar] [CrossRef]
- Bhowmik, S.; Bhowmick, S.; Maiti, K.; Chakra, A.; Shahi, P.; Jain, D.; Rajamannar, T. Two multicenter Phase I randomized trials to compare the bioequivalence and safety of a generic doxorubicin hydrochloride liposome injection with Doxil. Cancer Chemother. Pharmacol. 2018, 82, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Mathew, L.; Burney, M.; Nyshadham, P.; Coleman, R.L. Equivalency challenge: Evaluation of Lipodox® as the generic equivalent for Doxil® in a human ovarian cancer orthotropic mouse model. Gynecol. Oncol. 2016, 141, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Costales, A.B.; Jaffari, M.; Urbauer, D.L.; Frumovitz, M.; Kutac, C.K.; Tran, H.; Coleman, R.L. Is it equivalent? Evaluation of the clinical activity of single agent Lipodox® compared to single agent Doxil® in ovarian cancer treatment. J. Oncol. Pharm. Pract. 2016, 22, 599–604. [Google Scholar] [CrossRef]
- US Food and Drug Administration. Abraxane (Paclitaxel) Injection. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/021660s047lbl.pdf (accessed on 27 July 2022).
- Elzoghby, A.O.; Samy, W.M.; Elgindy, N.A. Albumin-based nanoparticles as potential controlled release drug delivery systems. J. Control. Release 2012, 157, 168–182. [Google Scholar] [CrossRef]
- Gradishar, W.J.; Tjulandin, S.; Davidson, N.; Shaw, H.; Desai, N.; Bhar, P.; Hawkins, M.; O’Shaughnessy, J. Phase III trial of nanoparticle albumin-bound paclitaxel compared with polyethylated castor oil-based paclitaxel in women with breast cancer. J. Clin. Oncol 2005, 23, 7794–7803. [Google Scholar] [CrossRef]
- European Medicines Agency. Abraxane. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/abraxane (accessed on 27 July 2022).
- Roy, V.; LaPlant, B.R.; Gross, G.G.; Bane, C.L.; Palmieri, F.M.; Group, N.C.C.T. Phase II trial of weekly nab (nanoparticle albumin-bound)-paclitaxel (nab-paclitaxel) (Abraxane) in combination with gemcitabine in patients with metastatic breast cancer (N0531). Ann. Oncol. 2009, 20, 449–453. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef]
- Goldstein, D.; El-Maraghi, R.H.; Hammel, P.; Heinemann, V.; Kunzmann, V.; Sastre, J.; Scheithauer, W.; Siena, S.; Tabernero, J.; Teixeira, L.; et al. nab-Paclitaxel plus gemcitabine for metastatic pancreatic cancer: Long-term survival from a phase III trial. J. Natl. Cancer Inst. 2015, 107, dju413. [Google Scholar] [CrossRef]
- Untch, M.; Jackisch, C.; Schneeweiss, A.; Conrad, B.; Aktas, B.; Denkert, C.; Eidtmann, H.; Wiebringhaus, H.; Kümmel, S.; Hilfrich, J.; et al. Nab-paclitaxel versus solvent-based paclitaxel in neoadjuvant chemotherapy for early breast cancer (GeparSepto-GBG 69): A randomised, phase 3 trial. Lancet Oncol. 2016, 17, 345–356. [Google Scholar] [CrossRef]
- Schmid, P.; Rugo, H.S.; Adams, S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Henschel, V.; Molinero, L.; Chui, S.Y.; et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): Updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 44–59. [Google Scholar] [CrossRef]
- Jain, M.M.; Gupte, S.U.; Patil, S.G.; Pathak, A.B.; Deshmukh, C.D.; Bhatt, N.; Haritha, C.; Govind Babu, K.; Bondarde, S.A.; Digumarti, R.; et al. Paclitaxel injection concentrate for nanodispersion versus nab-paclitaxel in women with metastatic breast cancer: A multicenter, randomized, comparative phase II/III study. Breast Cancer Res. Treat. 2016, 156, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Yadav, H.K.; Almokdad, A.A.; Sumia, I.; Debe, M.S. Polymer-based nanomaterials for drug-delivery carriers. In Nanocarriers for Drug Delivery; Elsevier: Amsterdam, The Netherlands, 2019; pp. 531–556. [Google Scholar]
- Majumder, N.; Das, N.G.; Das, S.K. Polymeric micelles for anticancer drug delivery. Ther. Deliv. 2020, 11, 613–635. [Google Scholar] [CrossRef] [PubMed]
- Madaan, A.; Singh, P.; Awasthi, A.; Verma, R.; Singh, A.T.; Jaggi, M.; Mishra, S.K.; Kulkarni, S.; Kulkarni, H. Efficiency and mechanism of intracellular paclitaxel delivery by novel nanopolymer-based tumor-targeted delivery system, Nanoxel(TM). Clin. Transl. Oncol. 2013, 15, 26–32. [Google Scholar] [CrossRef]
- Kim, S.C.; Kim, D.W.; Shim, Y.H.; Bang, J.S.; Oh, H.S.; Wan Kim, S.; Seo, M.H. In vivo evaluation of polymeric micellar paclitaxel formulation: Toxicity and efficacy. J. Control. Release 2001, 72, 191–202. [Google Scholar] [CrossRef]
- Ahn, H.K.; Jung, M.; Sym, S.J.; Shin, D.B.; Kang, S.M.; Kyung, S.Y.; Park, J.W.; Jeong, S.H.; Cho, E.K. A phase II trial of Cremorphor EL-free paclitaxel (Genexol-PM) and gemcitabine in patients with advanced non-small cell lung cancer. Cancer Chemother. Pharmacol. 2014, 74, 277–282. [Google Scholar] [CrossRef]
- Kim, D.W.; Kim, S.Y.; Kim, H.K.; Kim, S.W.; Shin, S.W.; Kim, J.S.; Park, K.; Lee, M.Y.; Heo, D.S. Multicenter phase II trial of Genexol-PM, a novel Cremophor-free, polymeric micelle formulation of paclitaxel, with cisplatin in patients with advanced non-small-cell lung cancer. Ann. Oncol. 2007, 18, 2009–2014. [Google Scholar] [CrossRef]
- Kim, T.Y.; Kim, D.W.; Chung, J.Y.; Shin, S.G.; Kim, S.C.; Heo, D.S.; Kim, N.K.; Bang, Y.J. Phase I and pharmacokinetic study of Genexol-PM, a cremophor-free, polymeric micelle-formulated paclitaxel, in patients with advanced malignancies. Clin. Cancer Res. 2004, 10, 3708–3716. [Google Scholar] [CrossRef]
- Zhang, X.; Jackson, J.K.; Burt, H.M. Development of amphiphilic diblock copolymers as micellar carriers of taxol. Int. J. Pharm. 1996, 132, 195–206. [Google Scholar] [CrossRef]
- European Medicines Agency. Apealea, INN-Paclitaxel. Available online: https://www.ema.europa.eu/en/documents/product-information/apealea-epar-product-information_en.pdf (accessed on 11 June 2022).
- Borgå, O.; Henriksson, R.; Bjermo, H.; Lilienberg, E.; Heldring, N.; Loman, N. Maximum tolerated dose and pharmacokinetics of paclitaxel micellar in patients with recurrent malignant solid tumours: A dose-escalation study. Adv. Ther. 2019, 36, 1150–1163. [Google Scholar] [CrossRef]
- Wang, F.; Porter, M.; Konstantopoulos, A.; Zhang, P.; Cui, H. Preclinical development of drug delivery systems for paclitaxel-based cancer chemotherapy. J. Control. Release 2017, 267, 100–118. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.; Chung, H.J.; Hong, J.W.; Yun, C.W.; Chung, H. Absorption mechanism of DHP107, an oral paclitaxel formulation that forms a hydrated lipidic sponge phase. Acta Pharmacol. Sin. 2017, 38, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.; Ko, M.K.; Park, Y.E.; Hong, J.W.; Lee, I.-H.; Chung, H.J.; Chung, H. Effect of paclitaxel content in the DHP107 oral formulation on oral bioavailability and antitumor activity. J. Drug Deliv. Sci. Technol. 2018, 48, 183–192. [Google Scholar] [CrossRef]
- Geever, L.M.; Devine, D.M.; Nugent, M.J.; Kennedy, J.E.; Lyons, J.G.; Higginbotham, C.L. The synthesis, characterisation, phase behaviour and swelling of temperature sensitive physically crosslinked poly (1-vinyl-2-pyrrolidinone)/poly (N-isopropylacrylamide) hydrogels. Eur. Polym. J. 2006, 42, 69–80. [Google Scholar] [CrossRef]
- Gagliardi, A.; Giuliano, E.; Venkateswararao, E.; Fresta, M.; Bulotta, S.; Awasthi, V.; Cosco, D. Biodegradable Polymeric Nanoparticles for Drug Delivery to Solid Tumors. Front. Pharmacol. 2021, 12, 601626. [Google Scholar] [CrossRef]
- Wacker, M. Nanocarriers for intravenous injection—The long hard road to the market. Int. J. Pharm. 2013, 457, 50–62. [Google Scholar] [CrossRef]
- O’Brien, M.E.; Wigler, N.; Inbar, M.; Rosso, R.; Grischke, E.; Santoro, A.; Catane, R.; Kieback, D.G.; Tomczak, P.; Ackland, S.P.; et al. Reduced cardiotoxicity and comparable efficacy in a phase III trial of pegylated liposomal doxorubicin HCl (CAELYX/Doxil) versus conventional doxorubicin for first-line treatment of metastatic breast cancer. Ann. Oncol. 2004, 15, 440–449. [Google Scholar] [CrossRef]
- Judson, I.; Radford, J.A.; Harris, M.; Blay, J.Y.; van Hoesel, Q.; le Cesne, A.; van Oosterom, A.T.; Clemons, M.J.; Kamby, C.; Hermans, C.; et al. Randomised phase II trial of pegylated liposomal doxorubicin (DOXIL/CAELYX) versus doxorubicin in the treatment of advanced or metastatic soft tissue sarcoma: A study by the EORTC Soft Tissue and Bone Sarcoma Group. Eur. J. Cancer 2001, 37, 870–877. [Google Scholar] [CrossRef]
- Batist, G.; Ramakrishnan, G.; Rao, C.S.; Chandrasekharan, A.; Gutheil, J.; Guthrie, T.; Shah, P.; Khojasteh, A.; Nair, M.K.; Hoelzer, K.; et al. Reduced cardiotoxicity and preserved antitumor efficacy of liposome-encapsulated doxorubicin and cyclophosphamide compared with conventional doxorubicin and cyclophosphamide in a randomized, multicenter trial of metastatic breast cancer. J. Clin. Oncol. 2001, 19, 1444–1454. [Google Scholar] [CrossRef]
- Harris, L.; Batist, G.; Belt, R.; Rovira, D.; Navari, R.; Azarnia, N.; Welles, L.; Winer, E.; Group, T.D.-S. Liposome-encapsulated doxorubicin compared with conventional doxorubicin in a randomized multicenter trial as first-line therapy of metastatic breast carcinoma. Cancer 2002, 94, 25–36. [Google Scholar] [CrossRef]
- Batist, G.; Harris, L.; Azarnia, N.; Lee, L.W.; Daza-Ramirez, P. Improved anti-tumor response rate with decreased cardiotoxicity of non-pegylated liposomal doxorubicin compared with conventional doxorubicin in first-line treatment of metastatic breast cancer in patients who had received prior adjuvant doxorubicin: Results of a retrospective analysis. Anticancer Drugs 2006, 17, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, N.K.; Desai, N.; Legha, S.; Soon-Shiong, P.; Theriault, R.L.; Rivera, E.; Esmaeli, B.; Ring, S.E.; Bedikian, A.; Hortobagyi, G.N.; et al. Phase I and pharmacokinetic study of ABI-007, a Cremophor-free, protein-stabilized, nanoparticle formulation of paclitaxel. Clin. Cancer Res. 2002, 8, 1038–1044. [Google Scholar] [PubMed]
- Martschick, A.; Sehouli, J.; Patzelt, A.; Richter, H.; Jacobi, U.; Oskay-Ozcelik, G.; Sterry, W.; Lademann, J. The pathogenetic mechanism of anthracycline-induced palmar-plantar erythrodysesthesia. Anticancer Res. 2009, 29, 2307–2313. [Google Scholar] [PubMed]
- Von Moos, R.; Thuerlimann, B.J.; Aapro, M.; Rayson, D.; Harrold, K.; Sehouli, J.; Scotte, F.; Lorusso, D.; Dummer, R.; Lacouture, M.E.; et al. Pegylated liposomal doxorubicin-associated hand-foot syndrome: Recommendations of an international panel of experts. Eur. J. Cancer 2008, 44, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Szebeni, J.; Muggia, F.; Gabizon, A.; Barenholz, Y. Activation of complement by therapeutic liposomes and other lipid excipient-based therapeutic products: Prediction and prevention. Adv. Drug Deliv. Rev. 2011, 63, 1020–1030. [Google Scholar] [CrossRef]
- Neun, B.W.; Barenholz, Y.; Szebeni, J.; Dobrovolskaia, M.A. Understanding the role of anti-PEG antibodies in the complement activation by doxil in vitro. Molecules 2018, 23, 1700. [Google Scholar] [CrossRef]
- Chen, E.; Chen, B.M.; Su, Y.C.; Chang, Y.C.; Cheng, T.L.; Barenholz, Y.; Roffler, S.R. Premature drug release from polyethylene glycol (PEG)-coated liposomal doxorubicin. ACS Nano 2020, 14, 7808–7822. [Google Scholar] [CrossRef]
- Bavli, Y.; Winkler, I.; Chen, B.M.; Roffler, S.; Cohen, R.; Szebeni, J.; Barenholz, Y. Doxebo (doxorubicin-free Doxil-like liposomes) is safe to use as a pre-treatment to prevent infusion reactions to PEGylated nanodrugs. J. Control. Release 2019, 306, 138–148. [Google Scholar] [CrossRef]
- Soloman, R.; Gabizon, A.A. Clinical pharmacology of liposomal anthracyclines: Focus on pegylated liposomal Doxorubicin. Clin. Lymphoma Myeloma 2008, 8, 21–32. [Google Scholar] [CrossRef]
- Scherphof, G.; Roerdink, F.; Waite, M.; Parks, J. Disintegration of phosphatidylcholine liposomes in plasma as a result of interaction with high-density lipoproteins. Biochim. Biophys. Acta 1978, 542, 296–307. [Google Scholar] [CrossRef]
- Kirby, C.; Clarke, J.; Gregoriadis, G. Cholesterol content of small unilamellar liposomes controls phospholipid loss to high density lipoproteins in the presence of serum. FEBS Lett. 1980, 111, 324–328. [Google Scholar] [CrossRef]
- Swenson, C.E.; Bolcsak, L.E.; Batist, G.; Guthrie, T.H.; Tkaczuk, K.H.; Boxenbaum, H.; Welles, L.; Chow, S.C.; Bhamra, R.; Chaikin, P. Pharmacokinetics of doxorubicin administered i.v. as Myocet (TLC D-99; liposome-encapsulated doxorubicin citrate) compared with conventional doxorubicin when given in combination with cyclophosphamide in patients with metastatic breast cancer. Anticancer Drugs 2003, 14, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Szebeni, J.; Muggia, F.M.; Alving, C.R. Complement activation by Cremophor EL as a possible contributor to hypersensitivity to paclitaxel: An in vitro study. J. Natl. Cancer Inst. 1998, 90, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Sparreboom, A.; Scripture, C.D.; Trieu, V.; Williams, P.J.; De, T.; Yang, A.; Beals, B.; Figg, W.D.; Hawkins, M.; Desai, N. Comparative preclinical and clinical pharmacokinetics of a cremophor-free, nanoparticle albumin-bound paclitaxel (ABI-007) and paclitaxel formulated in Cremophor (Taxol). Clin. Cancer Res. 2005, 11, 4136–4143. [Google Scholar] [CrossRef]
- Strebhardt, K.; Ullrich, A. Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat. Rev. Cancer 2008, 8, 473–480. [Google Scholar] [CrossRef]
- Gabizon, A.; Catane, R.; Uziely, B.; Kaufman, B.; Safra, T.; Cohen, R.; Martin, F.; Huang, A.; Barenholz, Y. Prolonged circulation time and enhanced accumulation in malignant exudates of doxorubicin encapsulated in polyethylene-glycol coated liposomes. Cancer Res. 1994, 54, 987–992. [Google Scholar]
- Koudelka, S.; Turánek, J. Liposomal paclitaxel formulations. J. Control. Release 2012, 163, 322–334. [Google Scholar] [CrossRef]
- Werner, M.E.; Cummings, N.D.; Sethi, M.; Wang, E.C.; Sukumar, R.; Moore, D.T.; Wang, A.Z. Preclinical evaluation of Genexol-PM, a nanoparticle formulation of paclitaxel, as a novel radiosensitizer for the treatment of non-small cell lung cancer. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 463–468. [Google Scholar] [CrossRef]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef]
- Yardley, D.A. nab-Paclitaxel mechanisms of action and delivery. J. Control. Release 2013, 170, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Simionescu, M.; Gafencu, A.; Antohe, F. Transcytosis of plasma macromolecules in endothelial cells: A cell biological survey. Microsc. Res. Tech. 2002, 57, 269–288. [Google Scholar] [CrossRef] [PubMed]
- John, T.A.; Vogel, S.M.; Tiruppathi, C.; Malik, A.B.; Minshall, R.D. Quantitative analysis of albumin uptake and transport in the rat microvessel endothelial monolayer. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L187–L196. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.W.; Lee, I.H.; Kwak, Y.H.; Park, Y.T.; Sung, H.C.; Kwon, I.C.; Chung, H. Efficacy and tissue distribution of DHP107, an oral paclitaxel formulation. Mol. Cancer Ther. 2007, 6, 3239–3247. [Google Scholar] [CrossRef] [PubMed]
- Murry, D.J.; Blaney, S.M. Clinical pharmacology of encapsulated sustained-release cytarabine. Ann. Pharmacother. 2000, 34, 1173–1178. [Google Scholar] [CrossRef]
- Silverman, L.; Barenholz, Y. In vitro experiments showing enhanced release of doxorubicin from Doxil® in the presence of ammonia may explain drug release at tumor site. Nanomedicine 2015, 11, 1841–1850. [Google Scholar] [CrossRef]
- Seynhaeve, A.L.B.; Dicheva, B.M.; Hoving, S.; Koning, G.A.; Ten Hagen, T.L.M. Intact Doxil is taken up intracellularly and released doxorubicin sequesters in the lysosome: Evaluated by in vitro/in vivo live cell imaging. J. Control. Release 2013, 172, 330–340. [Google Scholar] [CrossRef]
- Podhajcer, O.L.; Benedetti, L.; Girotti, M.R.; Prada, F.; Salvatierra, E.; Llera, A.S. The role of the matricellular protein SPARC in the dynamic interaction between the tumor and the host. Cancer Metastasis Rev. 2008, 27, 523–537. [Google Scholar] [CrossRef]
- Kiessling, F.; Fink, C.; Hansen, M.; Bock, M.; Sinn, H.; Schrenk, H.H.; Krix, M.; Egelhof, T.; Fusenig, N.E.; Delorme, S. Magnetic resonance imaging of nude mice with heterotransplanted high-grade squamous cell carcinomas: Use of a low-loaded, covalently bound Gd-Hsa conjugate as contrast agent with high tumor affinity. Investig. Radiol. 2002, 37, 193–198. [Google Scholar] [CrossRef]
- Chatterjee, M.; Ben-Josef, E.; Robb, R.; Vedaie, M.; Seum, S.; Thirumoorthy, K.; Palanichamy, K.; Harbrecht, M.; Chakravarti, A.; Williams, T.M. Caveolae-mediated endocytosis is critical for albumin cellular uptake and response to albumin-bound chemotherapy. Cancer Res. 2017, 77, 5925–5937. [Google Scholar] [CrossRef]
- Xue, X.; Liang, X.J. Overcoming drug efflux-based multidrug resistance in cancer with nanotechnology. Chin. J. Cancer 2012, 31, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Riganti, C.; Voena, C.; Kopecka, J.; Corsetto, P.A.; Montorfano, G.; Enrico, E.; Costamagna, C.; Rizzo, A.M.; Ghigo, D.; Bosia, A. Liposome-encapsulated doxorubicin reverses drug resistance by inhibiting P-glycoprotein in human cancer cells. Mol. Pharm. 2011, 8, 683–700. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.S.; Lamprecht, A. P-glycoprotein inhibition of drug resistant cell lines by nanoparticles. Drug Dev. Ind. Pharm. 2016, 42, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Mattingly, C.A.; Tseng, M.T.; Cho, M.J.; Liu, Y.; Adams, V.R.; Mumper, R.J. Doxorubicin and paclitaxel-loaded lipid-based nanoparticles overcome multidrug resistance by inhibiting P-glycoprotein and depleting ATP. Cancer Res. 2009, 69, 3918–3926. [Google Scholar] [CrossRef]
- Niazi, M.; Zakeri-Milani, P.; Najafi Hajivar, S.; Soleymani Goloujeh, M.; Ghobakhlou, N.; Shahbazi Mojarrad, J.; Valizadeh, H. Nano-based strategies to overcome p-glycoprotein-mediated drug resistance. Expert Opin. Drug Metab. Toxicol. 2016, 12, 1021–1033. [Google Scholar] [CrossRef]
- Zhao, M.; Li, H.; Ma, Y.; Gong, H.; Yang, S.; Fang, Q.; Hu, Z. Nanoparticle abraxane possesses impaired proliferation in A549 cells due to the underexpression of glucosamine 6-phosphate N-acetyltransferase 1 (GNPNAT1/GNA1). Int. J. Nanomed. 2017, 12, 1685–1697. [Google Scholar] [CrossRef]
- Yuan, H.; Guo, H.; Luan, X.; He, M.; Li, F.; Burnett, J.; Truchan, N.; Sun, D. Albumin nanoparticle of paclitaxel (abraxane) decreases while taxol increases breast cancer stem cells in treatment of triple negative breast cancer. Mol. Pharm. 2020, 17, 2275–2286. [Google Scholar] [CrossRef]
- Godlewska, M.; Majkowska-Pilip, A.; Stachurska, A.; Biernat, J.F.; Gaweł, D.; Nazaruk, E. Voltammetric and biological studies of folate-targeted non-lamellar lipid mesophases. Electrochim. Acta 2019, 299, 1–11. [Google Scholar] [CrossRef]
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the clinic: An update. Bioeng. Transl. Med. 2019, 4, e10143. [Google Scholar] [CrossRef]
- May, J.P.; Li, S.D. Hyperthermia-induced drug targeting. Expert Opin. Drug Deliv. 2013, 10, 511–527. [Google Scholar] [CrossRef]
- Pérez-Herrero, E.; Fernández-Medarde, A. Advanced targeted therapies in cancer: Drug nanocarriers, the future of chemotherapy. Eur. J. Pharm. Biopharm. 2015, 93, 52–79. [Google Scholar] [CrossRef] [PubMed]
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the clinic: An update post COVID-19 vaccines. Bioeng. Transl. Med. 2021, 6, e10246. [Google Scholar] [CrossRef] [PubMed]
- Elzoghby, A.O.; Abdelmoneem, M.A.; Hassanin, I.A.; Abd Elwakil, M.M.; Elnaggar, M.A.; Mokhtar, S.; Fang, J.Y.; Elkhodairy, K.A. Lactoferrin, a multi-functional glycoprotein: Active therapeutic, drug nanocarrier & targeting ligand. Biomaterials 2020, 263, 120355. [Google Scholar] [CrossRef] [PubMed]
- Shankaranarayanan, J.S.; Kanwar, J.R.; Al-Juhaishi, A.J.; Kanwar, R.K. Doxorubicin conjugated to immunomodulatory anticancer lactoferrin displays improved cytotoxicity overcoming prostate cancer chemo resistance and inhibits tumour development in TRAMP mice. Sci. Rep. 2016, 6, 32062. [Google Scholar] [CrossRef]
- Miao, D.; Jiang, M.; Liu, Z.; Gu, G.; Hu, Q.; Kang, T.; Song, Q.; Yao, L.; Li, W.; Gao, X.; et al. Co-administration of dual-targeting nanoparticles with penetration enhancement peptide for antiglioblastoma therapy. Mol. Pharm. 2014, 11, 90–101. [Google Scholar] [CrossRef]
- Junghanns, J.U.; Müller, R.H. Nanocrystal technology, drug delivery and clinical applications. Int. J. Nanomed. 2008, 3, 295–309. [Google Scholar] [CrossRef]
- Li, W.; Li, Z.; Wei, L.; Zheng, A. Evaluation of paclitaxel nanocrystals in vitro and in vivo. Drug Res. 2018, 68, 205–212. [Google Scholar] [CrossRef]
- Sesarman, A.; Muntean, D.; Abrudan, B.; Tefas, L.; Sylvester, B.; Licarete, E.; Rauca, V.; Luput, L.; Patras, L.; Banciu, M.; et al. Improved pharmacokinetics and reduced side effects of doxorubicin therapy by liposomal co-encapsulation with curcumin. J. Liposome Res. 2021, 31, 1–10. [Google Scholar] [CrossRef]
- Giacone, D.V.; Carvalho, V.F.M.; Costa, S.K.P.; Lopes, L.B. Evidence that P-glycoprotein inhibitor (Elacridar)-loaded nanocarriers improve epidermal targeting of an anticancer drug via absorptive cutaneous transporters inhibition. J. Pharm. Sci. 2018, 107, 698–705. [Google Scholar] [CrossRef]
- Carvalho, V.F.M.; Migotto, A.; Giacone, D.V.; de Lemos, D.P.; Zanoni, T.B.; Maria-Engler, S.S.; Costa-Lotufo, L.V.; Lopes, L.B. Co-encapsulation of paclitaxel and C6 ceramide in tributyrin-containing nanocarriers improve co-localization in the skin and potentiate cytotoxic effects in 2D and 3D models. Eur. J. Pharm. Sci. 2017, 109, 131–143. [Google Scholar] [CrossRef]
- Blair, H.A. Daunorubicin/cytarabine liposome: A review in acute myeloid Leukaemia. Drugs 2018, 78, 1903–1910. [Google Scholar] [CrossRef] [PubMed]
- Drugs.com. Prices, Coupons & Patient Assistance Programs. Available online: https://www.drugs.com/price-guide/ (accessed on 11 June 2022).
- Milewska, S.; Niemirowicz-Laskowska, K.; Siemiaszko, G.; Nowicki, P.; Wilczewska, A.Z.; Car, H. Current trends and challenges in pharmacoeconomic aspects of nanocarriers as drug delivery systems for cancer treatment. Int. J. Nanomed. 2021, 16, 6593–6644. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.L.; Calhoun, E.A. Pharmacoeconomics of liposomal anthracycline therapy. Semin. Oncol. 2004, 31, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Alba, E.; Ciruelos, E.; López, R.; López-Vega, J.M.; Lluch, A.; Martín, M.; Muñoz, M.; Sánchez-Rovira, P.; Seguí, M.; Liria, M.R.; et al. Cost-utility analysis of nanoparticle albumin-bound paclitaxel versus paclitaxel in monotherapy in pretreated metastatic breast cancer in Spain. Expert Rev. Pharmacoecon. Outcomes Res. 2013, 13, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, A.G.; Zotchev, S.B.; Dirsch, V.M.; Supuran, C.T.; Taskforce, I.N.P.S. Natural products in drug discovery: Advances and opportunities. Nat. Rev. Drug Discov. 2021, 20, 200–216. [Google Scholar] [CrossRef]
- Fox, J.L. FDA advisors okay NeXstar’s DaunoXome. Bio/Technology 1995, 13, 635–636. [Google Scholar] [CrossRef]
- Schwartsmann, G.; Brondani da Rocha, A.; Berlinck, R.G.; Jimeno, J. Marine organisms as a source of new anticancer agents. Lancet Oncol. 2001, 2, 221–225. [Google Scholar] [CrossRef]
- Betcher, D.L.; Burnham, N. Cytarabine. J. Pediatr. Oncol. Nurs. 1990, 7, 154–157. [Google Scholar] [CrossRef]
- Ando, K.; Mori, K.; Corradini, N.; Redini, F.; Heymann, D. Mifamurtide for the treatment of nonmetastatic osteosarcoma. Expert Opin. Pharmacother. 2011, 12, 285–292. [Google Scholar] [CrossRef]
- Anselmo, A.C.; Mitragotri, S. Nanoparticles in the clinic. Bioeng Transl Med. 2016, 1, 10–29. [Google Scholar] [CrossRef]
- Gidding, C.E.; Kellie, S.J.; Kamps, W.A.; de Graaf, S.S. Vincristine revisited. Crit. Rev. Oncol. Hematol. 1999, 29, 267–287. [Google Scholar] [CrossRef]
- Silverman, J.A.; Deitcher, S.R. Marqibo® (vincristine sulfate liposome injection) improves the pharmacokinetics and pharmacodynamics of vincristine. Cancer Chemother. Pharmacol. 2013, 71, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Carnevale, J.; Ko, A.H. MM-398 (nanoliposomal irinotecan): Emergence of a novel therapy for the treatment of advanced pancreatic cancer. Future Oncol. 2016, 12, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Apolinário, A.C.; Salata, G.C.; Bianco, A.F.; Fukumori, C.; Lopes, L.B. Abrindo a caixa de pandora dos nanomedicamentos: Há realmente muito mais ‘espaço lá embaixo’. Química Nova 2020, 43, 212–225. [Google Scholar] [CrossRef]
- European Medicines Agency. Nanotechnology. Available online: https://www.ema.europa.eu/en/glossary/nanotechnology (accessed on 27 July 2022).
- Bremer-Hoffmann, S.; Halamoda-Kenzaoui, B.; Borgos, S.E. Identification of regulatory needs for nanomedicines. J. Interdiscip. Nanomed. 2018, 3, 4–15. [Google Scholar] [CrossRef]
- US Food and Drug Administration. Drug Products, Including Biological Products, That Contain Nanomaterials. Available online: https://www.fda.gov/media/157812/download (accessed on 27 July 2022).
- Wibroe, P.P.; Ahmadvand, D.; Oghabian, M.A.; Yaghmur, A.; Moghimi, S.M. An integrated assessment of morphology, size, and complement activation of the PEGylated liposomal doxorubicin products Doxil®, Caelyx®, DOXOrubicin, and SinaDoxosome. J. Control. Release 2016, 221, 1–8. [Google Scholar] [CrossRef]
- Zhao, M.; Lei, C.; Yang, Y.; Bu, X.; Ma, H.; Gong, H.; Liu, J.; Fang, X.; Hu, Z.; Fang, Q. Abraxane, the nanoparticle formulation of paclitaxel can induce drug resistance by up-regulation of P-gp. PLoS ONE 2015, 10, e0131429. [Google Scholar] [CrossRef]
- Nichols, J.W.; Bae, Y.H. EPR: Evidence and fallacy. J. Control. Release 2014, 190, 451–464. [Google Scholar] [CrossRef]
- Luan, X.; Yuan, H.; Song, Y.; Hu, H.; Wen, B.; He, M.; Zhang, H.; Li, Y.; Li, F.; Shu, P.; et al. Reappraisal of anticancer nanomedicine design criteria in three types of preclinical cancer models for better clinical translation. Biomaterials 2021, 275, 120910. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Formulation | Administration Route | Therapeutic Applications | Recommended Dose | Maximum Tolerated Dose (MTD) |
|---|---|---|---|---|---|
| DOX | Adriamycin® | Intravenous infusion | A wide variety of tumors (hematologic, solid, and neural tumors) | 40–75 mg/m2 | * 500 mg/m² |
| Doxil®/Caelyx®/Lipodox® | AIDS-related Kaposi’s sarcoma, multiple myeloma, and ovarian and breast cancers | 20–50 mg/m2 | 120 mg/m2 | ||
| Myocet® | Metastatic breast cancer | 60–75 mg/m2 | 75–135 mg/m2 | ||
| PTX | Taxol® | Intravenous infusion | Ovarian and breast cancers | 135–175 mg/m2 | 240 mg/m2 |
| Abraxane® | Breast and pancreas cancers and NSCLC | 260 mg/m2 | 300 mg/m2 | ||
| PICN | Breast cancer | 260 mg/m2 | 325 mg/m2 | ||
| Genexol®-PM | Breast and pancreas cancers, NSCLC, AIDS-related Kaposi’s sarcoma | 300–390 mg/m2 | 390 mg/m2 | ||
| Nanoxel® | Breast cancer and NSCLC | 330 mg/m2 | 375 mg/m2 | ||
| Paclical®/Apealea® | Ovarian cancer | 250 mg/m2 | 250 mg/m2 | ||
| Lipusu® | NSCLC, ovarian and breast cancers | 175 mg/m2 | no data | ||
| Liporaxel® | Oral administration | Gastric cancer | 200 mg/m2 | 600 mg/m2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miguel, R.d.A.; Hirata, A.S.; Jimenez, P.C.; Lopes, L.B.; Costa-Lotufo, L.V. Beyond Formulation: Contributions of Nanotechnology for Translation of Anticancer Natural Products into New Drugs. Pharmaceutics 2022, 14, 1722. https://doi.org/10.3390/pharmaceutics14081722
Miguel RdA, Hirata AS, Jimenez PC, Lopes LB, Costa-Lotufo LV. Beyond Formulation: Contributions of Nanotechnology for Translation of Anticancer Natural Products into New Drugs. Pharmaceutics. 2022; 14(8):1722. https://doi.org/10.3390/pharmaceutics14081722
Chicago/Turabian StyleMiguel, Rodrigo dos A., Amanda S. Hirata, Paula C. Jimenez, Luciana B. Lopes, and Leticia V. Costa-Lotufo. 2022. "Beyond Formulation: Contributions of Nanotechnology for Translation of Anticancer Natural Products into New Drugs" Pharmaceutics 14, no. 8: 1722. https://doi.org/10.3390/pharmaceutics14081722